
Opioids and Vitamin C: Known Interactions and Potential for Redox-Signaling Crosstalk

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
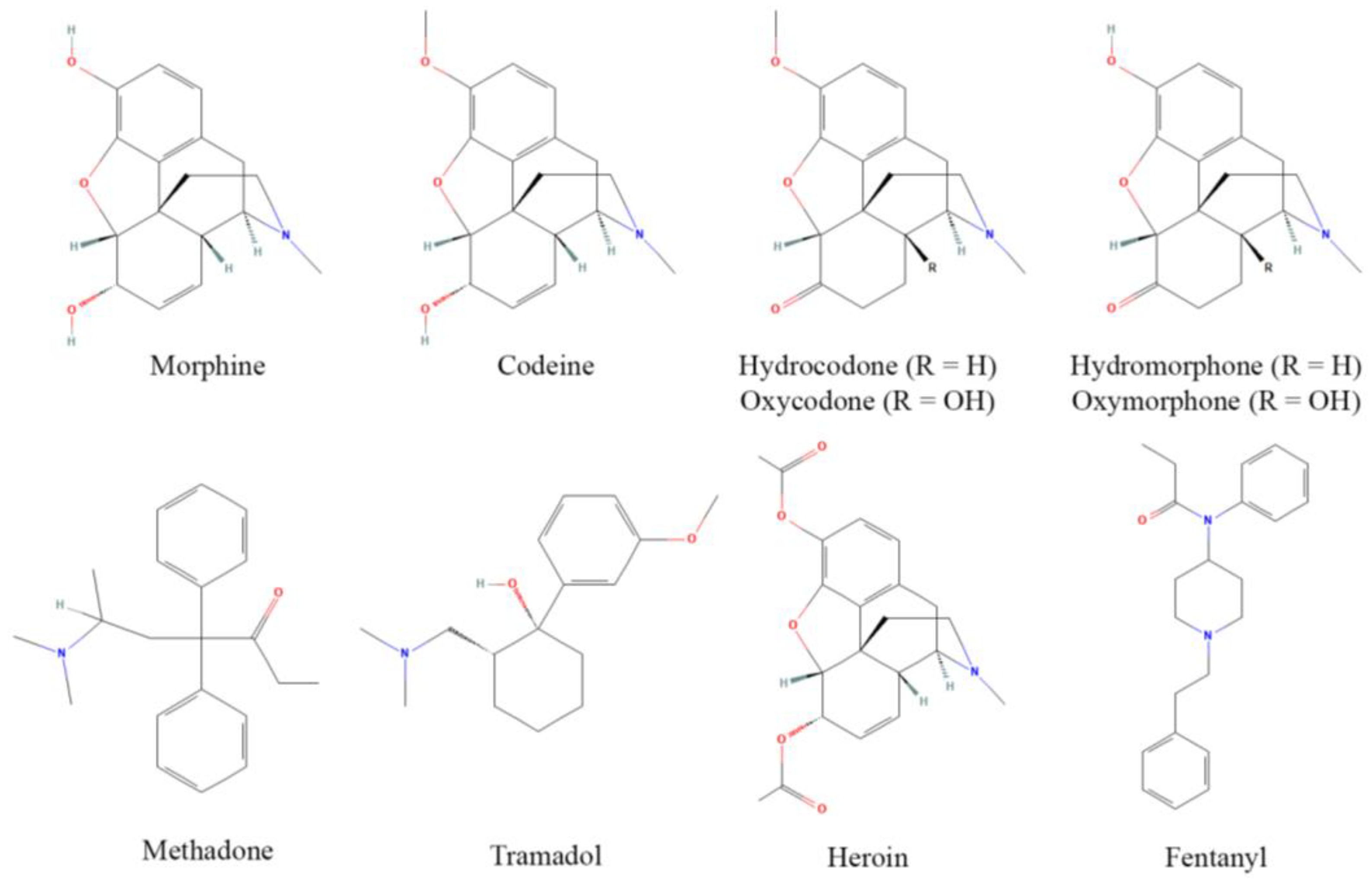
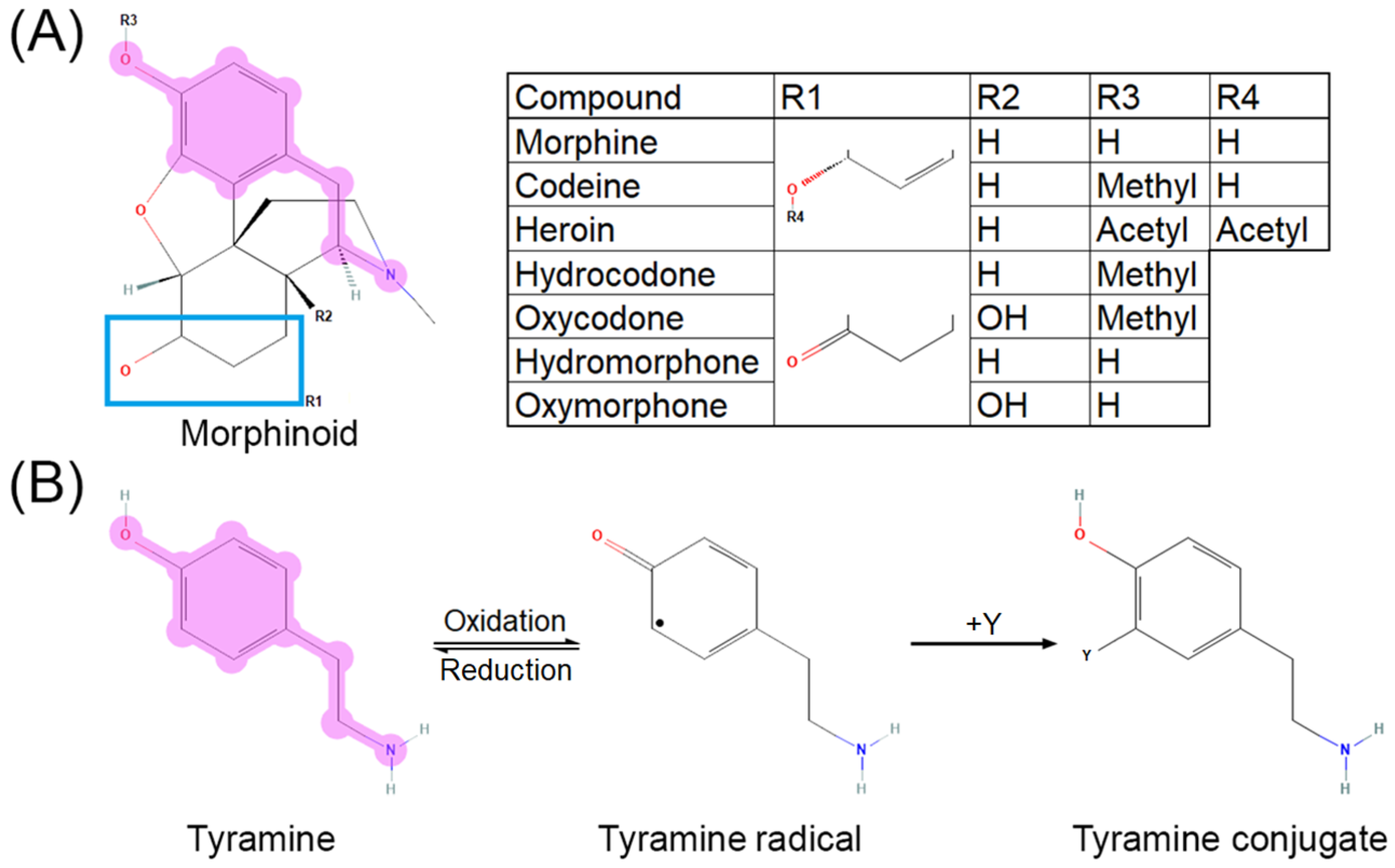
2. Opioid Activity and Metabolism
3. Vitamin C Activity and Metabolism
4. Opioids, Vitamin C, and Neurotransmission
5. Opioids, Vitamin C, and Direct Oxidative Stress Modulation
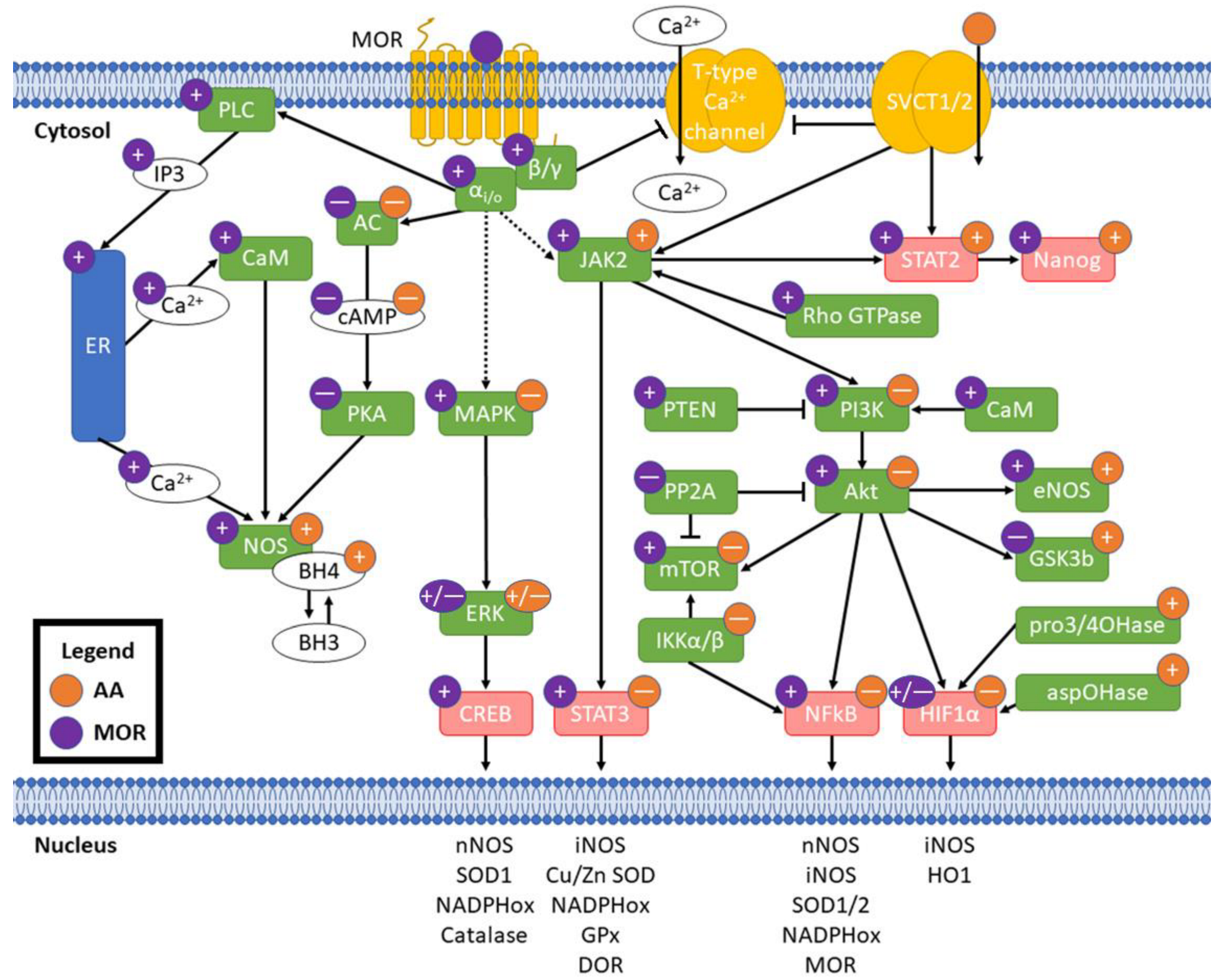
6. MOR Activity and NOS
7. Vitamin C and MOR: Potential for Crosstalk
8. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | ascorbic acid |
| AC | adenylate cyclase |
| Akt | protein kinase B |
| aspOHase | asparaginyl hydroxylase |
| BH3 | trihydrobiopterin |
| BH4 | tetrahydrobiopterin |
| CaM | calmodulin |
| cAMP | cyclic adenosine monophosphate |
| CAT | catalase |
| cNOS | constitutive nitric oxide synthase |
| CREB | cAMP response element-binding protein |
| DHA | dehydroascorbic acid |
| DOR | δ opioid receptor |
| eNOS | endothelial nitric oxide synthase |
| ER | endoplasmic reticulum |
| ERK | extracellular-regulated signaling kinase |
| GPCR | G-protein-coupled receptor |
| GPx | glutathione peroxidase |
| GSK3β | glycogen synthase kinase 3β |
| Gulo | L-gulonolactone oxidase |
| HIF1α | hypoxia-induced factor 1α |
| IKK | IκB kinase |
| IP3 | inositol-3-phosphate |
| JAK | janus kinase |
| LC | locus ceruleus |
| MAPK | mitogen-activated protein kinase |
| MOR | μ opioid receptor |
| mTOR | mammalian target of rapamycin |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| nNOS | neuronal nitric oxide synthase |
| NO | nitric oxide |
| OIRD | opioid-induced respiratory disorder |
| ONOO- | peroxynitrite |
| PI3K | phosphoinositol-3-kinase |
| PKA | protein kinase A |
| PLC | phospholipase C |
| PP2A | protein phosphatase 2A |
| pro3/4OHase | prolyl 3- and prolyl 4-hydroxylases |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| SOD | superoxide dismutase |
| STAT1/2/3 | signal transducer and activator of transcription 1/2/3 |
| SVCT1/2 | sodium-dependent vitamin C transporters 1/2 |
| TH | tyrosine hydroxylase |
| TPH | tryptophan hydroxylase |
| VTA | ventral tegmental area |
| αi/o | Gαi/o subunit |
| β/γ | Gβ/γ subunit |
References
- Swegle, J.M.; Logemann, C. Management of common opioid-induced adverse effects. Am. Fam. Physician 2006, 74, 1347–1354. [Google Scholar] [PubMed]
- Benyamin, R.; Trescot, A.M.; Datta, S.; Buenaventura, R.; Adlaka, R.; Sehgal, N.; Glaser, S.E.; Vallejo, R. Opioid complications and side effects. Pain Physician 2008, 11, S105–S120. [Google Scholar] [CrossRef] [PubMed]
- Porreca, F.; Ossipov, M.H. Nausea and Vomiting Side Effects with Opioid Analgesics during Treatment of Chronic Pain: Mechanisms, Implications, and Management Options. Pain Med. 2009, 10, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Vowles, K.E.; McEntee, M.L.; Julnes, P.S.; Frohe, T.; Ney, J.P.; van der Goes, D.N. Rates of opioid misuse, abuse, and addiction in chronic pain. Pain 2015, 156, 569–576. [Google Scholar] [CrossRef] [Green Version]
- Boom, M.; Niesters, M.; Sarton, E.; Aarts, L.; Smith, T.W.; Dahan, A. Non-Analgesic Effects of Opioids: Opioid-induced Respiratory Depression. Curr. Pharm. Des. 2012, 18, 5994–6004. [Google Scholar] [CrossRef]
- Talwar, D.; McConnachie, A.; Welsh, P.; Upton, M.; O'Reilly, D.; Smith, G.D.; Watt, G.; Sattar, N. Which Circulating Antioxidant Vitamins Are Confounded by Socioeconomic Deprivation? The MIDSPAN Family Study. PLoS ONE 2010, 5, e11312. [Google Scholar] [CrossRef]
- Bloodbook Blood Test Results—Normal Ranges. Available online: www.bloodbook.com/ranges.html (accessed on 4 June 2022).
- Bhagavan, N.V.; Ha, C.-E. Protein and Amino Acid Metabolism. In Essentials of Medical Biochemistry; Elsevier: Amsterdam, The Netherlands, 2015; pp. 227–268. [Google Scholar]
- Majewska, M.D.; Bell, J.A.; London, E.D. Regulation of the NMDA receptor by redox phenomena: Inhibitory role of ascorbate. Brain Res. 1990, 537, 328–332. [Google Scholar] [CrossRef]
- Moores, J. Vitamin C: A wound healing perspective. Br. J. Community Nurs. 2013, 18, S6–S11. [Google Scholar] [CrossRef]
- Cerullo, G.; Negro, M.; Parimbelli, M.; Pecoraro, M.; Perna, S.; Liguori, G.; Rondanelli, M.; Cena, H.; D’Antona, G. The Long History of Vitamin C: From Prevention of the Common Cold to Potential Aid in the Treatment of COVID-19. Front. Immunol. 2020, 11, 574029. [Google Scholar] [CrossRef]
- Zhou, J.; Si, P.; Ruan, Z.; Ma, S.; Yan, X.; Sun, L.; Peng, F.; Yuan, H.; Cai, D.; Ding, D.; et al. Primary studies on heroin abuse and injury induced by oxidation and lipoperoxidation. Chin. Med. J. 2001, 114, 297–302. [Google Scholar]
- Zhou, J.F.; Yan, X.F.; Ruan, Z.R.; Peng, F.Y.; Cai, D.; Yuan, H.; Sun, L.; Ding, D.Y.; Xu, S.S. Heroin abuse and nitric oxide, oxidation, peroxidation, lipoperoxidation. Biomed. Environ. Sci. 2000, 13, 131–139. [Google Scholar] [PubMed]
- Johnston, P.; Chahl, L.A. Chronic treatment with ascorbic acid inhibits the morphine withdrawal response in guinea-pigs. Neurosci. Lett. 1992, 135, 23–27. [Google Scholar] [CrossRef]
- Alaei, H.A.; Ramshini, E.; Talkhooncheh, M.; Shahidani, S. The effect of vitamin C on morphine self-administration in rats. Adv. Biomed. Res. 2014, 3, 178. [Google Scholar] [CrossRef]
- Evangelou, A.; Kalfakakou, V.; Georgakas, P.; Koutras, V.; Vezyraki, P.; Iliopoulou, L.; Vadalouka, A. Ascorbic acid (vitamin C) effects on withdrawal syndrome of heroin abusers. In Vivo 2000, 14, 363–366. [Google Scholar]
- Zelfand, E. Vitamin C, Pain and Opioid Use Disorder. Integr. Med. 2020, 19, 18–29. [Google Scholar]
- Scott, J.; Winfield, A.; Kennedy, E.; Bond, C. Laboratory study of the effects of citric and ascorbic acids on injections prepared with brown heroin. Int. J. Drug Policy 2000, 11, 417–422. [Google Scholar] [CrossRef]
- Andersen, J.M.; Bogen, I.L.; Karinen, R.; Brochmann, G.W.; Mørland, J.; Vindenes, V.; Boix, F. Does the preparation for intravenous administration affect the composition of heroin injections? A controlled laboratory study. Addiction 2021, 116, 3104–3112. [Google Scholar] [CrossRef]
- Kanazi, G.E.; El-Khatib, M.F.; Karam, V.G.Y.; Hanna, J.E.; Masri, B.; Aouad, M.T. Effect of vitamin C on morphine use after laparoscopic cholecystectomy: A randomized controlled trial. Can. J. Anaesth. 2012, 59, 538–543. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Roffey, D.; Dion, C.-A.; Arab, A.; Wai, E.K. Effect of Perioperative Vitamin C Supplementation on Postoperative Pain and the Incidence of Chronic Regional Pain Syndrome. Clin. J. Pain 2016, 32, 179–185. [Google Scholar] [CrossRef]
- Tunay, D.L.; Ilgınel, M.T.; Ünlügenç, H.; Tunay, M.; Karacaer, F.; Biricik, E. Comparison of the effects of preoperative melatonin or vitamin C administration on postoperative analgesia. Bosn. J. Basic Med. Sci. 2020, 20, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Chaitanya, N.C.; Muthukrishnan, A.; Krishnaprasad, C.; Sanjuprasanna, G.; Pillay, P.; Mounika, B. An Insight and Update on the Analgesic Properties of Vitamin C. J. Pharm. Bioallied Sci. 2018, 10, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.C.; McCall, C. The role of vitamin C in the treatment of pain: New insights. J. Transl. Med. 2017, 15, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- E Willette, R.; Thomas, B.L.; Barnett, G. Inhibition of morphine analgesia by ascorbate. Res. Commun. Chem. Pathol. Pharmacol. 1983, 42, 485–491. [Google Scholar] [CrossRef]
- Khanna, N.; Sharma, S.K. Megadoses of vitamin C prevent the development of tolerance and physical dependence on morphine in mice. Life Sci. 1983, 33, 401–404. [Google Scholar] [CrossRef]
- Alaei, H.; Esmaeili, M.; Nasimi, A.; Pourshanazari, A. Ascorbic acid decreases morphine self-administration and withdrawal symptoms in rats. Pathophysiology 2005, 12, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Enrico, P.; A Mura, M.; Esposito, G.; Serra, P.; Migheli, R.; De Natale, G.; Desole, M.S.; Miele, M.; Miele, E. Effect of naloxone on morphine-induced changes in striatal dopamine metabolism and glutamate, ascorbic acid and uric acid release in freely moving rats. Brain Res. 1998, 797, 94–102. [Google Scholar] [CrossRef]
- Desole, M.S.; Esposito, G.; Fresu, L.; Migheli, R.; Enrico, P.; Mura, M.A.; De Natale, G.; Miele, E.; Miele, M. Effects of morphine treatment and withdrawal on striatal and limbic monoaminergic activity and ascorbic acid oxidation in the rat. Brain Res. 1996, 723, 154–161. [Google Scholar] [CrossRef]
- Enrico, P.; Esposito, G.; Mura, M.A.; Fresu, L.; De Natale, G.; Miele, E.; Desole, M.S.; Miele, M. Effect of morphine on striatal dopamine metabolism and ascorbic acid and uric acid release in freely moving rats. Brain Res. 1997, 745, 173–182. [Google Scholar] [CrossRef]
- Ordan, B.A.; Cvejic, S.; Devi, L.A. Opioids and Their Complicated Receptor Complexes. Neuropsychopharmacology 2000, 23, S5–S18. [Google Scholar] [CrossRef]
- Ballantyne, J.C.; Sullivan, M.D. Discovery of endogenous opioid systems: What it has meant for the clinician's understanding of pain and its treatment. Pain 2017, 158, 2290–2300. [Google Scholar] [CrossRef]
- Pasternak, G.; Pan, Y.-X. Mu opioid receptors in pain management. Acta Anaesthesiol. Taiwanica 2011, 49, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Valentino, R.J.; Volkow, N.D. Untangling the complexity of opioid receptor function. Neuropsychopharmacology 2018, 43, 2514–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef] [PubMed]
- Human Protein Atlas. Available online: https://www.proteinatlas.org/ (accessed on 15 May 2022).
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, N.; Smith, H.S.; Manchikanti, L. Peripherally acting opioids and clinical implications for pain control. Pain Physician 2011, 14, 249–258. [Google Scholar] [CrossRef]
- Connor, M.; Christie, M. Opioid receptor signalling mechanisms. Clin. Exp. Pharmacol. Physiol. 1999, 26, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Gonzalez-Zulueta, M.; Huang, H.; Herring, W.J.; Ahn, S.; Ginty, D.D.; Dawson, V.L.; Dawson, T.M. Dynamic regulation of neuronal NO synthase transcription by calcium influx through a CREB family transcription factor-dependent mechanism. Proc. Natl. Acad. Sci. USA 2000, 97, 8617–8622. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Odom, D.T.; Koo, S.-H.; Conkright, M.D.; Canettieri, G.; Best, J.; Chen, H.; Jenner, R.; Herbolsheimer, E.; Jacobsen, E.; et al. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 4459–4464. [Google Scholar] [CrossRef] [Green Version]
- Listos, J.; Łupina, M.; Talarek, S.; Mazur, A.; Orzelska-Górka, J.; Kotlińska, J. The Mechanisms Involved in Morphine Addiction: An Overview. Int. J. Mol. Sci. 2019, 20, 4302. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.H. Opioid metabolism. In Mayo Clinic Proceedings; Elsevier: Amsterdam, The Netherlands, 2009; pp. 613–624. [Google Scholar]
- Stone, A.N.; Mackenzie, P.; Galetin, A.; Houston, J.B.; Miners, J.O. Isoform selectivity and kinetics of morphine 3- and 6-glucuronidation by human udp-glucuronosyltransferases: Evidence for atypical glucuronidation kinetics by ugt2b7. Drug Metab. Dispos. 2003, 31, 1086–1089. [Google Scholar] [CrossRef] [Green Version]
- Veith, A.; Moorthy, B. Role of cytochrome P450s in the generation and metabolism of reactive oxygen species. Curr. Opin. Toxicol. 2018, 7, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Feltrin, C.; Farias, I.V.; Sandjo, L.P.; Reginatto, F.H.; Simões, C.M.O. Effects of Standardized Medicinal Plant Extracts on Drug Metabolism Mediated by CYP3A4 and CYP2D6 Enzymes. Chem. Res. Toxicol. 2020, 33, 2408–2419. [Google Scholar] [CrossRef] [PubMed]
- Klimas, R.; Mikus, G. Morphine-6-glucuronide is responsible for the analgesic effect after morphine administration: A quantitative review of morphine, morphine-6-glucuronide, and morphine-3-glucuronide. Br. J. Anaesth. 2014, 113, 935–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Kang, Y.S.; Bickel, U.; Pardridge, W.M. Blood-brain barrier permeability to morphine-6-glucuronide is markedly reduced compared with morphine. Drug Metab. Dispos. 1997, 25, 768–771. [Google Scholar]
- Samer, C.F.; Daali, Y.; Wagner, M.; Hopfgartner, G.; Eap, C.B.; Rebsamen, M.C.; Rossier, M.F.; Hochstrasser, D.; Dayer, P.; Desmeules, J.A. The effects of CYP2D6 and CYP3A activities on the pharmacokinetics of immediate release oxycodone. Br. J. Pharmacol. 2010, 160, 907–918. [Google Scholar] [CrossRef]
- Balyan, R.; Mecoli, M.; Venkatasubramanian, R.; Chidambaran, V.; Kamos, N.; Clay, S.; Moore, D.L.; Mavi, J.; Glover, C.D.; Szmuk, P.; et al. CYP2D6 pharmacogenetic and oxycodone pharmacokinetic association study in pediatric surgical patients. Pharmacogenomics 2017, 18, 337–348. [Google Scholar] [CrossRef] [Green Version]
- Owen, J.A.; Nakatsu, K. Diacetylmorphine (heroin) hydrolases in human blood. Can. J. Physiol. Pharmacol. 1983, 61, 870–875. [Google Scholar] [CrossRef]
- Lockridge, O.; Mottershaw-Jackson, N.; Eckerson, H.W.; La Du, B.N. Hydrolysis of diacetylmorphine (heroin) by human serum cholinesterase. J. Pharmacol. Exp. Ther. 1980, 215, 1–8. [Google Scholar]
- Garbuz, O.; Gulea, A.; Dyniewicz, J.; Zablocka, B.; Lipkowski, A.W. The non-opioid receptor, antioxidant properties of morphine and the opioid peptide analog biphalin. Peptides 2015, 63, 1–3. [Google Scholar] [CrossRef]
- Gülçin, I.; Beydemir, S.; Alici, H.A.; Elmastaş, M.; Büyükokuroğlu, M.E. In vitro antioxidant properties of morphine. Pharmacol. Res. 2004, 49, 59–66. [Google Scholar] [CrossRef]
- Nilsen-Moe, A.; Reinhardt, C.R.; Glover, S.D.; Liang, L.; Hammes-Schiffer, S.; Hammarström, L.; Tommos, C. Proton-Coupled Electron Transfer from Tyrosine in the Interior of a de novo Protein: Mechanisms and Primary Proton Acceptor. J. Am. Chem. Soc. 2020, 142, 11550–11559. [Google Scholar] [CrossRef] [PubMed]
- Radi, R. Protein Tyrosine Nitration: Biochemical Mechanisms and Structural Basis of Functional Effects. Acc. Chem. Res. 2013, 46, 550–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyndale, R.F.; Droll, K.P.; Sellers, E. Genetically deficient CYP2D6 metabolism provides protection against oral opiate dependence. Pharmacogenetics 1997, 7, 375–379. [Google Scholar] [CrossRef]
- Samer, C.; Daali, Y.; Wagner, M.; Hopfgartner, G.; Eap, C.; Rebsamen, M.; Rossier, M.; Hochstrasser, D.; Dayer, P.; Desmeules, J. Genetic polymorphisms and drug interactions modulating CYP2D6 and CYP3A activities have a major effect on oxycodone analgesic efficacy and safety. J. Cereb. Blood Flow Metab. 2010, 160, 919–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toki, S.; Yamano, S. Production of Morphinone as a Metabolite of Morphine and Its Physiological Role. YAKUGAKU ZASSHI 1999, 119, 249–267. [Google Scholar] [CrossRef] [Green Version]
- Yamano, S.; Takahashi, A.; Todaka, T.; Toki, S. In vivo and in vitro formation of morphinone from morphine in rat. Xenobiotica 1997, 27, 645–656. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Amatore, C.; Arbault, S.; Ferreira, D.C.M.; Tapsoba, I.; Verchier, Y. Vitamin C stimulates or attenuates reactive oxygen and nitrogen species (ROS, RNS) production depending on cell state: Quantitative amperometric measurements of oxidative bursts at PLB-985 and RAW 264.7 cells at the single cell level. J. Electroanal. Chem. 2008, 615, 34–44. [Google Scholar] [CrossRef]
- Cobley, J.N.; McHardy, H.; Morton, J.P.; Nikolaidis, M.G.; Close, G.L. Influence of vitamin C and vitamin E on redox signaling: Implications for exercise adaptations. Free Radic. Biol. Med. 2015, 84, 65–76. [Google Scholar] [CrossRef] [Green Version]
- Paciolla, C.; Fortunato, S.; Dipierro, N.; Paradiso, A.; De Leonardis, S.; Mastropasqua, L.; de Pinto, M.C. Vitamin C in Plants: From Functions to Biofortification. Antioxidants 2019, 8, 519. [Google Scholar] [CrossRef] [Green Version]
- Yang, H. Conserved or Lost: Molecular Evolution of the Key Gene GULO in Vertebrate Vitamin C Biosynthesis. Biochem. Genet. 2013, 51, 413–425. [Google Scholar] [CrossRef]
- Jiao, Y.; Zhang, J.; Yan, J.; Stuart, J.; Gibson, G.; Lu, L.; Willaims, R.; Wang, Y.J.; Gu, W. Differential gene expression between wild-type and Gulo-deficient mice supplied with vitamin C. Genet. Mol. Biol. 2011, 34, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Curran, C.P.; Nebert, D.W.; Patel, K.V.; Williams, M.T.; Vorhees, C.V. Effect of vitamin C deficiency during postnatal development on adult behavior: Functional phenotype ofGulo(−/−)knockout mice. Genes Brain Behav. 2012, 11, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Aumailley, L.; Warren, A.; Garand, C.; Dubois, M.J.; Paquet, E.R.; Le Couteur, D.G.; Marette, A.; Cogger, V.C.; Lebel, M. Vitamin C modulates the metabolic and cytokine profiles, alleviates hepatic endoplasmic reticulum stress, and increases the life span of Gulo−/− mice. Aging 2016, 8, 458–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shyu, K.-G.; Chang, C.-C.; Yeh, Y.-C.; Sheu, J.-R.; Chou, D.-S. Mechanisms of Ascorbyl Radical Formation in Human Platelet-Rich Plasma. BioMed Res. Int. 2014, 2014, 614506. [Google Scholar] [CrossRef] [PubMed]
- ScienceLab.com Material Safety Data Sheet Ascorbic Acid MSDS 2022. Available online: https://sciencelab.com/ (accessed on 12 April 2022).
- Levine, M.; Rumsey, S.C.; Daruwala, R.; Park, J.B.; Wang, Y. Criteria and Recommendations for Vitamin C Intake. JAMA 1999, 281, 1415–1423. [Google Scholar] [CrossRef]
- Mandl, J.; Szarka, A.; Bánhegyi, G. Vitamin C: Update on physiology and pharmacology. J. Cereb. Blood Flow Metab. 2009, 157, 1097–1110. [Google Scholar] [CrossRef] [Green Version]
- Buettner, G.R.; Jurkiewicz, B.A. Catalytic metals, ascorbate and free radicals: Combinations to avoid. Radiat. Res. 1996, 145, 532–541. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Cobb, C.E.; Hill, K.E.; Burk, R.F.; May, J.M. Mitochondrial Uptake and Recycling of Ascorbic Acid. Arch. Biochem. Biophys. 2001, 387, 143–153. [Google Scholar] [CrossRef]
- Lykkesfeldt, J.; Tveden-Nyborg, P. The Pharmacokinetics of Vitamin C. Nutrients 2019, 11, 2412. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Schellhorn, H.E. New Developments and Novel Therapeutic Perspectives for Vitamin C. J. Nutr. 2007, 137, 2171–2184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rock, C.L.; Jacob, R.A.; Bowen, P. Update on the Biological Characteristics of the Antioxidant Micronutrients: Vitamin C, Vitamin E, and the Carotenoids. J. Am. Diet. Assoc. 1996, 96, 693–702. [Google Scholar] [CrossRef]
- Lane, D.J.; Richardson, D.R. The active role of vitamin C in mammalian iron metabolism: Much more than just enhanced iron absorption! Free Radic. Biol. Med. 2014, 75, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Ballaz, S.J.; Rebec, G.V. Neurobiology of vitamin C: Expanding the focus from antioxidant to endogenous neuromodulator. Pharmacol. Res. 2019, 146, 104321. [Google Scholar] [CrossRef] [PubMed]
- Jacob, R.A.; Sotoudeh, G. Vitamin C Function and Status in Chronic Disease. Nutr. Clin. Care 2002, 5, 66–74. [Google Scholar] [CrossRef]
- Figueroa-Méndez, R.; Rivas-Arancibia, S. Vitamin C in Health and Disease: Its Role in the Metabolism of Cells and Redox State in the Brain. Front. Physiol. 2015, 6, 397. [Google Scholar] [CrossRef] [Green Version]
- Mou, L.; Lankford-Turner, P.; Leander, M.V.; Bissonnette, R.P.; Donahoe, R.M.; Royal, W. RXR-induced TNF-α suppression is reversed by morphine in activated U937 cells. J. Neuroimmunol. 2004, 147, 99–105. [Google Scholar] [CrossRef]
- Gu, P.F.; Wu, C.F.; Yang, J.Y.; Shang, Y.; Hou, Y.; Bi, X.L.; Dai, F. Differential effects of drug-induced ascorbic acid release in the striatum and nucleus accumbens of freely moving rats. Neurosci. Lett. 2006, 399, 79–84. [Google Scholar] [CrossRef]
- Cisternas, P.; Silva-Alvarez, C.; Martínez, F.; Fernández, E.; Ferrada, L.; Oyarce, K.; Salazar, K.; Bolanos, J.P.; Nualart, F. The oxidized form of vitamin C, dehydroascorbic acid, regulates neuronal energy metabolism. J. Neurochem. 2014, 129, 663–671. [Google Scholar] [CrossRef]
- Johnston, C.S.; Corte, C.; Swan, P.D. Marginal vitamin C status is associated with reduced fat oxidation during submaximal exercise in young adults. Nutr. Metab. 2006, 3, 35. [Google Scholar] [CrossRef] [Green Version]
- Chambial, S.; Dwivedi, S.; Shukla, K.K.; John, P.J.; Sharma, P. Vitamin C in Disease Prevention and Cure: An Overview. Indian J. Clin. Biochem. 2013, 28, 314–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosten, T.R.; George, T.P. The Neurobiology of Opioid Dependence: Implications for Treatment. Sci. Pract. Perspect. 2002, 1, 13–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bubier, J.A.; He, H.; Philip, V.M.; Roy, T.; Hernandez, C.M.; Bernat, R.; Donohue, K.D.; O’Hara, B.F.; Chesler, E.J. Genetic variation regulates opioid-induced respiratory depression in mice. Sci. Rep. 2020, 10, 14970. [Google Scholar] [CrossRef]
- Hayes, J.A.; Kottick, A.; Picardo, M.C.D.; Halleran, A.D.; Smith, R.D.; Smith, G.D.; Saha, M.S.; Del Negro, C.A. Transcriptome of neonatal preBötzinger complex neurones in Dbx1 reporter mice. Sci. Rep. 2017, 7, 8669. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.C.; Ellenberger, H.H.; Ballanyi, K.; Richter, D.W.; Feldman, J.L. Pre-Bötzinger Complex: A Brainstem Region that May Generate Respiratory Rhythm in Mammals. Science 1991, 254, 726–729. [Google Scholar] [CrossRef]
- Raehal, K.M.; Bohn, L.M. β-Arrestins: Regulatory Role and Therapeutic Potential in Opioid and Cannabinoid Receptor-Mediated Analgesia. Arrestins-Pharmacol. Ther. Potential 2014, 219, 427–443. [Google Scholar] [CrossRef] [Green Version]
- Raehal, K.M.; Walker, J.K.L.; Bohn, L. Morphine Side Effects in β-Arrestin 2 Knockout Mice. J. Pharmacol. Exp. Ther. 2005, 314, 1195–1201. [Google Scholar] [CrossRef] [Green Version]
- Bohn, L.M.; Gainetdinov, R.; Lin, F.-T.; Lefkowitz, R.J.; Caron, M.G. μ-Opioid receptor desensitization by β-arrestin-2 determines morphine tolerance but not dependence. Nature 2000, 408, 720–723. [Google Scholar] [CrossRef]
- Stahl, E.L.; Schmid, C.L.; Acevedo-Canabal, A.; Read, C.; Grim, T.W.; Kennedy, N.M.; Bannister, T.D.; Bohn, L.M. G protein signaling–biased mu opioid receptor agonists that produce sustained G protein activation are noncompetitive agonists. Proc. Natl. Acad. Sci. USA 2021, 118, e2102178118. [Google Scholar] [CrossRef]
- Gillis, A.; Kliewer, A.; Kelly, E.; Henderson, G.; Christie, M.J.; Schulz, S.; Canals, M. Critical Assessment of G Protein-Biased Agonism at the μ-Opioid Receptor. Trends Pharmacol. Sci. 2020, 41, 947–959. [Google Scholar] [CrossRef]
- Stanczyk, M.A.; Kandasamy, R. Biased agonism: The quest for the analgesic holy grail. PAIN Rep. 2018, 3, e650. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, G.W.; Pan, Y.-X. Mu Opioids and Their Receptors: Evolution of a Concept. Pharmacol. Rev. 2013, 65, 1257–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agus, D.B.; Gambhir, S.S.; Pardridge, W.M.; Spielholz, C.; Baselga, J.; Vera, J.C.; Golde, D.W. Vitamin C crosses the blood-brain barrier in the oxidized form through the glucose transporters. J. Clin. Investig. 1997, 100, 2842–2848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, D.J.R.; Lawen, A. The Glutamate Aspartate Transporter (GLAST) Mediates l-Glutamate-Stimulated Ascorbate-Release via Swelling-Activated Anion Channels in Cultured Neonatal Rodent Astrocytes. Cell Biophys. 2013, 65, 107–119. [Google Scholar] [CrossRef]
- Lane, D.J.R.; Lawen, A. Ascorbate and plasma membrane electron transport—Enzymes vs efflux. Free Radic. Biol. Med. 2009, 47, 485–495. [Google Scholar] [CrossRef]
- Sekiguchi, F.; Kawabata, A. T-type Calcium Channels: Functional Regulation and Implication in Pain Signaling. J. Pharmacol. Sci. 2013, 122, 244–250. [Google Scholar] [CrossRef] [Green Version]
- Ward, M.S.; Lamb, J.; May, J.M.; Harrison, F.E. Behavioral and monoamine changes following severe vitamin C deficiency. J. Neurochem. 2013, 124, 363–375. [Google Scholar] [CrossRef] [Green Version]
- Yen, G.-C.; Hsieh, C.-L. Antioxidant Effects of Dopamine and Related Compounds. Biosci. Biotechnol. Biochem. 1997, 61, 1646–1649. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef] [PubMed]
- Colamartino, M.; Santoro, M.; Duranti, G.; Sabatini, S.; Ceci, R.; Testa, A.; Padua, L.; Cozzi, R. Evaluation of Levodopa and Carbidopa Antioxidant Activity in Normal Human Lymphocytes In Vitro: Implication for Oxidative Stress in Parkinson’s Disease. Neurotox. Res. 2015, 27, 106–117. [Google Scholar] [CrossRef]
- Álvarez-Diduk, R.; Galano, A. Adrenaline and Noradrenaline: Protectors against Oxidative Stress or Molecular Targets? J. Phys. Chem. B 2015, 119, 3479–3491. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.A.; Chae, S.; Lee, K.H.; Zhang, R.; Jung, M.S.; You, H.J.; Kim, J.S.; Hyun, J.W. Antioxidant effect of homogenetisic acid on hydrogen peroxide induced oxidative stress in human lung fibroblast cells. Biotechnol. Bioprocess Eng. 2005, 10, 556–563. [Google Scholar] [CrossRef]
- Boundy, V.A.; Gold, S.J.; Messer, C.J.; Chen, J.; Son, J.H.; Joh, T.H.; Nestler, E.J. Regulation of Tyrosine Hydroxylase Promoter Activity by Chronic Morphine in TH9.0-LacZ Transgenic Mice. J. Neurosci. 1998, 18, 9989–9995. [Google Scholar] [CrossRef]
- Beitner-Johnson, D.; Nestler, E.J. Morphine and Cocaine Exert Common Chronic Actions on Tyrosine Hydroxylase in Dopaminergic Brain Reward Regions. J. Neurochem. 1991, 57, 344–347. [Google Scholar] [CrossRef]
- Bariselli, S.; Glangetas, C.; Tzanoulinou, S.; Bellone, C. Ventral tegmental area subcircuits process rewarding and aversive experiences. J. Neurochem. 2016, 139, 1071–1080. [Google Scholar] [CrossRef]
- Boadle-Bibe, M.C.; Johannessen, J.N.; Narasimhachari, N.; Phan, T.-H. Activation of cortical tryptophan hydroxylase by acute morphine treatment: Blockade by 6-hydroxydopamine. Eur. J. Pharmacol. 1987, 139, 193–204. [Google Scholar] [CrossRef]
- Bhat, R.S.; Bhaskaran, M.; Mongia, A.; Hitosugi, N.; Singhal, P.C. Morphine-induced macrophage apoptosis: Oxidative stress and strategies for modulation. J. Leukoc. Biol. 2004, 75, 1131–1138. [Google Scholar] [CrossRef]
- Cai, Y.; Yang, L.; Hu, G.; Chen, X.; Niu, F.; Yuan, L.; Liu, H.; Xiong, H.; Arikkath, J.; Buch, S. Regulation of morphine-induced synaptic alterations: Role of oxidative stress, ER stress, and autophagy. J. Cell Biol. 2016, 215, 245–258. [Google Scholar] [CrossRef] [Green Version]
- Skrabalova, J.; Drastichova, Z.; Novotny, J. Morphine as a Potential Oxidative Stress-Causing Agent. Mini-Rev. Org. Chem. 2013, 10, 367–372. [Google Scholar] [CrossRef] [Green Version]
- Wilcox, B.J.; Ritenour-Rodgers, K.J.; Asser, A.S.; Baumgart, L.E.; Baumgart, M.A.; Boger, D.L.; DeBlassio, J.L.; Delong, M.A.; Glufke, U.; Henz, M.E.; et al. N-Acylglycine Amidation: Implications for the Biosynthesis of Fatty Acid Primary Amides. Biochemistry 1999, 38, 3235–3245. [Google Scholar] [CrossRef] [PubMed]
- Lipina, C.; Hundal, H.S. Modulation of cellular redox homeostasis by the endocannabinoid system. Open Biol. 2016, 6, 150276. [Google Scholar] [CrossRef] [Green Version]
- Al-Azzam, N.; Teegala, L.R.; Pokhrel, S.; Ghebreigziabher, S.; Chachkovskyy, T.; Thodeti, S.; Gavilanes, I.; Covington, K.; Thodeti, C.K.; Paruchuri, S. Transient Receptor Potential Vanilloid channel regulates fibroblast differentiation and airway remodeling by modulating redox signals through NADPH Oxidase 4. Sci. Rep. 2020, 10, 9827. [Google Scholar] [CrossRef] [PubMed]
- Farfariello, V.; Amantini, C.; Santoni, G. Transient receptor potential vanilloid 1 activation induces autophagy in thymocytes through ROS-regulated AMPK and Atg4C pathways. J. Leukoc. Biol. 2012, 92, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Salarian, A.; Kadkhodaee, M.; Zahmatkesh, M.; Seifi, B.; Bakhshi, E.; Akhondzadeh, S.; Adeli, S.; Askari, H.; Arbabi, M. Opioid Use Disorder Induces Oxidative Stress and Inflammation: The Attenuating Effect of Methadone Maintenance Treatment. Iran. J. Psychiatry 2018, 13, 46–54. [Google Scholar]
- Ajayi, A.F.; Akhigbe, R.E. Codeine-induced sperm DNA damage is mediated predominantly by oxidative stress rather than apoptosis. Redox Rep. 2020, 25, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, H.M.; Mahmoud, A.M. Chronic exposure to the opioid tramadol induces oxidative damage, inflammation and apoptosis, and alters cerebral monoamine neurotransmitters in rats. Biomed. Pharmacother. 2019, 110, 239–247. [Google Scholar] [CrossRef]
- Motaghinejad, M.; Karimian, M.; Motaghinejad, O.; Shabab, B.; Yazdani, I.; Fatima, S. Protective effects of various dosage of Curcumin against morphine induced apoptosis and oxidative stress in rat isolated hippocampus. Pharmacol. Rep. 2015, 67, 230–235. [Google Scholar] [CrossRef]
- Samarghandian, S.; Afshari, R.; Farkhondeh, T. Effect of long-term treatment of morphine on enzymes, oxidative stress indices and antioxidant status in male rat liver. Int. J. Clin. Exp. Med. 2014, 7, 1449–1453. [Google Scholar]
- Abdel-Zaher, A.O.; Mostafa, M.G.; Farghaly, H.S.; Hamdy, M.M.; Abdel-Hady, R.H. Role of oxidative stress and inducible nitric oxide synthase in morphine-induced tolerance and dependence in mice. Effect of alpha-lipoic acid. Behav. Brain Res. 2013, 247, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Bu, Q.; Yang, Y.; Yan, G.; Hu, Z.; Hu, C.; Duan, J.; Lv, L.; Zhou, J.; Zhao, J.; Shao, X.; et al. Proteomic analysis of the nucleus accumbens in rhesus monkeys of morphine dependence and withdrawal intervention. J. Proteom. 2012, 75, 1330–1342. [Google Scholar] [CrossRef] [PubMed]
- Nyssen, P.; Mouithys-Mickalad, A.; Minguet, G.; Sauvage, E.; Wouters, J.; Franck, T.; Hoebeke, M. Morphine, a potential inhibitor of myeloperoxidase activity. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2236–2244. [Google Scholar] [CrossRef] [PubMed]
- Kramarenko, G.G.; Hummel, S.G.; Martin, S.M.; Buettner, G.R. Ascorbate Reacts with Singlet Oxygen to Produce Hydrogen Peroxide. Photochem. Photobiol. 2006, 82, 1634–1637. [Google Scholar] [CrossRef]
- Buettner, G.R.; Jurkiewicz, B.A. Ascorbate Radical: A Valuable Marker of Oxidative Stress. In Analysis of Free Radicals in Biological Systems; Birkhäuser Basel: Basel, Switherland, 1995; pp. 145–164. [Google Scholar]
- Brennan, L.A.; Morris, G.M.; Wasson, G.R.; Hannigan, B.M.; Barnett, Y.A. The effect of vitamin C or vitamin E supplementation on basal and H2O2-induced DNA damage in human lymphocytes. Br. J. Nutr. 2000, 84, 195–202. [Google Scholar] [CrossRef] [Green Version]
- Kao, P.-F.; Lee, W.-S.; Liu, J.-C.; Chan, P.; Tsai, J.-C.; Hsu, Y.-H.; Chang, W.-Y.; Cheng, T.-H.; Liao, S.-S. Downregulation of superoxide dismutase activity and gene expression in cultured rat brain astrocytes after incubation with vitamin C. Pharmacology 2003, 69, 1–6. [Google Scholar] [CrossRef]
- Chen, L.H.; Thacker, R.R. An increase in glutathione peroxidase activity induced by high supplementation of vitamin C in rats. Nutr. Res. 1984, 4, 657–664. [Google Scholar] [CrossRef]
- Chen, X.; Touyz, R.M.; Park, J.B.; Schiffrin, E.L. Antioxidant Effects of Vitamins C and E Are Associated With Altered Activation of Vascular NADPH Oxidase and Superoxide Dismutase in Stroke-Prone SHR. Hypertension 2001, 38, 606–611. [Google Scholar] [CrossRef] [Green Version]
- Sureda, A.; Batle, J.M.; Tauler, P.; Ferrer, M.D.; Tur, J.A.; Pons, A. Vitamin C supplementation influences the antioxidant response and nitric oxide handling of erythrocytes and lymphocytes to diving apnea. Eur. J. Clin. Nutr. 2006, 60, 838–846. [Google Scholar] [CrossRef]
- Mahmoudabadi, M.M.S.; Rahbar, A.R. Effect of EPA and Vitamin C on Superoxide Dismutase, Glutathione Peroxidase, Total Antioxidant Capacity and Malondialdehyde in Type 2 Diabetic Patients. Oman Med. J. 2014, 29, 39–45. [Google Scholar] [CrossRef]
- Sedighe, B.; Maryam, B.; Fahimeh, A.; Somayyeh, A. Bigom T Effect of Vitamin C on Salivary Superoxide Dismutase Ac-tivity in Smokers. Aftrican J. Biotechnol. 2011, 10, 7267–7270. [Google Scholar]
- Helen, A.; Vijayammal, P.L. Vitamin C Supplementation on Hepatic Oxidative Stress Induced by Cigarette Smoke. J. Appl. Toxicol. 1997, 17, 289–295. [Google Scholar] [CrossRef]
- Salami, S.A.; Salahdeen, H.M.; Moronkola, O.T.; Murtala, B.A.; Raji, Y. Vitamin C supplementation during chronic variable stress exposure modulates contractile functions of testicular artery and sperm parameters in male Wistar rats. Middle East Fertil. Soc. J. 2020, 25, 8. [Google Scholar] [CrossRef]
- Khassaf, M.; McArdle, A.; Esanu, C.; Vasilaki, A.; Griffiths, R.D.; Brodie, D.A.; Jackson, M.J. Effect of Vitamin C Supplements on Antioxidant Defence and Stress Proteins in Human Lymphocytes and Skeletal Muscle. J. Physiol. 2003, 549, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Ueta, E.; Tadokoro, Y.; Yamamoto, T.; Yamane, C.; Suzuki, E.; Nanba, E.; Otsuka, Y.; Kurata, T. The Effect of Cigarette Smoke Exposure and Ascorbic Acid Intake on Gene Expression of Antioxidant Enzymes and Other Related Enzymes in the Livers and Lungs of Shionogi Rats with Osteogenic Disorders. Toxicol. Sci. 2003, 73, 339–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaya, F.; Belin, S.; Diamantidis, G.; Fontes, M. Ascorbic acid is a regulator of the intracellular cAMP concentration: Old molecule, new functions? FEBS Lett. 2008, 582, 3614–3618. [Google Scholar] [CrossRef] [Green Version]
- Rahman, F.; Al Frouh, F.; Bordignon, B.; Fraterno, M.; Landrier, J.-F.; Peiretti, F.; Fontes, M. Ascorbic acid is a dose-dependent inhibitor of adipocyte differentiation, probably by reducing cAMP pool. Front. Cell Dev. Biol. 2014, 2, 29. [Google Scholar] [CrossRef]
- Doulas, N.L.; Constantopoulos, A.; Litsios, B. Effect of Ascorbic Acid on Guinea Pig Adrenal Adenylate Cyclase Activity and Plasma Cortisol. J. Nutr. 1987, 117, 1108–1114. [Google Scholar] [CrossRef]
- Schattauer, S.S.; Bedini, A.; Summers, F.; Reilly-Treat, A.; Andrews, M.M.; Land, B.B.; Chavkin, C. Reactive oxygen species (ROS) generation is stimulated by κ opioid receptor activation through phosphorylated c-Jun N-terminal kinase and inhibited by p38 mitogen-activated protein kinase (MAPK) activation. J. Biol. Chem. 2019, 294, 16884–16896. [Google Scholar] [CrossRef]
- Su, X.; Shen, Z.; Yang, Q.; Sui, F.; Pu, J.; Ma, J.; Ma, S.; Yao, D.; Ji, M.; Hou, P. Vitamin C kills thyroid cancer cells through ROS-dependent inhibition of MAPK/ERK and PI3K/AKT pathways via distinct mechanisms. Theranostics 2019, 9, 4461–4473. [Google Scholar] [CrossRef]
- Su, X.; Li, P.; Han, B.; Jia, H.; Liang, Q.; Wang, H.; Gu, M.; Cai, J.; Li, S.; Zhou, Y.; et al. Vitamin C sensitizes BRAFV600E thyroid cancer to PLX4032 via inhibiting the feedback activation of MAPK/ERK signal by PLX4032. J. Exp. Clin. Cancer Res. 2021, 40, 34. [Google Scholar] [CrossRef]
- Rosas, M.; Porru, S.; Sabariego, M.; Piludu, M.A.; Giorgi, O.; Corda, M.G.; Acquas, E. Effects of morphine on place conditioning and ERK1/2 phosphorylation in the nucleus accumbens of psychogenetically selected Roman low- and high-avoidance rats. Psychopharmacology 2018, 235, 59–69. [Google Scholar] [CrossRef]
- De Freitas, B.G.; Pereira, L.M.; Santa-Cecília, F.V.; Hösch, N.G.; Picolo, G.; Cury, Y.; Zambelli, V.O. Mitogen-Activated Protein Kinase Signaling Mediates Morphine Induced-Delayed Hyperalgesia. Front. Neurosci. 2019, 13, 1018. [Google Scholar] [CrossRef]
- Ferrada, L.; Magdalena, R.; Barahona, M.; Ramírez, E.; Sanzana, C.; Gutiérrez, J.; Nualart, F. Two Distinct Faces of Vitamin C: AA vs. DHA. Antioxidants 2021, 10, 215. [Google Scholar] [CrossRef]
- Park, H.J.; Ock, S.M.; Kim, H.J.; Park, H.J.; Lee, Y.B.; Choi, J.M.; Cho, C.S.; Lee, J.Y.; Cho, B.K.; Cho, D.H. Vitamin C attenuates ERK signalling to inhibit the regulation of collagen production by LL-37 in human dermal fibroblasts. Exp. Dermatol. 2010, 19, e258–e264. [Google Scholar] [CrossRef]
- Ulrich-Merzenich, G.; Zeitler, H.; Panek, D.; Bokemeyer, D.; Vetter, H. Vitamin C promotes human endothelial cell growth via the ERK-signaling pathway. Eur. J. Nutr. 2007, 46, 87–94. [Google Scholar] [CrossRef]
- Su, Z.; Blazing, M.A.; Fan, D.; George, S.E. The Calmodulin-Nitric Oxide Synthase Interaction. J. Biol. Chem. 1995, 270, 29117–29122. [Google Scholar] [CrossRef] [Green Version]
- Cadet, P.; Bilfinger, T.V.; Fimiani, C.; Peter, D.; Stefano, G.B. Human Vascular and Cardiac Endothelia Express Mu Opiate Receptor Transcripts. Endothelium 2000, 7, 185–191. [Google Scholar] [CrossRef]
- Toda, N.; Kishioka, S.; Hatano, Y.; Toda, H. Interactions between morphine and nitric oxide in various organs. J. Anesthesia 2009, 23, 554–568. [Google Scholar] [CrossRef]
- Stefano, G.B.; Mantione, K.; Capellan, L.; Casares, F.M.; Challenger, S.; Ramin, R.; Samuel, J.M.; Snyder, C.; Kream, R.M. Morphine stimulates nitric oxide release in human mitochondria. J. Bioenerg. Biomembr. 2015, 47, 409–417. [Google Scholar] [CrossRef]
- D’Uscio, L.V.; Milstien, S.; Richardson, D.; Smith, L.; Katusic, Z.S. Long-Term Vitamin C Treatment Increases Vascular Tetrahydrobiopterin Levels and Nitric Oxide Synthase Activity. Circ. Res. 2003, 92, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Kuzkaya, N.; Weissmann, N.; Harrison, D.G.; Dikalov, S. Interactions of Peroxynitrite, Tetrahydrobiopterin, Ascorbic Acid, and Thiols. J. Biol. Chem. 2003, 278, 22546–22554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckman, J.S.; Koppenol, W.H. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and ugly. Am. J. Physiol. Cell Physiol. 1996, 271, C1424–C1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric Oxide and Peroxynitrite in Health and Disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [Green Version]
- Muscoli, C.; Cuzzocrea, S.; Ndengele, M.M.; Mollace, V.; Porreca, F.; Fabrizi, F.; Esposito, E.; Masini, E.; Matuschak, G.M.; Salvemini, D. Therapeutic manipulation of peroxynitrite attenuates the development of opiate-induced antinociceptive tolerance in mice. J. Clin. Investig. 2007, 117, 3530–3539. [Google Scholar] [CrossRef] [PubMed]
- Batthyány, C.; Bartesaghi, S.; Mastrogiovanni, M.; Lima, A.; Demicheli, V.; Radi, R. Tyrosine-Nitrated Proteins: Proteomic and Bioanalytical Aspects. Antioxid. Redox Signal. 2017, 26, 313–328. [Google Scholar] [CrossRef]
- Straaten, H.M.O.-V.; Man, A.M.S.-D.; De Waard, M.C. Vitamin C revisited. Crit. Care 2014, 18, 460. [Google Scholar] [CrossRef] [Green Version]
- Pelletier, S.; Duhamel, F.; Coulombe, P.; Popoff, M.R.; Meloche, S. Rho Family GTPases Are Required for Activation of Jak/STAT Signaling by G Protein-Coupled Receptors. Mol. Cell. Biol. 2003, 23, 1316–1333. [Google Scholar] [CrossRef] [Green Version]
- Gross, E.R.; Hsu, A.K.; Gross, G.J. The JAK/STAT pathway is essential for opioid-induced cardioprotection: JAK2 as a mediator of STAT3, Akt, and GSK-3β. Am. J. Physiol. Circ. Physiol. 2006, 291, H827–H834. [Google Scholar] [CrossRef] [Green Version]
- Han, Z.; Zhang, Z.; Guan, Y.; Chen, B.; Yu, M.; Zhang, L.; Fang, J.; Gao, Y.; Guo, Z. New insights into Vitamin C function: Vitamin C induces JAK2 activation through its receptor-like transporter SVCT2. Int. J. Biol. Macromol. 2021, 173, 379–398. [Google Scholar] [CrossRef]
- Chen, J.-X.; Huang, K.-M.; Liu, M.; Jiang, J.-X.; Liu, J.-P.; Zhang, Y.-X.; Yang, C.; Xin, W.-J.; Zhang, X.-Q. Activation of TLR4/STAT3 signaling in VTA contributes to the acquisition and maintenance of morphine-induced conditioned place preference. Behav. Brain Res. 2017, 335, 151–157. [Google Scholar] [CrossRef]
- Sun, X.; Wu, S.; Xing, D. The reactive oxygen species-Src-Stat3 pathway provokes negative feedback inhibition of apoptosis induced by high-fluence low-power laser irradiation. FEBS J. 2010, 277, 4789–4802. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Xu, J.; Ji, W.; Wang, L.; Wang, A. Upregulation of TMEFF2 is involved in the antiproliferative effects of vitamin C and tyrphostin AG490 on GES-1 and AGS cells. Oncol. Lett. 2018, 17, 652–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puram, S.V.; Yeung, C.M.; Jahani-Asl, A.; Lin, C.; De La Iglesia, N.; Konopka, G.; Jackson-Grusby, L.; Bonni, A. STAT3-iNOS Signaling Mediates EGFRvIII-Induced Glial Proliferation and Transformation. J. Neurosci. 2012, 32, 7806–7818. [Google Scholar] [CrossRef] [PubMed]
- Queval, C.J.; Song, O.-R.; Deboosere, N.; Delorme, V.; Debrie, A.-S.; Iantomasi, R.; Veyron-Churlet, R.; Jouny, S.; Redhage, K.; Deloison, G.; et al. STAT3 Represses Nitric Oxide Synthesis in Human Macrophages upon Mycobacterium tuberculosis Infection. Sci. Rep. 2016, 6, 29297. [Google Scholar] [CrossRef]
- Rouillard, A.D.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermott, M.G.; Ma’Ayan, A. The harmonizome: A collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016, 2016, baw100. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Jiang, L.; Wang, J.; Chai, Z.; Xiong, W. Morphine promotes angiogenesis by activating PI3K/Akt/HIF-1α pathway and upregulating VEGF in hepatocellular carcinoma. J. Gastrointest. Oncol. 2021, 12, 1761–1772. [Google Scholar] [CrossRef]
- Hahn-Windgassen, A.; Nogueira, V.; Chen, C.-C.; Skeen, J.E.; Sonenberg, N.; Hay, N. Akt Activates the Mammalian Target of Rapamycin by Regulating Cellular ATP Level and AMPK Activity. J. Biol. Chem. 2005, 280, 32081–32089. [Google Scholar] [CrossRef] [Green Version]
- Li, C.-J.; Elsasser, T.H.; Kahl, S. AKT/eNOS signaling module functions as a potential feedback loop in the growth hormone signaling pathway. J. Mol. Signal. 2009, 4, 1. [Google Scholar] [CrossRef] [Green Version]
- Fang, Q.; Naidu, K.A.; A Naidu, K.; Zhao, H.; Sun, M.; Dan, H.C.; Nasir, A.; E Kaiser, H.; Cheng, J.Q.; Nicosia, S.V.; et al. Ascorbyl stearate inhibits cell proliferation and tumor growth in human ovarian carcinoma cells by targeting the PI3K/AKT pathway. Anticancer Res. 2006, 26, 203–209. [Google Scholar]
- Yang, L.; Wang, S.; Sung, B.; Lim, G.; Mao, J. Morphine Induces Ubiquitin-Proteasome Activity and Glutamate Transporter Degradation. J. Biol. Chem. 2008, 283, 21703–21713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, S.; Browning, E.A.; Hong, N.; DeBolt, K.; Sorokina, E.M.; Liu, W.; Birnbaum, M.; Fisher, A.B. Membrane depolarization is the trigger for PI3K/Akt activation and leads to the generation of ROS. Am. J. Physiol. Circ. Physiol. 2012, 302, H105–H114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koundouros, N.; Poulogiannis, G. Phosphoinositide 3-Kinase/Akt Signaling and Redox Metabolism in Cancer. Front. Oncol. 2018, 8, 160. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, J.; Yang, C.; Huang, X.; Han, M.; Kang, F.; Li, J. Morphine promotes the malignant biological behavior of non-small cell lung cancer cells through the MOR/Src/mTOR pathway. Cancer Cell Int. 2021, 21, 622. [Google Scholar] [CrossRef]
- Xu, J.-T.; Zhao, J.-Y.; Zhao, X.; Ligons, D.; Tiwari, V.; Atianjoh, F.E.; Lee, C.-Y.; Liang, L.; Zang, W.; Njoku, D.; et al. Opioid receptor–triggered spinal mTORC1 activation contributes to morphine tolerance and hyperalgesia. J. Clin. Investig. 2014, 124, 592–603. [Google Scholar] [CrossRef] [Green Version]
- Mazei-Robison, M.S.; Koo, J.W.; Friedman, A.K.; Lansink, C.S.; Robison, A.J.; Vinish, M.; Krishnan, V.; Kim, S.; Siuta, M.A.; Galli, A.; et al. Role for mTOR Signaling and Neuronal Activity in Morphine-Induced Adaptations in Ventral Tegmental Area Dopamine Neurons. Neuron 2011, 72, 977–990. [Google Scholar] [CrossRef] [Green Version]
- Delgoffe, G.M. PP2A's restraint of mTOR is critical for T(reg) cell activity. Nat. Immunol. 2016, 17, 478–479. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.A.; Eid, R.M.; Hanafi, M.Y. Vitamin C and E chronic supplementation differentially affect hepatic insulin signaling in rats. Life Sci. 2018, 194, 196–204. [Google Scholar] [CrossRef]
- Zahmatkesh, M.; Kadkhodaee, M.; Salarian, A.; Seifi, B.; Adeli, S. Impact of opioids on oxidative status and related signaling pathways: An integrated view. J. Opioid Manag. 2017, 13, 241–251. [Google Scholar] [CrossRef]
- Lingappan, K. NF-κB in oxidative stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Cárcamo, J.M.; Pedraza, A.; Bórquez-Ojeda, A.O.; Golde, D.W. Vitamin C Suppresses TNFα-Induced NFκB Activation by Inhibiting IκBα Phosphorylation. Biochemistry 2002, 41, 12995–13002. [Google Scholar] [CrossRef] [PubMed]
- Das, K.C.; Lewis-Molock, Y.; White, C.W. Activation of NF-kappa B and elevation of MnSOD gene expression by thiol reducing agents in lung adenocarcinoma (A549) cells. Am. J. Physiol. Cell. Mol. Physiol. 1995, 269, L588–L602. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Fang, F.; Clair, D.K.S.; Josson, S.; Sompol, P.; Spasojevic, I.; Clair, W.H.S. Suppression of RelB-mediated manganese superoxide dismutase expression reveals a primary mechanism for radiosensitization effect of 1α,25-dihydroxyvitamin D3 in prostate cancer cells. Mol. Cancer Ther. 2007, 6, 2048–2056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- I Rojo, A.; Salinas, M.; Martín, D.; Perona, R.; Cuadrado, A. Regulation of Cu/Zn-Superoxide Dismutase Expression via the Phosphatidylinositol 3 Kinase/Akt Pathway and Nuclear Factor- B. J. Neurosci. 2004, 24, 7324–7334. [Google Scholar] [CrossRef] [Green Version]
- Guo, G.; Bhat, N.R. Hypoxia/Reoxygenation Differentially Modulates NF-κB Activation and iNOS Expression in Astrocytes and Microglia. Antioxid. Redox Signal. 2006, 8, 911–918. [Google Scholar] [CrossRef]
- Nakata, S.; Tsutsui, M.; Shimokawa, H.; Yamashita, T.; Tanimoto, A.; Tasaki, H.; Ozumi, K.; Sabanai, K.; Morishita, T.; Suda, O.; et al. Statin Treatment Upregulates Vascular Neuronal Nitric Oxide Synthase Through Akt/NF-κB Pathway. Arter. Thromb. Vasc. Biol. 2007, 27, 92–98. [Google Scholar] [CrossRef] [Green Version]
- Yao, K.-S.; O'Dwyer, P.J. Involvement of NF-κB in the induction of NAD(P)H: Quinone oxidoreductase (DT-diaphorase) by hypoxia, oltipraz and mitomycin C. Biochem. Pharmacol. 1995, 49, 275–282. [Google Scholar] [CrossRef]
- Kraus, J.; Börner, C.; Giannini, E.; Höllt, V. The Role of Nuclear Factor κB in Tumor Necrosis Factor-Regulated Transcription of the Human μ-Opioid Receptor Gene. Mol. Pharmacol. 2003, 64, 876–884. [Google Scholar] [CrossRef]
- Nennig, S.; Schank, J. The Role of NFkB in Drug Addiction: Beyond Inflammation. Alcohol Alcohol. 2017, 52, 172–179. [Google Scholar] [CrossRef]
- Cárcamo, J.M.; Pedraza, A.; Bórquez-Ojeda, O.; Zhang, B.; Sanchez, R.; Golde, D.W. Vitamin C Is a Kinase Inhibitor: Dehydroascorbic Acid Inhibits IκBα Kinase β. Mol. Cell. Biol. 2004, 24, 6645–6652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dan, H.C.; Ebbs, A.; Pasparakis, M.; Van Dyke, T.; Bassères, D.; Baldwin, A.S. Akt-dependent Activation of mTORC1 Complex Involves Phosphorylation of mTOR (Mammalian Target of Rapamycin) by IκB Kinase α (IKKα). J. Biol. Chem. 2014, 289, 25227–25240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muellner, M.K.; Schreier, S.M.; Schmidbauer, B.; Moser, M.; Quehenberger, P.; Kapiotis, S.; Goldenberg, H.; Laggner, H. Vitamin C inhibits NO-induced stabilization of HIF-1α in HUVECs. Free Radic. Res. 2010, 44, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Balasubramanian, S.; Wang, J.; Chandrashekhar, Y.; Charboneau, R.; Barke, R. Morphine inhibits VEGF expression in myocardial ischemia. Surgery 2003, 134, 336–344. [Google Scholar] [CrossRef]
- Takabuchi, S.; Hirota, K.; Oda, S.; Nishi, K.; Oda, T.; Shingu, K.; Adachi, T.; Fukuda, K. Opioid receptor stimulation does not affect cellular hypoxia-induced gene responses mediated by hypoxia-inducible factor 1 in cultured cell lines. J. Anesthesia 2005, 19, 263–265. [Google Scholar] [CrossRef]
- Daijo, H.; Kai, S.; Tanaka, T.; Wakamatsu, T.; Kishimoto, S.; Suzuki, K.; Harada, H.; Takabuchi, S.; Adachi, T.; Fukuda, K.; et al. Fentanyl activates hypoxia-inducible factor 1 in neuronal SH-SY5Y cells and mice under non-hypoxic conditions in a μ-opioid receptor-dependent manner. Eur. J. Pharmacol. 2011, 667, 144–152. [Google Scholar] [CrossRef] [Green Version]
- Koodie, L.; Ramakrishnan, S.; Roy, S. Morphine Suppresses Tumor Angiogenesis through a HIF-1α/p38MAPK Pathway. Am. J. Pathol. 2010, 177, 984–997. [Google Scholar] [CrossRef]
- Tang, X.; Chen, Y.; Ran, H.; Jiang, Y.; He, B.; Wang, B.; Kong, S.; Wang, H. Systemic Morphine Treatment Derails Normal Uterine Receptivity, Leading to Embryo Implantation Failure in Mice1. Biol. Reprod. 2015, 92, 118. [Google Scholar] [CrossRef]
- Cheng, L.; Yu, H.; Yan, N.; Lai, K.; Xiang, M. Hypoxia-Inducible Factor-1α Target Genes Contribute to Retinal Neuroprotection. Front. Cell. Neurosci. 2017, 11, 20. [Google Scholar] [CrossRef]
- Masson, N.; Ratcliffe, P. HIF prolyl and asparaginyl hydroxylases in the biological response to intracellular O2 levels. J. Cell Sci. 2003, 116, 3041–3049. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, R.; Salloum, F.N.; Fisher, B.J.; Kukreja, R.C.; FowlerIII, A.A. Hypoxia Inducible Factor-1 Activation by Prolyl 4-Hydroxylase-2 Gene Silencing Attenuates Myocardial Ischemia Reperfusion Injury. Circ. Res. 2006, 98, 133–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mylonis, I.; Simos, G.; Paraskeva, E. Hypoxia-Inducible Factors and the Regulation of Lipid Metabolism. Cells 2019, 8, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- al Sagair, O.A. Effect of morphine sulphate on total lipids and triglycerides contents in serum and brain regions of rat. Med. Islamic World Sci. 2005, 15, 117–125. [Google Scholar]
- Li, L.; Wang, J.; Li, D.; Zhang, H. Morphine increases myocardial triacylglycerol through regulating adipose triglyceride lipase S406 phosphorylation. Life Sci. 2021, 283, 119866. [Google Scholar] [CrossRef]
- Drummond, G.B.; Lafferty, B. Oxygen saturation decreases acutely when opioids are given during anaesthesia. Br. J. Anaesth. 2010, 104, 661–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiyatkin, E.A. Respiratory depression and brain hypoxia induced by opioid drugs: Morphine, oxycodone, heroin, and fentanyl. Neuropharmacology 2019, 151, 219–226. [Google Scholar] [CrossRef]
- Pattinson, K.T.S. Opioids and the control of respiration. Br. J. Anaesth. 2008, 100, 747–758. [Google Scholar] [CrossRef] [Green Version]
- Bouillon, T.; Bruhn, J.; Roepcke, H.; Hoeft, A. Opioid-induced respiratory depression is associated with increased tidal volume variability. Eur. J. Anaesthesiol. 2003, 20, 127–133. [Google Scholar] [CrossRef]
- Dong, T.W.; MacLeod, D.B.; Santoro, A.; Augustine, Z.; Barth, S.; Cooter, M.; E Moon, R. A Methodology to Explore Ventilatory Chemosensitivity and Opioid-Induced Respiratory Depression Risk. J. Appl. Physiol. 2020, 129, 500–507. [Google Scholar] [CrossRef]
- Kor, J.J.; Sprung, J.; Khanna, A.K.; Weingarten, T.N. Continuous Monitoring Detected Respiratory Depressive Episodes in Proximity to Adverse Respiratory Events During the PRODIGY Trial. J. Patient Saf. 2022, 10–97. [Google Scholar] [CrossRef]
- Stone, L.S.; German, J.P.; Kitto, K.F.; Fairbanks, C.A.; Wilcox, G.L. Morphine and Clonidine Combination Therapy Improves Therapeutic Window in Mice: Synergy in Antinociceptive but Not in Sedative or Cardiovascular Effects. PLoS ONE 2014, 9, e109903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baehr, C.A.; Kelcher, A.H.; Khaimraj, A.; Reed, D.E.; Pandit, S.G.; Aucoin, D.; Averick, S.; Pravetoni, M. Monoclonal Antibodies Counteract Opioid-Induced Behavioral and Toxic Effects in Mice and Rats. J. Pharmacol. Exp. Ther. 2020, 375, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Ding, X.; Funk, G.; Greer, J.J. Ampakine CX717 Protects against Fentanyl-induced Respiratory Depression and Lethal Apnea in Rats. Anesthesiology 2009, 110, 1364–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baby, S.M.; Gruber, R.B.; Young, A.P.; MacFarlane, P.M.; Teppema, L.J.; Lewis, S.J. Bilateral carotid sinus nerve transection exacerbates morphine-induced respiratory depression. Eur. J. Pharmacol. 2018, 834, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Kim, D.-I.; Oh, T.G.; Pao, G.M.; Kim, J.-H.; Palmiter, R.D.; Banghart, M.R.; Lee, K.-F.; Evans, R.M.; Han, S. Neural basis of opioid-induced respiratory depression and its rescue. Proc. Natl. Acad. Sci. USA 2021, 118, e2022134118. [Google Scholar] [CrossRef]
- Osborne, R.; Joel, S.; Grebenik, K.; Trew, D.; Slevin, M. The pharmacokinetics of morphine and morphine glucuronides in kidney failure. Clin. Pharmacol. Ther. 1993, 54, 158–167. [Google Scholar] [CrossRef]
- Osborne, R.J.; Joel, S.P.; Slevin, M.L. Morphine intoxication in renal failure: The role of morphine-6-glucuronide. BMJ 1986, 292, 1548–1549. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Newman, M.; Connery, H.; Boyd, J. Opioids and Vitamin C: Known Interactions and Potential for Redox-Signaling Crosstalk. Antioxidants 2022, 11, 1267. https://doi.org/10.3390/antiox11071267
Newman M, Connery H, Boyd J. Opioids and Vitamin C: Known Interactions and Potential for Redox-Signaling Crosstalk. Antioxidants. 2022; 11(7):1267. https://doi.org/10.3390/antiox11071267
Chicago/Turabian StyleNewman, Mackenzie, Heather Connery, and Jonathan Boyd. 2022. "Opioids and Vitamin C: Known Interactions and Potential for Redox-Signaling Crosstalk" Antioxidants 11, no. 7: 1267. https://doi.org/10.3390/antiox11071267
APA StyleNewman, M., Connery, H., & Boyd, J. (2022). Opioids and Vitamin C: Known Interactions and Potential for Redox-Signaling Crosstalk. Antioxidants, 11(7), 1267. https://doi.org/10.3390/antiox11071267