
3β-Hydroxy-5β-hydroxy-B-norcholestane-6β-carboxaldehyde (SEC-B) Induces Proinflammatory Activation of Human Endothelial Cells Associated with Nitric Oxide Production and Endothelial Nitric Oxide Synthase/Caveolin-1 Dysregulation

 , , , , ,
, , , , ,  ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatments
2.2. WST-8 Cell Viability Assay
2.3. Wound Healing Assay (Scratch Assay)
2.4. Intracellular ROS Evaluation
2.5. Measurement of NO Production
2.6. ELISA Immunoassay
2.7. Cell Extracts and Western Immunoblotting Analysis
2.8. Caveolin-1 (Cav-1) Immunofluorescence
2.9. Real-Time PCR
2.10. Flow-Cytometric Analysis of Supravital Propidium Iodide (PI) Staining and ICAM-1 Expression
2.11. Cell Adhesion Assay
2.12. Statistical Analysis
3. Results
3.1. Effect of SEC-B on HMEC-1 Cell Viability and Damage
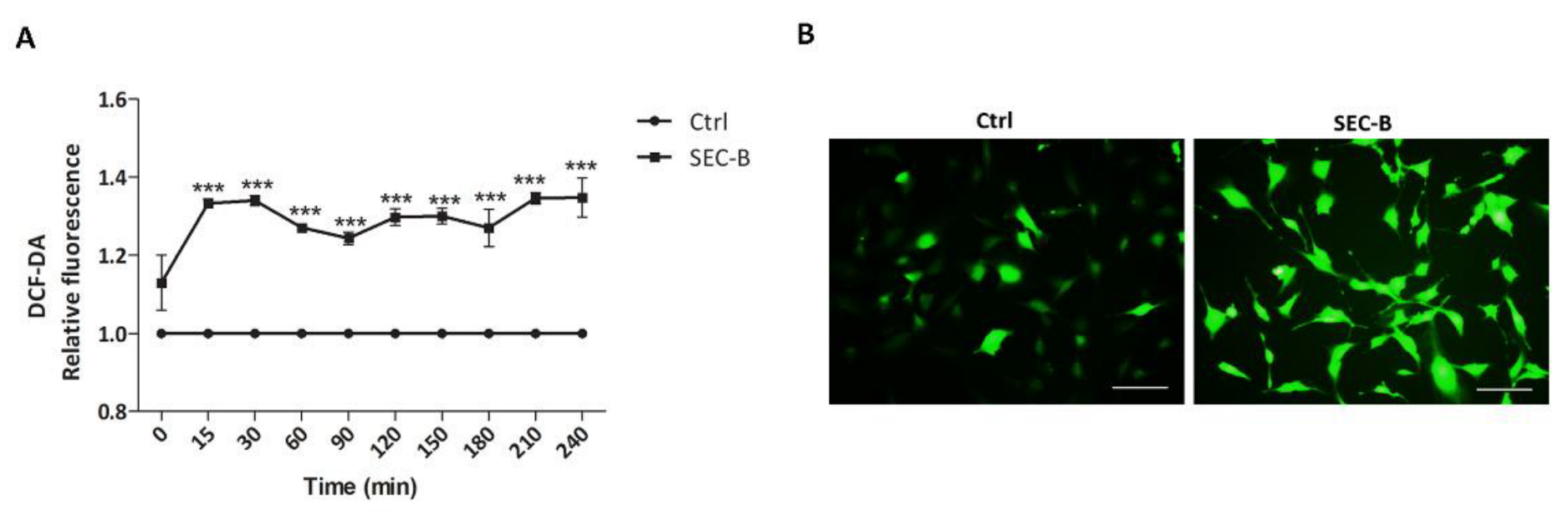
3.2. SEC-B Increases Intracellular ROS Production
3.3. SEC-B Increases Intracellular NO Content and Modulates eNOS/Cav-1 Expression
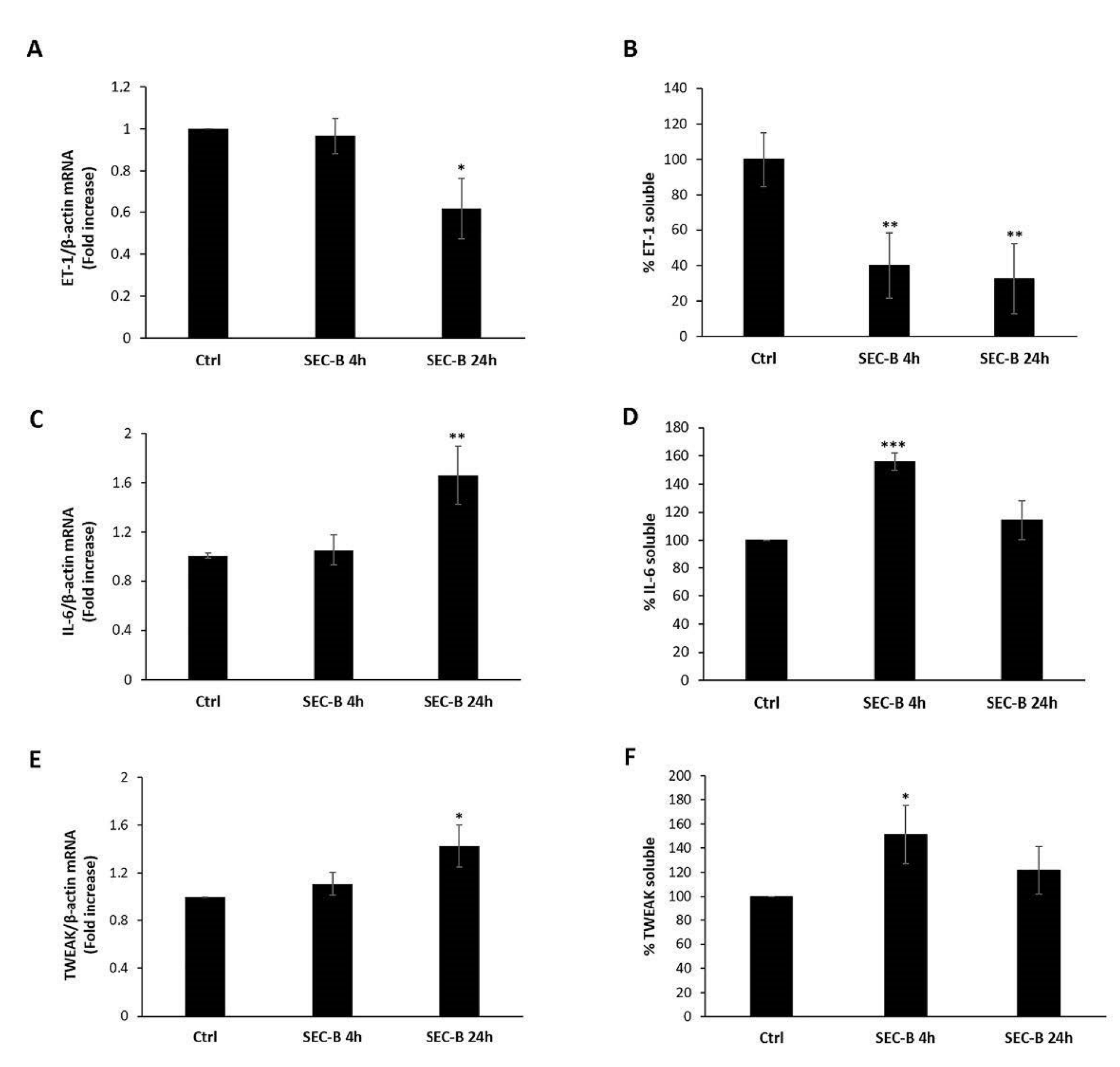
3.4. SEC-B Down-Regulates ET-1 and Up-Regulate Pro-Inflammatory Cytokines IL-6 and TWEAK Expression
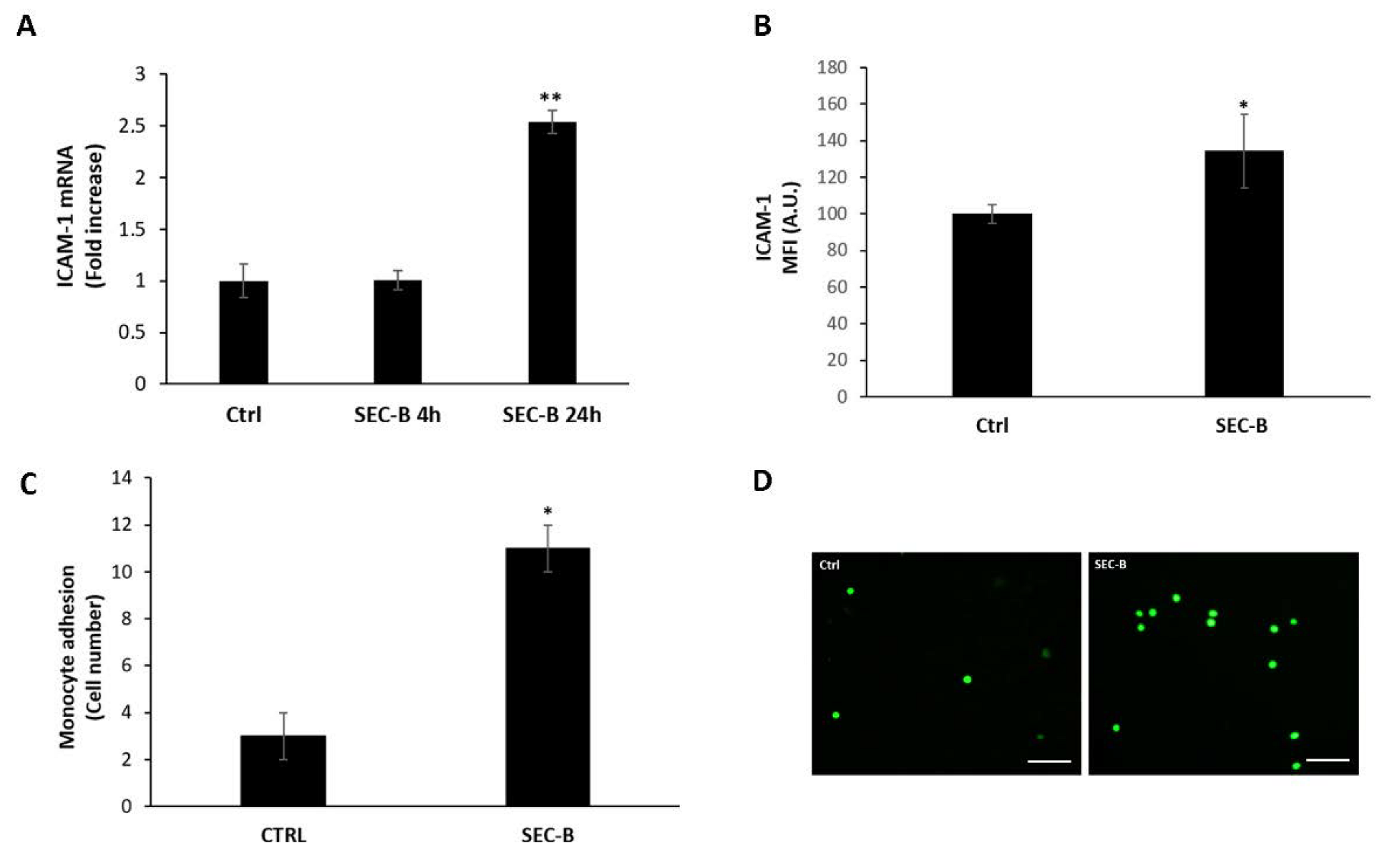
3.5. SEC-B Enhances ICAM-1 Expression and Adhesion of U937 Cells
3.6. Effect of NO Inhibition on SEC-B-Induced HMEC-1 Cell Dysfunction
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rodrigues, S.F.; Granger, D.N. Blood Cells and Endothelial Barrier Function. Tissue Barriers 2015, 3, e978720. [Google Scholar] [CrossRef]
- Hsieh, H.-J.; Liu, C.-A.; Huang, B.; Tseng, A.H.; Wang, D.L. Shear-Induced Endothelial Mechanotransduction: The Interplay between Reactive Oxygen Species (ROS) and Nitric Oxide (NO) and the Pathophysiological Implications. J. Biomed. Sci. 2014, 21, 3. [Google Scholar] [CrossRef]
- Baldelli, S.; Ciccarone, F.; Limongi, D.; Checconi, P.; Palamara, A.T.; Ciriolo, M.R. Glutathione and Nitric Oxide: Key Team Players in Use and Disuse of Skeletal Muscle. Nutrients 2019, 11, 2318. [Google Scholar] [CrossRef]
- Rigamonti, E.; Touvier, T.; Clementi, E.; Manfredi, A.A.; Brunelli, S.; Rovere-Querini, P. Requirement of Inducible Nitric Oxide Synthase for Skeletal Muscle Regeneration after Acute Damage. J. Immunol. 2013, 190, 1767–1777. [Google Scholar] [CrossRef]
- Simons, K.; Ikonen, E. Functional Rafts in Cell Membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef]
- Simons, K.; Toomre, D. Lipid Rafts and Signal Transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef]
- Chen, Z.; Oliveira, S.D.; Zimnicka, A.M.; Jiang, Y.; Sharma, T.; Chen, S.; Lazarov, O.; Bonini, M.G.; Haus, J.M.; Minshall, R.D. Reciprocal Regulation of ENOS and Caveolin-1 Functions in Endothelial Cells. Mol. Biol. Cell 2018, 29, 1190–1202. [Google Scholar] [CrossRef]
- Gargiulo, S.; Gamba, P.; Testa, G.; Leonarduzzi, G.; Poli, G. The Role of Oxysterols in Vascular Ageing. J. Physiol. 2016, 594, 2095–2113. [Google Scholar] [CrossRef]
- Iuliano, L. Pathways of Cholesterol Oxidation via Non-Enzymatic Mechanisms. Chem. Phys. Lipids 2011, 164, 457–468. [Google Scholar] [CrossRef]
- Vurusaner, B.; Gamba, P.; Testa, G.; Gargiulo, S.; Biasi, F.; Zerbinati, C.; Iuliano, L.; Leonarduzzi, G.; Basaga, H.; Poli, G. Survival Signaling Elicited by 27-Hydroxycholesterol through the Combined Modulation of Cellular Redox State and ERK/Akt Phosphorylation. Free Radic. Biol. Med. 2014, 77, 376–385. [Google Scholar] [CrossRef]
- Wung, B.S.; Ni, C.W.; Wang, D.L. ICAM-1 Induction by TNFalpha and IL-6 Is Mediated by Distinct Pathways via Rac in Endothelial Cells. J. Biomed. Sci. 2005, 12, 91–101. [Google Scholar] [CrossRef]
- Harada, N.; Nakayama, M.; Nakano, H.; Fukuchi, Y.; Yagita, H.; Okumura, K. Pro-Inflammatory Effect of TWEAK/Fn14 Interaction on Human Umbilical Vein Endothelial Cells. Biochem. Biophys. Res. Commun. 2002, 299, 488–493. [Google Scholar] [CrossRef]
- Liu, H.; Lin, D.; Xiang, H.; Chen, W.; Zhao, S.; Peng, H.; Yang, J.; Chen, P.; Chen, S.; Lu, H. The Role of Tumor Necrosis Factor-like Weak Inducer of Apoptosis in Atherosclerosis via Its Two Different Receptors. Exp. Ther. Med. 2017, 14, 891–897. [Google Scholar] [CrossRef]
- Dohi, T.; Burkly, L.C. The TWEAK/Fn14 Pathway as an Aggravating and Perpetuating Factor in Inflammatory Diseases: Focus on Inflammatory Bowel Diseases. J. Leukoc. Biol. 2012, 92, 265–279. [Google Scholar] [CrossRef]
- Wang, K.; Bermúdez, E.; Pryor, W.A. The Ozonation of Cholesterol: Separation and Identification of 2,4-Dinitrophenylhydrazine Derivatization Products of 3 Beta-Hydroxy-5-Oxo-5,6-Secocholestan-6-Al. Steroids 1993, 58, 225–229. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A New Mathematical Model for Relative Quantification in Real-Time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Canonico, B.; Cesarini, E.; Salucci, S.; Luchetti, F.; Falcieri, E.; Di Sario, G.; Palma, F.; Papa, S. Defective Autophagy, Mitochondrial Clearance and Lipophagy in Niemann-Pick Type B Lymphocytes. PLoS ONE 2016, 11, e0165780. [Google Scholar] [CrossRef]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative Stress and Reactive Oxygen Species in Endothelial Dysfunction Associated with Cardiovascular and Metabolic Diseases. Vascul. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef]
- Qian, J.; Fulton, D. Post-Translational Regulation of Endothelial Nitric Oxide Synthase in Vascular Endothelium. Front. Physiol. 2013, 4, 347. [Google Scholar] [CrossRef]
- Belhassen, L.; Feron, O.; Kaye, D.M.; Michel, T.; Kelly, R.A. Regulation by CAMP of Post-Translational Processing and Subcellular Targeting of Endothelial Nitric-Oxide Synthase (Type 3) in Cardiac Myocytes. J. Biol. Chem. 1997, 272, 11198–11204. [Google Scholar] [CrossRef]
- Marasciulo, F.L.; Montagnani, M.; Potenza, M.A. Endothelin-1: The Yin and Yang on Vascular Function. Curr. Med. Chem. 2006, 13, 1655–1665. [Google Scholar] [CrossRef] [PubMed]
- Kubes, P.; Suzuki, M.; Granger, D.N. Nitric Oxide: An Endogenous Modulator of Leukocyte Adhesion. Proc. Natl. Acad. Sci. USA 1991, 88, 4651–4655. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.-S.; Lindberg, R.A.; Zhou, H.-L.; Haroon, Z.A.; Dewhirst, M.W.; Hausladen, A.; Juang, Y.-L.; Stamler, J.S.; Greenberg, C.S. Endothelial Cell-Surface Tissue Transglutaminase Inhibits Neutrophil Adhesion by Binding and Releasing Nitric Oxide. Sci. Rep. 2017, 7, 16163. [Google Scholar] [CrossRef] [PubMed]
- Davignon, J.; Ganz, P. Role of Endothelial Dysfunction in Atherosclerosis. Circulation 2004, 109, III27–III32. [Google Scholar] [CrossRef] [PubMed]
- Widmer, R.J.; Lerman, A. Endothelial Dysfunction and Cardiovascular Disease. Glob. Cardiol. Sci. Pract. 2014, 2014, 291–308. [Google Scholar] [CrossRef]
- Shiroto, T.; Romero, N.; Sugiyama, T.; Sartoretto, J.L.; Kalwa, H.; Yan, Z.; Shimokawa, H.; Michel, T. Caveolin-1 Is a Critical Determinant of Autophagy, Metabolic Switching, and Oxidative Stress in Vascular Endothelium. PLoS ONE 2014, 9, e87871. [Google Scholar] [CrossRef]
- Fielding, C.J.; Bist, A.; Fielding, P.E. Caveolin MRNA Levels Are Up-Regulated by Free Cholesterol and down-Regulated by Oxysterols in Fibroblast Monolayers. Proc. Natl. Acad. Sci. USA 1997, 94, 3753–3758. [Google Scholar] [CrossRef]
- Bernatchez, P.; Sharma, A.; Bauer, P.M.; Marin, E.; Sessa, W.C. A Noninhibitory Mutant of the Caveolin-1 Scaffolding Domain Enhances ENOS-Derived NO Synthesis and Vasodilation in Mice. J. Clin. Investig. 2011, 121, 3747–3755. [Google Scholar] [CrossRef]
- Michel, J.B.; Feron, O.; Sacks, D.; Michel, T. Reciprocal Regulation of Endothelial Nitric-Oxide Synthase by Ca2+-Calmodulin and Caveolin. J. Biol. Chem. 1997, 272, 15583–15586. [Google Scholar] [CrossRef]
- Napoli, C.; Ignarro, L.J. Nitric Oxide and Pathogenic Mechanisms Involved in the Development of Vascular Diseases. Arch. Pharm. Res. 2009, 32, 1103–1108. [Google Scholar] [CrossRef]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric Oxide and Peroxynitrite in Health and Disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef]
- Terasaka, N.; Westerterp, M.; Koetsveld, J.; Fernández-Hernando, C.; Yvan-Charvet, L.; Wang, N.; Sessa, W.C.; Tall, A.R. ATP-Binding Cassette Transporter G1 and High-Density Lipoprotein Promote Endothelial NO Synthesis through a Decrease in the Interaction of Caveolin-1 and Endothelial NO Synthase. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2219–2225. [Google Scholar] [CrossRef] [PubMed]
- Terasaka, N.; Yu, S.; Yvan-Charvet, L.; Wang, N.; Mzhavia, N.; Langlois, R.; Pagler, T.; Li, R.; Welch, C.L.; Goldberg, I.J.; et al. ABCG1 and HDL Protect against Endothelial Dysfunction in Mice Fed a High-Cholesterol Diet. J. Clin. Investig. 2008, 118, 3701–3713. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Wong, S.L.; Lau, C.W.; Huang, Y.; Yu, C.-M. Oxidized LDL at Low Concentration Promotes In-Vitro Angiogenesis and Activates Nitric Oxide Synthase through PI3K/Akt/ENOS Pathway in Human Coronary Artery Endothelial Cells. Biochem. Biophys. Res. Commun. 2011, 407, 44–48. [Google Scholar] [CrossRef]
- Afonso, V.; Champy, R.; Mitrovic, D.; Collin, P.; Lomri, A. Reactive Oxygen Species and Superoxide Dismutases: Role in Joint Diseases. Jt. Bone Spine 2007, 74, 324–329. [Google Scholar] [CrossRef]
- Gheddouchi, S.; Mokhtari-Soulimane, N.; Merzouk, H.; Bekhti, F.; Soulimane, F.; Guermouche, B.; Meziane Tani, A.; Narce, M. Low SOD Activity Is Associated with Overproduction of Peroxynitrite and Nitric Oxide in Patients with Acute Coronary Syndrome. Nitric Oxide Biol. Chem. 2015, 49, 40–46. [Google Scholar] [CrossRef]
- Sharma, J.N.; Al-Omran, A.; Parvathy, S.S. Role of Nitric Oxide in Inflammatory Diseases. Inflammopharmacology 2007, 15, 252–259. [Google Scholar] [CrossRef]
- Beauchamp, M.H.; Sennlaub, F.; Speranza, G.; Gobeil, F.J.; Checchin, D.; Kermorvant-Duchemin, E.; Abran, D.; Hardy, P.; Lachapelle, P.; Varma, D.R.; et al. Redox-Dependent Effects of Nitric Oxide on Microvascular Integrity in Oxygen-Induced Retinopathy. Free Radic. Biol. Med. 2004, 37, 1885–1894. [Google Scholar] [CrossRef]
- Wedgwood, S.; Black, S.M. Endothelin-1 Decreases Endothelial NOS Expression and Activity through ETA Receptor-Mediated Generation of Hydrogen Peroxide. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 288, L480–L487. [Google Scholar] [CrossRef]
- He, Y.; Kwan, W.C.; Steinbrecher, U.P. Effects of Oxidized Low Density Lipoprotein on Endothelin Secretion by Cultured Endothelial Cells and Macrophages. Atherosclerosis 1996, 119, 107–118. [Google Scholar] [CrossRef]
- Gao, M.; Zeng, Y.; Guan, Y.; Hu, Z.; Zhong, D.; Shen, X.; Zhang, L.; Xu, Z.; Gong, W.; Zhang, Y.; et al. Activation of Liver X Receptor Attenuates Endothelin-1 Expression in Vascular Endothelial Cells. Int. J. Biochem. Cell Biol. 2012, 44, 2299–2307. [Google Scholar] [CrossRef] [PubMed]
- Taniyama, Y.; Griendling, K.K. Reactive Oxygen Species in the Vasculature: Molecular and Cellular Mechanisms. Hypertens. Dallas Tex 1979 2003, 42, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bian, Y.; Zhang, R.; Liu, X.; Ni, L.; Ma, B.; Zeng, R.; Zhao, Z.; Song, X.; Liu, C. Melatonin Alleviates Cigarette Smoke-Induced Endothelial Cell Pyroptosis through Inhibiting ROS/NLRP3 Axis. Biochem. Biophys. Res. Commun. 2019, 519, 402–408. [Google Scholar] [CrossRef]
- Gutiérrez-Muñoz, C.; Méndez-Barbero, N.; Svendsen, P.; Sastre, C.; Fernández-Laso, V.; Quesada, P.; Egido, J.; Escolá-Gil, J.C.; Martín-Ventura, J.L.; Moestrup, S.K.; et al. CD163 Deficiency Increases Foam Cell Formation and Plaque Progression in Atherosclerotic Mice. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 14960–14976. [Google Scholar] [CrossRef] [PubMed]
- Alfaidi, M.; Wilson, H.; Daigneault, M.; Burnett, A.; Ridger, V.; Chamberlain, J.; Francis, S. Neutrophil Elastase Promotes Interleukin-1β Secretion from Human Coronary Endothelium. J. Biol. Chem. 2015, 290, 24067–24078. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Feng, H.; Guo, S.; Han, Y.; Chen, X. Danshenol A Inhibits TNF-α-Induced Expression of Intercellular Adhesion Molecule-1 (ICAM-1) Mediated by NOX4 in Endothelial Cells. Sci. Rep. 2017, 7, 12953. [Google Scholar] [CrossRef]
- Aguilar, G.; Córdova, F.; Koning, T.; Sarmiento, J.; Boric, M.P.; Birukov, K.; Cancino, J.; Varas-Godoy, M.; Soza, A.; Alves, N.G.; et al. TNF-α-Activated ENOS Signaling Increases Leukocyte Adhesion through the S-Nitrosylation Pathway. Am. J. Physiol. Heart Circ. Physiol. 2021, 321, H1083–H1095. [Google Scholar] [CrossRef]
- Liu, G.; Place, A.T.; Chen, Z.; Brovkovych, V.M.; Vogel, S.M.; Muller, W.A.; Skidgel, R.A.; Malik, A.B.; Minshall, R.D. ICAM-1-Activated Src and ENOS Signaling Increase Endothelial Cell Surface PECAM-1 Adhesivity and Neutrophil Transmigration. Blood 2012, 120, 1942–1952. [Google Scholar] [CrossRef]
- Martinelli, R.; Gegg, M.; Longbottom, R.; Adamson, P.; Turowski, P.; Greenwood, J. ICAM-1-Mediated Endothelial Nitric Oxide Synthase Activation via Calcium and AMP-Activated Protein Kinase Is Required for Transendothelial Lymphocyte Migration. Mol. Biol. Cell 2009, 20, 995–1005. [Google Scholar] [CrossRef]
- Fielding, C.A.; McLoughlin, R.M.; McLeod, L.; Colmont, C.S.; Najdovska, M.; Grail, D.; Ernst, M.; Jones, S.A.; Topley, N.; Jenkins, B.J. IL-6 Regulates Neutrophil Trafficking during Acute Inflammation via STAT3. J. Immunol. 2008, 181, 2189–2195. [Google Scholar] [CrossRef]
- Wung, B.S.; Hsu, M.C.; Wu, C.C.; Hsieh, C.W. Resveratrol Suppresses IL-6-Induced ICAM-1 Gene Expression in Endothelial Cells: Effects on the Inhibition of STAT3 Phosphorylation. Life Sci. 2005, 78, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Didion, S.P. Cellular and Oxidative Mechanisms Associated with Interleukin-6 Signaling in the Vasculature. Int. J. Mol. Sci. 2017, 18, 2563. [Google Scholar] [CrossRef]
- Ali, M.H.; Schlidt, S.A.; Chandel, N.S.; Hynes, K.L.; Schumacker, P.T.; Gewertz, B.L. Endothelial Permeability and IL-6 Production during Hypoxia: Role of ROS in Signal Transduction. Am. J. Physiol. 1999, 277, L1057–L1065. [Google Scholar] [CrossRef] [PubMed]
- Karbach, S.; Wenzel, P.; Waisman, A.; Munzel, T.; Daiber, A. ENOS Uncoupling in Cardiovascular Diseases—the Role of Oxidative Stress and Inflammation. Curr. Pharm. Des. 2014, 20, 3579–3594. [Google Scholar] [CrossRef] [PubMed]
- Kofler, S.; Nickel, T.; Weis, M. Role of Cytokines in Cardiovascular Diseases: A Focus on Endothelial Responses to Inflammation. Clin. Sci. Lond. Engl. 1979 2005, 108, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Wajant, H. The TWEAK-Fn14 System as a Potential Drug Target. Br. J. Pharmacol. 2013, 170, 748–764. [Google Scholar] [CrossRef] [PubMed]
- Lynch, C.N.; Wang, Y.C.; Lund, J.K.; Chen, Y.W.; Leal, J.A.; Wiley, S.R. TWEAK Induces Angiogenesis and Proliferation of Endothelial Cells. J. Biol. Chem. 1999, 274, 8455–8459. [Google Scholar] [CrossRef]
- Saas, P.; Boucraut, J.; Walker, P.R.; Quiquerez, A.L.; Billot, M.; Desplat-Jego, S.; Chicheportiche, Y.; Dietrich, P.Y. TWEAK Stimulation of Astrocytes and the Proinflammatory Consequences. Glia 2000, 32, 102–107. [Google Scholar] [CrossRef]
- Stephan, D.; Sbai, O.; Wen, J.; Couraud, P.-O.; Putterman, C.; Khrestchatisky, M.; Desplat-Jégo, S. TWEAK/Fn14 Pathway Modulates Properties of a Human Microvascular Endothelial Cell Model of Blood Brain Barrier. J. Neuroinflamm. 2013, 10, 9. [Google Scholar] [CrossRef]
- Muñoz-García, B.; Moreno, J.A.; López-Franco, O.; Sanz, A.B.; Martín-Ventura, J.L.; Blanco, J.; Jakubowski, A.; Burkly, L.C.; Ortiz, A.; Egido, J.; et al. Tumor Necrosis Factor-like Weak Inducer of Apoptosis (TWEAK) Enhances Vascular and Renal Damage Induced by Hyperlipidemic Diet in ApoE-Knockout Mice. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 2061–2068. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nasoni, M.G.; Benedetti, S.; Crinelli, R.; Palma, F.; Canonico, B.; Monittola, F.; Zerbinati, C.; Iuliano, L.; Luchetti, F. 3β-Hydroxy-5β-hydroxy-B-norcholestane-6β-carboxaldehyde (SEC-B) Induces Proinflammatory Activation of Human Endothelial Cells Associated with Nitric Oxide Production and Endothelial Nitric Oxide Synthase/Caveolin-1 Dysregulation. Antioxidants 2022, 11, 1148. https://doi.org/10.3390/antiox11061148
Nasoni MG, Benedetti S, Crinelli R, Palma F, Canonico B, Monittola F, Zerbinati C, Iuliano L, Luchetti F. 3β-Hydroxy-5β-hydroxy-B-norcholestane-6β-carboxaldehyde (SEC-B) Induces Proinflammatory Activation of Human Endothelial Cells Associated with Nitric Oxide Production and Endothelial Nitric Oxide Synthase/Caveolin-1 Dysregulation. Antioxidants. 2022; 11(6):1148. https://doi.org/10.3390/antiox11061148
Chicago/Turabian StyleNasoni, Maria Gemma, Serena Benedetti, Rita Crinelli, Francesco Palma, Barbara Canonico, Francesca Monittola, Chiara Zerbinati, Luigi Iuliano, and Francesca Luchetti. 2022. "3β-Hydroxy-5β-hydroxy-B-norcholestane-6β-carboxaldehyde (SEC-B) Induces Proinflammatory Activation of Human Endothelial Cells Associated with Nitric Oxide Production and Endothelial Nitric Oxide Synthase/Caveolin-1 Dysregulation" Antioxidants 11, no. 6: 1148. https://doi.org/10.3390/antiox11061148