
Mitochondrial Oxidative Stress Promotes Cardiac Remodeling in Myocardial Infarction through the Activation of Endoplasmic Reticulum Stress

 , , and
, , and
Abstract
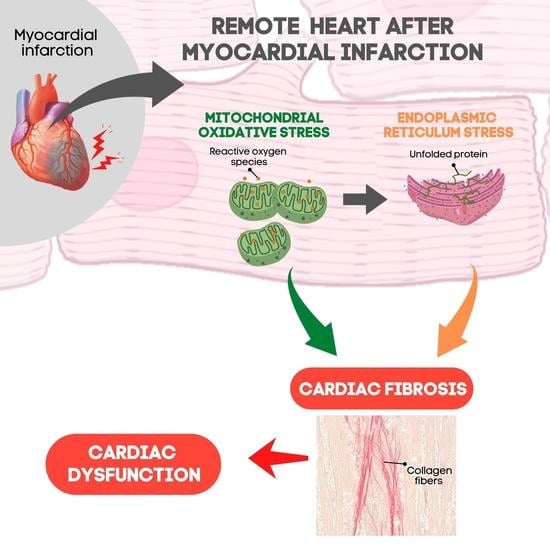
:
1. Introduction
2. Materials and Methods
2.1. Animal Model
2.2. Cardiac Function Measurement
2.3. Morphological and Histological Evaluation
2.4. Measurement of Cardiac Reactive Oxygen Species (ROS) Levels
2.5. Western Blot Analysis
2.6. Clinical Study
2.7. Cardiac Function
2.8. Myocardial Extracellular Volume Fraction
2.9. Circulating Plasma Levels of Myeloperoxidase (MPO) and BiP
2.10. Analysis of Biobank Cardiac Samples. Cardiac Immunohistochemistry of BiP
2.11. Cardiac Collagen Content
2.12. Statistical Analysis
3. Results
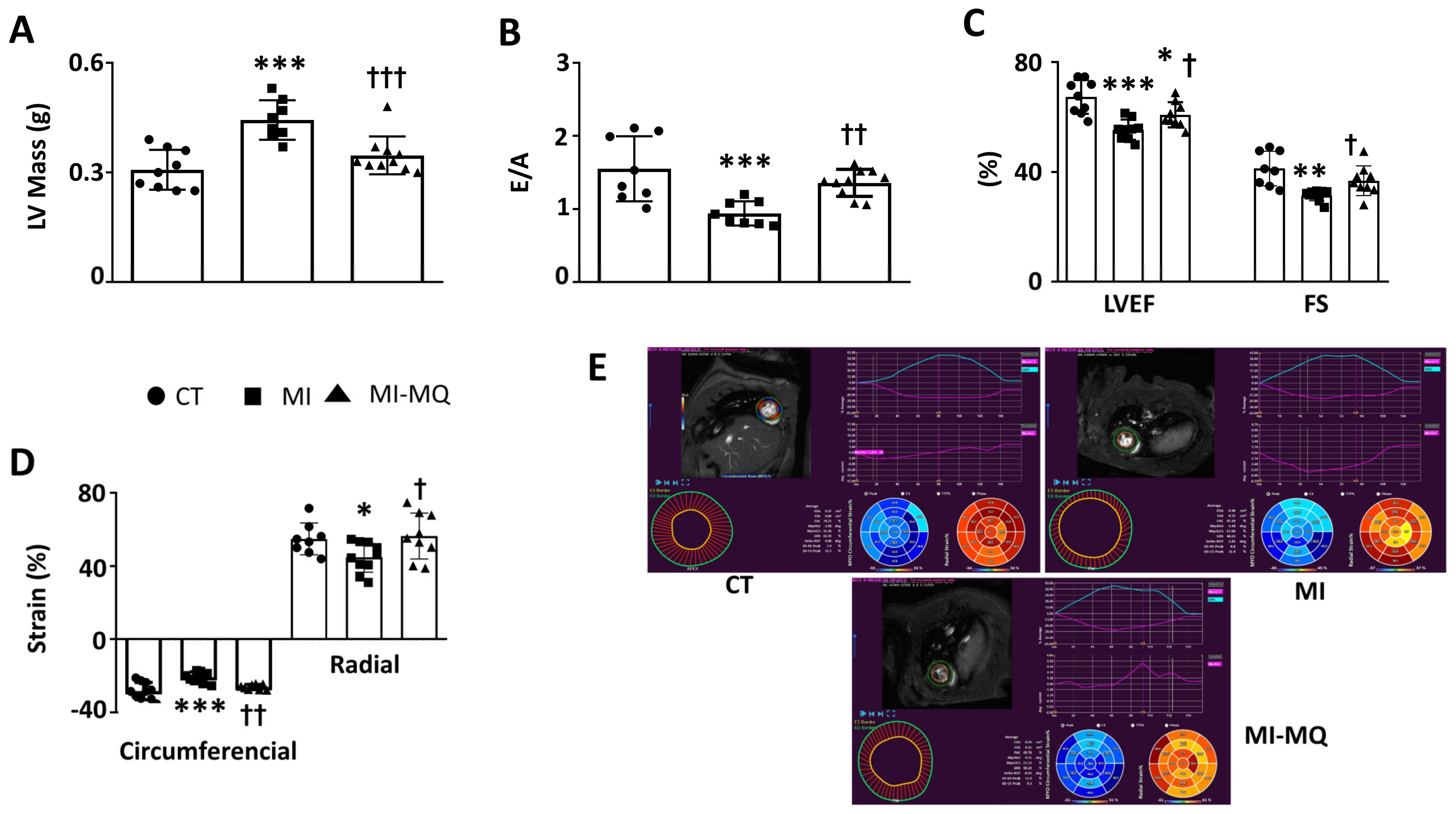
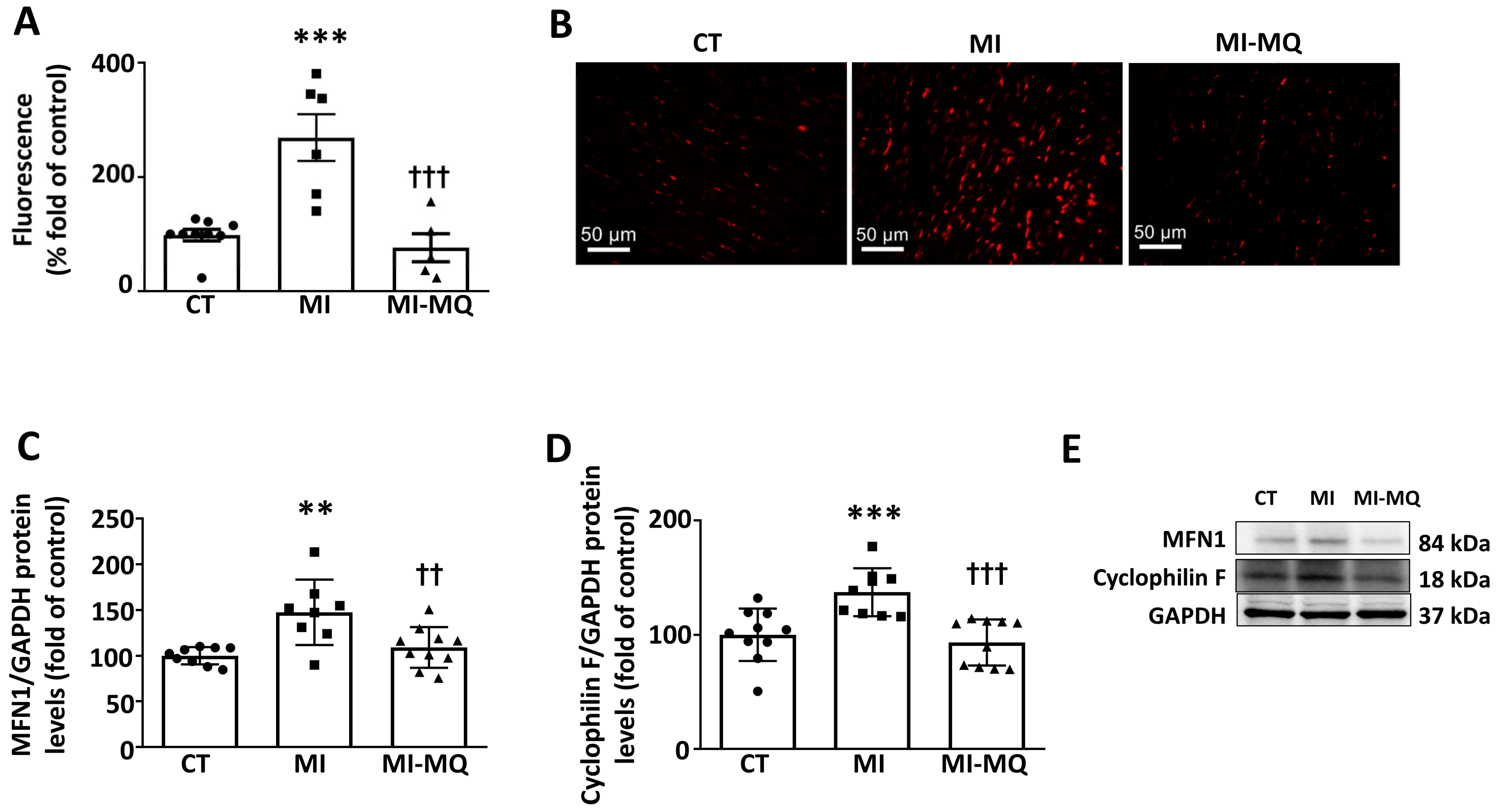
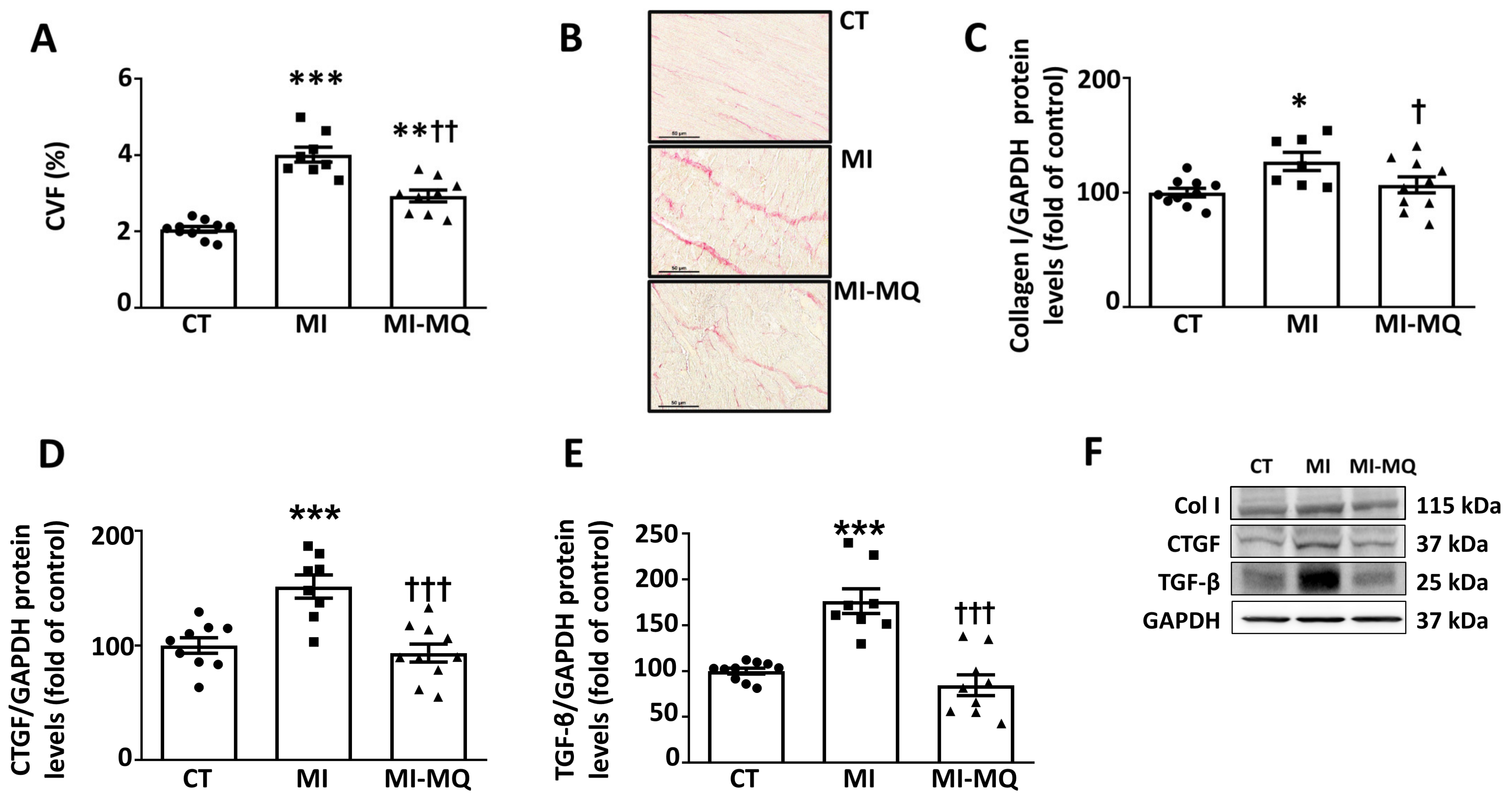
3.1. Animal Study
3.2. Clinical Study
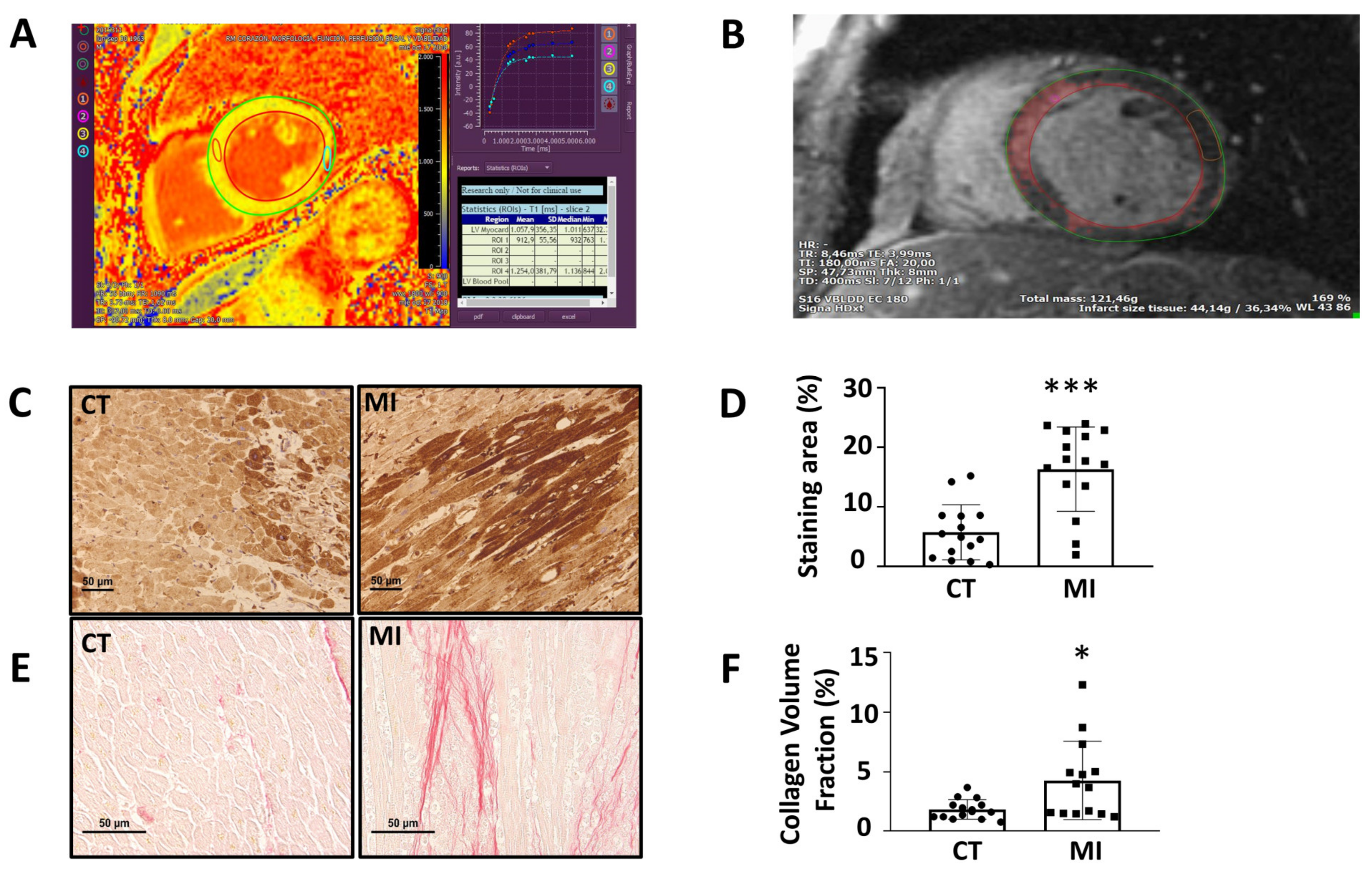
3.3. Analysis of Biobank Cardiac Samples
4. Discussion
5. Conclusions
6. Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Doeleman, L.C. Stop prolonging resuscitations in drowned patients with asystole. Resuscitation 2016, 106, e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talman, V.; Ruskoaho, H. Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration. Cell Tissue Res. 2016, 365, 563–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scalise, R.F.M.; De Sarro, R.; Caracciolo, A.; Lauro, R.; Squadrito, F.; Carerj, S.; Bitto, A.; Micari, A.; Bella, G.D.; Costa, F.; et al. Fibrosis after Myocardial Infarction: An Overview on Cellular Processes, Molecular Pathways, Clinical Evaluation and Prognostic Value. Med. Sci. 2021, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- Garg, P.; Broadbent, D.A.; Swoboda, P.P.; Foley, J.R.J.; Fent, G.J.; Musa, T.A.; Ripley, D.P.; Erhayiem, B.; Dobson, L.E.; McDiarmid, A.K.; et al. Extra-cellular expansion in the normal, non-infarcted myocardium is associated with worsening of regional myocardial function after acute myocardial infarction. J. Cardiovasc. Magn. Reson. 2017, 19, 73. [Google Scholar] [CrossRef]
- Marin-Royo, G.; Ortega-Hernandez, A.; Martinez-Martinez, E.; Jurado-Lopez, R.; Luaces, M.; Islas, F.; Gomez-Garre, D.; Delgado-Valero, B.; Lagunas, E.; Ramchandani, B.; et al. The Impact of Cardiac Lipotoxicity on Cardiac Function and Mirnas Signature in Obese and Non-Obese Rats with Myocardial Infarction. Sci. Rep. 2019, 9, 444. [Google Scholar] [CrossRef] [Green Version]
- Tse, G.; Yan, B.P.; Chan, Y.W.; Tian, X.Y.; Huang, Y. Reactive Oxygen Species, Endoplasmic Reticulum Stress and Mitochondrial Dysfunction: The Link with Cardiac Arrhythmogenesis. Front. Physiol. 2016, 7, 313. [Google Scholar] [CrossRef] [Green Version]
- Sheeran, F.L.; Pepe, S. Energy deficiency in the failing heart: Linking increased reactive oxygen species and disruption of oxidative phosphorylation rate. Biochim. Biophys. Acta 2006, 1757, 543–552. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.K.; Nilius, B.; Kim, N.; Ko, K.S.; Rhee, B.D.; Han, J. Cardiac Response to Oxidative Stress Induced by Mitochondrial Dysfunction. Rev. Physiol. Biochem. Pharmacol. 2016, 170, 101–127. [Google Scholar] [CrossRef]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [Green Version]
- Braakman, I.; Hebert, D.N. Protein folding in the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 2013, 5, a013201. [Google Scholar] [CrossRef] [Green Version]
- Bravo, R.; Parra, V.; Gatica, D.; Rodriguez, A.E.; Torrealba, N.; Paredes, F.; Wang, Z.V.; Zorzano, A.; Hill, J.A.; Jaimovich, E.; et al. Endoplasmic reticulum and the unfolded protein response: Dynamics and metabolic integration. Int. Rev. Cell Mol. Biol. 2013, 301, 215–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuzaki, S.; Hiratsuka, T.; Taniguchi, M.; Shingaki, K.; Kubo, T.; Kiya, K.; Fujiwara, T.; Kanazawa, S.; Kanematsu, R.; Maeda, T.; et al. Physiological ER Stress Mediates the Differentiation of Fibroblasts. PLoS ONE 2015, 10, e0123578. [Google Scholar] [CrossRef] [Green Version]
- Souza-Neto, F.V.; Jimenez-Gonzalez, S.; Delgado-Valero, B.; Jurado-Lopez, R.; Genty, M.; Romero-Miranda, A.; Rodriguez, C.; Nieto, M.L.; Martinez-Martinez, E.; Cachofeiro, V. The Interplay of Mitochondrial Oxidative Stress and Endoplasmic Reticulum Stress in Cardiovascular Fibrosis in Obese Rats. Antioxidants 2021, 10, 1274. [Google Scholar] [CrossRef] [PubMed]
- ESC Committee for Practice Guidelines (CPG); Bax, J.J.; Baumgartner, H.; Ceconi, C.; Dean, V.; Deaton, C.; Fagard, R.; Funck-Brentano, C.; Hasdai, D.; Hoes, A.; et al. Third universal definition of myocardial infarction. J. Am. Coll. Cardiol. 2012, 60, 1581–1598. [Google Scholar] [CrossRef] [Green Version]
- Lang, R.M.; Badano, L.P.; Mor-Avi, V.; Afilalo, J.; Armstrong, A.; Ernande, L.; Flachskampf, F.A.; Foster, E.; Goldstein, S.A.; Kuznetsova, T.; et al. Recommendations for cardiac chamber quantification by echocardiography in adults: An update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. Eur. Heart J. Cardiovasc. Imaging 2015, 16, 233–271. [Google Scholar] [CrossRef]
- Haaf, P.; Garg, P.; Messroghli, D.R.; Broadbent, D.A.; Greenwood, J.P.; Plein, S. Cardiac T1 Mapping and Extracellular Volume (ECV) in clinical practice: A comprehensive review. J. Cardiovasc. Magn. Reson. 2016, 18, 89. [Google Scholar] [CrossRef] [Green Version]
- Reiter, U.; Reiter, C.; Krauter, C.; Fuchsjager, M.; Reiter, G. Cardiac magnetic resonance T1 mapping. Part 2: Diagnostic potential and applications. Eur. J. Radiol. 2018, 109, 235–247. [Google Scholar] [CrossRef] [Green Version]
- Gulati, A.; Japp, A.G.; Raza, S.; Halliday, B.P.; Jones, D.A.; Newsome, S.; Ismail, N.A.; Morarji, K.; Khwaja, J.; Spath, N.; et al. Absence of Myocardial Fibrosis Predicts Favorable Long-Term Survival in New-Onset Heart Failure. Circ. Cardiovasc. Imaging 2018, 11, e007722. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Liu, X.; Yang, F.; Wang, J.; Xu, Y.; Fang, T.; Pu, L.; Zhou, X.; Han, Y.; Chen, Y. Prognostic value of myocardial extracellular volume fraction evaluation based on cardiac magnetic resonance T1 mapping with T1 long and short in hypertrophic cardiomyopathy. Eur. Radiol. 2021, 31, 4557–4567. [Google Scholar] [CrossRef]
- Raiker, N.; Vullaganti, S.; Collins, J.D.; Allen, B.D.; Choudhury, L. Myocardial tissue characterization by gadolinium-enhanced cardiac magnetic resonance imaging for risk stratification of adverse events in hypertrophic cardiomyopathy. Int. J. Cardiovasc. Imaging 2020, 36, 1147–1156. [Google Scholar] [CrossRef]
- Fernandez-Jimenez, R.; Barreiro-Perez, M.; Martin-Garcia, A.; Sanchez-Gonzalez, J.; Aguero, J.; Galan-Arriola, C.; Garcia-Prieto, J.; Diaz-Pelaez, E.; Vara, P.; Martinez, I.; et al. Dynamic Edematous Response of the Human Heart to Myocardial Infarction: Implications for Assessing Myocardial Area at Risk and Salvage. Circulation 2017, 136, 1288–1300. [Google Scholar] [CrossRef] [PubMed]
- Oyenuga, A.O.; Couper, D.; Matsushita, K.; Boerwinkle, E.; Folsom, A.R. Association of monocyte myeloperoxidase with incident cardiovascular disease: The Atherosclerosis Risk in Communities Study. PLoS ONE 2018, 13, e0205310. [Google Scholar] [CrossRef] [PubMed]
- Brennan, M.L.; Penn, M.S.; Van Lente, F.; Nambi, V.; Shishehbor, M.H.; Aviles, R.J.; Goormastic, M.; Pepoy, M.L.; McErlean, E.S.; Topol, E.J.; et al. Prognostic value of myeloperoxidase in patients with chest pain. N. Engl. J. Med. 2003, 349, 1595–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, M.J. Myeloperoxidase-derived oxidation: Mechanisms of biological damage and its prevention. J. Clin. Biochem. Nutr. 2011, 48, 8–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, E.Y.; Ghosn, M.G.; Khan, M.A.; Gramze, N.L.; Brunner, G.; Nabi, F.; Nambi, V.; Nagueh, S.F.; Nguyen, D.T.; Graviss, E.A.; et al. Myocardial Extracellular Volume Fraction Adds Prognostic Information Beyond Myocardial Replacement Fibrosis. Circ. Cardiovasc. Imaging 2019, 12, e009535. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Gonzalez, S.; Marin-Royo, G.; Jurado-Lopez, R.; Bartolome, M.V.; Romero-Miranda, A.; Luaces, M.; Islas, F.; Nieto, M.L.; Martinez-Martinez, E.; Cachofeiro, V. The Crosstalk between Cardiac Lipotoxicity and Mitochondrial Oxidative Stress in the Cardiac Alterations in Diet-Induced Obesity in Rats. Cells 2020, 9, 451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sia, Y.T.; Parker, T.G.; Liu, P.; Tsoporis, J.N.; Adam, A.; Rouleau, J.L. Improved post-myocardial infarction survival with probucol in rats: Effects on left ventricular function, morphology, cardiac oxidative stress and cytokine expression. J. Am. Coll. Cardiol. 2002, 39, 148–156. [Google Scholar] [CrossRef] [Green Version]
- Van der Pol, A.; van Gilst, W.H.; Voors, A.A.; van der Meer, P. Treating oxidative stress in heart failure: Past, present and future. Eur. J. Heart Fail. 2019, 21, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Hori, M.; Nishida, K. Oxidative stress and left ventricular remodelling after myocardial infarction. Cardiovasc. Res. 2009, 81, 457–464. [Google Scholar] [CrossRef] [Green Version]
- Starkov, A.A. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann. N. Y. Acad. Sci. 2008, 1147, 37–52. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Yu, D.; Hu, Y.; Zhang, P.; Yang, Y.; Hu, Q.; Li, M. Angiotensin II upregulates cyclophilin A by enhancing ROS production in rat cardiomyocytes. Mol. Med. Rep. 2018, 18, 4349–4355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, J.C.B.; Campos, J.C.; Qvit, N.; Qi, X.; Bozi, L.H.M.; Bechara, L.R.G.; Lima, V.M.; Queliconi, B.B.; Disatnik, M.H.; Dourado, P.M.M.; et al. A selective inhibitor of mitofusin 1-betaIIPKC association improves heart failure outcome in rats. Nat. Commun. 2019, 10, 329. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.R.; Burke, N.; Dongworth, R.K.; Kalkhoran, S.B.; Dyson, A.; Vicencio, J.M.; Dorn, G.W., II; Yellon, D.M.; Hausenloy, D.J. Hearts deficient in both Mfn1 and Mfn2 are protected against acute myocardial infarction. Cell Death Dis. 2016, 7, e2238. [Google Scholar] [CrossRef] [PubMed]
- Schrepfer, E.; Scorrano, L. Mitofusins, from Mitochondria to Metabolism. Mol. Cell 2016, 61, 683–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pryds, K.; Larsen, A.H.; Hansen, M.S.; Grondal, A.Y.K.; Tougaard, R.S.; Hansson, N.H.; Clemmensen, T.S.; Logstrup, B.B.; Wiggers, H.; Kim, W.Y.; et al. Myocardial strain assessed by feature tracking cardiac magnetic resonance in patients with a variety of cardiovascular diseases—A comparison with echocardiography. Sci. Rep. 2019, 9, 11296. [Google Scholar] [CrossRef]
- Liu, R.M.; Desai, L.P. Reciprocal regulation of TGF-beta and reactive oxygen species: A perverse cycle for fibrosis. Redox Biol. 2015, 6, 565–577. [Google Scholar] [CrossRef] [Green Version]
- Jain, M.; Rivera, S.; Monclus, E.A.; Synenki, L.; Zirk, A.; Eisenbart, J.; Feghali-Bostwick, C.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial reactive oxygen species regulate transforming growth factor-beta signaling. J. Biol. Chem. 2013, 288, 770–777. [Google Scholar] [CrossRef] [Green Version]
- Dean, R.G.; Balding, L.C.; Candido, R.; Burns, W.C.; Cao, Z.; Twigg, S.M.; Burrell, L.M. Connective tissue growth factor and cardiac fibrosis after myocardial infarction. J. Histochem. Cytochem. 2005, 53, 1245–1256. [Google Scholar] [CrossRef]
- Guan, C.; Zhang, H.F.; Wang, Y.J.; Chen, Z.T.; Deng, B.Q.; Qiu, Q.; Chen, S.X.; Wu, M.X.; Chen, Y.X.; Wang, J.F. The Downregulation of ADAM17 Exerts Protective Effects against Cardiac Fibrosis by Regulating Endoplasmic Reticulum Stress and Mitophagy. Oxid. Med. Cell Longev. 2021, 2021, 5572088. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Su, Z.; Tan, X.; Su, Y.; Chen, Y.; Shu, X.; Liang, S.; Wang, J.; Xie, S. Effect of Endoplasmic Reticulum Stress and Autophagy in the Regulation of Post-infarct Cardiac Repair. Arch. Med. Res. 2018, 49, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, Y.; Tang, Z.; Xu, J.; Ma, M.; Pan, S.; Qiu, C.; Guan, G.; Wang, J. Matrine attenuates cardiac fibrosis by affecting ATF6 signaling pathway in diabetic cardiomyopathy. Eur. J. Pharmacol. 2017, 804, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Kitakaze, M. ER stress in cardiovascular disease. J. Mol. Cell Cardiol. 2010, 48, 1105–1110. [Google Scholar] [CrossRef]
- Bourdier, G.; Flore, P.; Sanchez, H.; Pepin, J.L.; Belaidi, E.; Arnaud, C. High-intensity training reduces intermittent hypoxia-induced ER stress and myocardial infarct size. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H279–H289. [Google Scholar] [CrossRef] [Green Version]
- Fribley, A.; Zhang, K.; Kaufman, R.J. Regulation of apoptosis by the unfolded protein response. Methods Mol. Biol. 2009, 559, 191–204. [Google Scholar] [CrossRef] [Green Version]
- Xin, Y.; Wu, W.; Qu, J.; Wang, X.; Lei, S.; Yuan, L.; Liu, X. Inhibition of Mitofusin-2 Promotes Cardiac Fibroblast Activation via the PERK/ATF4 Pathway and Reactive Oxygen Species. Oxid. Med. Cell Longev. 2019, 2019, 3649808. [Google Scholar] [CrossRef] [Green Version]
- Stoner, M.W.; McTiernan, C.F.; Scott, I.; Manning, J.R. Calreticulin expression in human cardiac myocytes induces ER stress-associated apoptosis. Physiol. Rep. 2020, 8, e14400. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.; Oka, T.; Hunter, B.; Robinson, A.; Papp, S.; Nakamura, K.; Srisakuldee, W.; Nickel, B.E.; Light, P.E.; Dyck, J.R.; et al. Calreticulin induces dilated cardiomyopathy. PLoS ONE 2013, 8, e56387. [Google Scholar] [CrossRef] [Green Version]
- Owusu, B.Y.; Zimmerman, K.A.; Murphy-Ullrich, J.E. The role of the endoplasmic reticulum protein calreticulin in mediating TGF-beta-stimulated extracellular matrix production in fibrotic disease. J. Cell Commun. Signal. 2018, 12, 289–299. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.Y.; Liu, X.H.; Hu, W.C.; Rong, F.; Wu, X.D. The calcineurin-myocyte enhancer factor 2c pathway mediates cardiac hypertrophy induced by endoplasmic reticulum stress in neonatal rat cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1499–H1509. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.K.; Balasubramanian, S.; Zavadzkas, J.A.; Jeffords, L.B.; Rivers, W.T.; Zile, M.R.; Mukherjee, R.; Spinale, F.G.; Kuppuswamy, D. Calpain inhibition preserves myocardial structure and function following myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1744–H1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Thompson, J.; Hu, Y.; Lesnefsky, E.J.; Willard, B.; Chen, Q. Calpain-mediated protein targets in cardiac mitochondria following ischemia-reperfusion. Sci. Rep. 2022, 12, 138. [Google Scholar] [CrossRef] [PubMed]
- Wan, F.; Letavernier, E.; Le Saux, C.J.; Houssaini, A.; Abid, S.; Czibik, G.; Sawaki, D.; Marcos, E.; Dubois-Rande, J.L.; Baud, L.; et al. Calpastatin overexpression impairs postinfarct scar healing in mice by compromising reparative immune cell recruitment and activation. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1883–H1893. [Google Scholar] [CrossRef] [Green Version]
- Kudo-Sakamoto, Y.; Akazawa, H.; Ito, K.; Takano, J.; Yano, M.; Yabumoto, C.; Naito, A.T.; Oka, T.; Lee, J.K.; Sakata, Y.; et al. Calpain-dependent cleavage of N-cadherin is involved in the progression of post-myocardial infarction remodeling. J. Biol. Chem. 2014, 289, 19408–19419. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BASAL | FOLLOW-UP | |
|---|---|---|
| BMI (kg/m2) | 28.7 ± 3.5 | 28.1 ± 3.0 |
| IVST (mm) | 11.5 ± 0.22 | 10.9 ± 1.41 * |
| PWT (mm) | 10.8 ± 1.08 | 9.8 ± 1.01 *** |
| EDD (mm) | 47.8 ± 4.5 | 48.6 ± 6.2 * |
| ESD (mm) | 33.6 ± 5.6 | 32.1 ± 7.9 |
| LVEF (%) | 57.6 ± 7.9 | 55.9 ± 9.2 |
| E/A | 0.96 ± 0.34 | 1.05 ± 0.45 |
| ECV (%) | 43.4 ± 15.9 | 42.7 ± 12.7 |
| LV Mass (g) | 99.9 ± 15.5 | 96.1 ± 17.9 |
| Infarcted Mass (g) | 20.2 ± 16.1 | 15.2 ± 10.4 *** |
| MPO (ng/mL) | 41.0 ± 22.2 | 75.4 ± 56.5 ** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Souza-Neto, F.V.; Islas, F.; Jiménez-González, S.; Luaces, M.; Ramchandani, B.; Romero-Miranda, A.; Delgado-Valero, B.; Roldan-Molina, E.; Saiz-Pardo, M.; Cerón-Nieto, M.Á.; et al. Mitochondrial Oxidative Stress Promotes Cardiac Remodeling in Myocardial Infarction through the Activation of Endoplasmic Reticulum Stress. Antioxidants 2022, 11, 1232. https://doi.org/10.3390/antiox11071232
Souza-Neto FV, Islas F, Jiménez-González S, Luaces M, Ramchandani B, Romero-Miranda A, Delgado-Valero B, Roldan-Molina E, Saiz-Pardo M, Cerón-Nieto MÁ, et al. Mitochondrial Oxidative Stress Promotes Cardiac Remodeling in Myocardial Infarction through the Activation of Endoplasmic Reticulum Stress. Antioxidants. 2022; 11(7):1232. https://doi.org/10.3390/antiox11071232
Chicago/Turabian StyleSouza-Neto, Francisco V., Fabian Islas, Sara Jiménez-González, María Luaces, Bunty Ramchandani, Ana Romero-Miranda, Beatriz Delgado-Valero, Elena Roldan-Molina, Melchor Saiz-Pardo, Mª Ángeles Cerón-Nieto, and et al. 2022. "Mitochondrial Oxidative Stress Promotes Cardiac Remodeling in Myocardial Infarction through the Activation of Endoplasmic Reticulum Stress" Antioxidants 11, no. 7: 1232. https://doi.org/10.3390/antiox11071232