Abstract
Opioids are among the most widely used classes of pharmacologically active compounds both clinically and recreationally. Beyond their analgesic efficacy via μ opioid receptor (MOR) agonism, a prominent side effect is central respiratory depression, leading to systemic hypoxia and free radical generation. Vitamin C (ascorbic acid; AA) is an essential antioxidant vitamin and is involved in the recycling of redox cofactors associated with inflammation. While AA has been shown to reduce some of the negative side effects of opioids, the underlying mechanisms have not been explored. The present review seeks to provide a signaling framework under which MOR activation and AA may interact. AA can directly quench reactive oxygen and nitrogen species induced by opioids, yet this activity alone does not sufficiently describe observations. Downstream of MOR activation, confounding effects from AA with STAT3, HIF1α, and NF-κB have the potential to block production of antioxidant proteins such as nitric oxide synthase and superoxide dismutase. Further mechanistic research is necessary to understand the underlying signaling crosstalk of MOR activation and AA in the amelioration of the negative, potentially fatal side effects of opioids.
Keywords:
vitamin C; opioids; mu opioid receptor; oxidative stress; signaling; metabolism; crosstalk 1. Introduction
Opioids are among the most widely used classes of pharmacologically active compounds both clinically and recreationally. While opioids have been invaluable in the clinic for analgesia, they produce numerous side effects and are highly addictive [1,2,3,4]. Their principal binding at μ opioid receptors (MOR) causes potentially fatal opioid induced respiratory depression (OIRD) by inactivation of the pre-Bötzinger complex [5], leading to decreased breathing, systemic hypoxia, oxidative stress, and free radical generation. Ascorbic acid (AA; vitamin C), the antioxidant vitamin at the highest circulating concentration in the human body (compared to vitamins A, D, E, and derivatives) [6,7], has critical roles in processes such as neurotransmission [8,9], wound healing [10], and immune homeostasis [11] and may be able to quench aberrant oxidative signaling associated with opioid exposures. The low toxicity of vitamin C makes it an ideal investigational adjuvant to address some of the negative effects of opioid administration. This review seeks to address the current state of in vitro and in vivo co-administration research associated with opioids and vitamin C, while identifying potential underlying mechanisms of interactions. Additionally, this review identifies current gaps in the literature associated with intracellular redox-signaling mechanisms between vitamin C and opioids and presents focused paths forward for research in the field.
The role of vitamin C on opioid administration, specifically morphine as the classical opioid, has been briefly investigated both clinically and in model systems. Few studies have been carried out in humans, but some small cohorts have shown reduced circulating vitamin C levels in chronic opioid users [12,13]. Treatment with vitamin C has been shown to decrease opioid withdrawal symptoms in guinea pigs [14], rats [15], and humans [16,17]. AA has been described as a solubilizing agent for street heroin base, although the amount of AA required is far below the amounts used in laboratory and clinical research settings [18,19]. In post-operative humans, administration of vitamin C has been shown to lower morphine consumption [20], nausea, and overall pain scores [17,21,22]. The ability of vitamin C to potentiate analgesia from morphine has been noted in humans [17,23,24], as well as mice [25,26] and rats [27], measured most often via tail-flick or hot plate test. Overall, brain concentrations of AA have been shown to be increased after morphine administration in rats [28,29,30], but effects in other organ systems are currently unknown. While most studies on the effects of vitamin C and morphine (or similar opioids) in humans are focused on changes in withdrawal symptoms in abuse settings and post-surgery recovery outcomes, much work is left to be done to understand the potential critical interaction mechanisms that may lead to these integrated physiological responses.
2. Opioid Activity and Metabolism
Opioids, originally derived from the opium poppy plant, are one of the largest and most potent classes of pharmacologically active compounds, and are readily used for their antinociceptive properties, although they are often abused due to their euphoric effects at higher doses. The discovery of MOR and endogenous opioid peptides in the 1970s [31] has led to a revolution in opioid design, discovery, and implementation [32]. Commonly used opioids include prescription drugs such as morphine, codeine, hydrocodone, oxycodone, hydromorphone, oxymorphone, methadone, and tramadol, as well as heroin and fentanyl, which have major roles as drugs of abuse (although fentanyl is still used clinically) (Figure 1). These act as either partial or full agonists of the MOR, the focal receptor for antinociception and analgesia, and principally bind surface opioid receptors [33]. Opioids frequently have partial affinity for κ and δ opioid receptors as well, yet analgesic contributions from these receptors are contested [34].
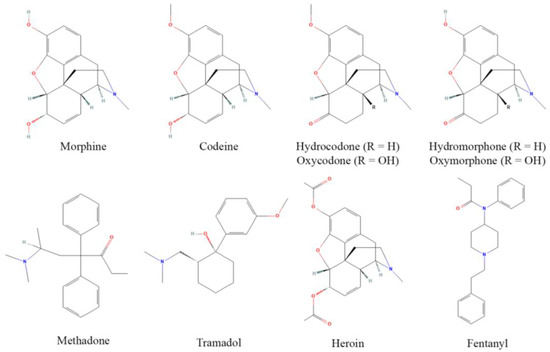
Figure 1.
Structures of common opioids. Some prominent clinical opioids include morphine, codeine, hydrocodone, oxycodone, hydromorphone, oxymorphone, methadone, and tramadol, while heroin and fentanyl are most often subject to illicit use. Structures are derived from [35].
Opioid receptors are present in nearly all tissues and cell types [36,37], and are especially upregulated in the nervous system, but also have critical roles in the gastrointestinal tract, respiratory tract, and cardiovascular system where they modulate inflammatory signaling [38]. MOR is a typical G-protein-coupled receptor (GPCR), acting via an intracellular inhibitory G protein cascade (Gi/Go), preferentially through Go and Gi2, but also Gi1, Gi3, Gz, and G16 [39]. After ligand binding, dissociation of the G-protein complex inhibits adenylate cyclase (AC) activity, causing decreased intracellular levels of cyclic adenosine monophosphate (cAMP) [26] and downstream reductions in protein kinase A activity, as well as activation of the mitogen-activated protein kinase (MAPK) cascade; these actions cause CREB translocation to the nucleus to alter transcription, including redox-relevant genes encoding for neuronal nitric oxide synthase (nNOS) [40], NAD(P)H oxidases, catalase (CAT), and superoxide dismutase (SOD) [41]. While the Gα complex activates phospholipase C, generating inositol-3-phosphate (IP3) and thus inducing intracellular calcium release from the endoplasmic reticulum, the Gβ/γ complex blocks plasma membrane calcium channels, leading to an overall increase in intracellular calcium signaling [39,42].
The cytochrome P450 system is primarily responsible for most opioid metabolism, through CYP3A4 and CYP2D6, for species such as codeine, hydrocodone, oxycodone, fentanyl, methadone, and tramadol [43]. Morphine, however, does not go through first-pass metabolism and is instead directly glucuronidated by UGT2B7 into morphine-3-glucuronide or morphine-6-glucuronide [43,44]. Its major metabolites, morphine-3-glucuronide and morphine-6-glucuronide, are active at MOR. Major metabolism occurs in the liver, heart, and kidneys. While UGT2B7 has not been described to promote free radical formation as reactive oxygen species (ROS) or reactive nitrogen species (RNS), CYP3A4 and CYP2D6 are established as contributing to ROS generation [45]. These CYP enzymes are notably responsible for the majority of all drug metabolism [46]. Opioid metabolism is confounded by pharmacokinetic and pharmacodynamic interactions that warrant consideration for signaling transduction research. For example, morphine-6-gluconuride has a higher affinity for MOR than morphine [47], yet cannot pass through the blood–brain barrier as efficiently due to higher polarity [48]. O-demethylation of the anisole group in oxycodone to oxymorphone, hydrocodone to hydromorphone, and codeine to morphine via CYP2D6 [49,50], as well as deacetylation of the phenylacetate in heroin to 6-monoacetylmorphine by esterases [51,52], exposes a tyramine-like phenol moiety (Figure 2A) that can undergo oxidative transformation to quench free radicals [53,54] via a neutral radical intermediate [55] through the mechanism shown in Figure 2B (adapted from [56]). Common polymorphisms in CYP2D6 lead to decreased enzyme activity and therefore decreased conversion of the aforementioned opioids to their active metabolites [57,58], thus potentially shunting the parent compounds toward alternative metabolic pathways. Morphinone derivatives can also undergo secondary elimination via conjugation to glutathione, although the MOR affinities of these metabolites have not been characterized [59,60].
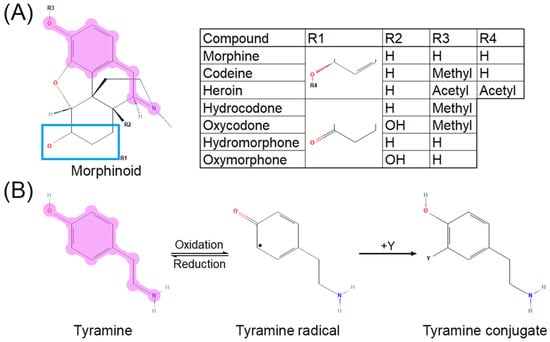
Figure 2.
Tyramine moiety in morphinoid pharmacophore. (A) Morphinoid pharmacophore with tyramine moiety highlighted. (B) Reaction mechanism for oxidation of tyramine (adapted with permission from Ref. [56]. 2013, American Chemical Society). Reversible oxidation of tyramine (left) leads to a neutral, reactive radical intermediate species (middle) which can quench free radicals such as superoxide, hydroxide radical, nitrite radical “Y”, yielding a substituted phenol (right). Reaction of tyrosine, a tyramine-containing amino acid, with the nitrite radical or peroxynitrite forms nitrotyrosine, a residue disruptive to protein–protein interactions.
3. Vitamin C Activity and Metabolism
Vitamin C is a well-known antioxidant that helps protect cells against the detrimental effects of free radicals such as ROS and RNS [61,62,63]. Vitamin C is synthesized in plants and protists from galactose and from gulonolactone in many vertebrates (e.g., mice, rats, rabbits, cats, dogs, and sheep), yet humans, primates, bats, guinea pigs, and some bony fish (notably zebrafish) possess pseudogenes or entirely lack the gene encoding for L-gulonolactone oxidase (Gulo), the enzyme required to produce it [64,65]. Gulo knockout mice (derived on a C57BL/6 background) have been established to investigate dietary AA-dependent processes [66,67,68], yet Gulo has not been ablated in other vitamin C-synthesizing mammals, such as rats and rabbits.
At physiological pH, AA exists as its conjugate base and prominently serves as a direct antioxidant via ascorbyl radical [69] or as an enzyme cofactor. Vitamin C is well-tolerated in humans with zero reported deaths; its LD50 is estimated around 3.3 g/kg–11.9 g/kg in rodents [70]. The low toxicity of AA is attributable to high water solubility, allowing rapid clearance upon administration; however, excess vitamin C can result in oxalate production, thus administration is contraindicated for individuals with kidney stones [71]. AA functions in numerous hydroxylation reactions, often Fe2+-dependent, such as oxidation of lysine and proline residues in collagen to form mature fibrils, DNA repair, and activation of hypoxia-induced factors (e.g., HIF1α) [72]. It can function as a pro-oxidant species [73], yet this biological role has not been fully established. The oxidized form of AA, dehydroascorbic acid (DHA), can be recycled back into vitamin C at the expense of NADPH or glutathione [74].
The underlying mechanisms of vitamin C transport, under normal conditions or oxidative stress, have not been fully elucidated. While import of AA primarily occurs via SVCT1 and SVCT2, two sodium-dependent cotransporters, DHA is imported through GLUT transporters and recycled into vitamin C [75]. It is important to note that GLUT-mediated transport of DHA is competitively inhibited by glucose as they share the same transporter [76]. Vitamin C is absorbed in the intestine through a saturable, dose-dependent active transport process, where it is converted to DHA and then reduced back into AA once it has entered epithelial tissue/cells. Reabsorbed excess vitamin C travels through the renal tubules and is excreted in the urine [77].
Efflux mechanisms have yet to be determined, but are unlikely to be a function of simple diffusion despite a high intracellular:extracellular concentration gradient caused by the polarity and hydrophilicity of AA. DHA is structurally unstable in physiological conditions [78] and may be further hydrolyzed to diketogulonic acid [79], which can then be decomposed to metabolites such as oxalic acid [80]. Efflux may occur as a function of membrane disruption resulting from oxidative stress, and it has been hypothesized that transport-mediated cellular swelling may activate volume sensitive anion channels in the basolateral membrane thus enabling the export of AA [81]. Vitamin C levels after morphine administration have been noted to be higher in the striatum, but not nucleus accumbens in one study [82], yet higher in the nucleus accumbens than the striatum in another [83], suggesting that cell-specific efflux mechanisms may be present.
Vitamin C acts as a cofactor in various metabolic reactions, including iron metabolism [78], neuronal energy metabolism [84], and lipid metabolism [85]. Vitamin C is both a major electron donor directly to Fe2+ and also modulates iron metabolism through stimulation of ferritin synthesis, which inhibits ferritin degradation by lysosomes and reduces efflux of cellular iron [78]. Moreover, vitamin C also facilitates the transformation of cholesterol into bile acids through modulation of microsomal 7α-hydroxylation catabolism of cholesterol in the liver, and increases the rate of hydroxylation reactions through the maintenance of metal ions in their reduced state to optimize hydroxylase and oxygenase function [86]. In the brain, DHA taken up by neurons inhibits glycolysis, activates the pentose phosphate pathway which produces NADPH, oxidizes glutathione, and stimulates lactate production and uptake in the neurons [84].
4. Opioids, Vitamin C, and Neurotransmission
When morphine or other agonists bind to MOR, the mesolimbic reward system is activated. GABA interneurons in the ventral tegmental area (VTA), more specifically the rostromedial tegmental nucleus, are inhibited, leading to a disinhibition of dopaminergic neurons, causing release of dopamine to the nucleus accumbens to produce feelings of euphoria [87]. GABA input may also be inhibited in the nucleus accumbens itself, as well as in the periaqueductal gray and raphe magnus, where this affect is thought to contribute to the analgesia that occurs with opioid use [34]. In concert with VTA dopamine release, suppression of norepinephrinergic neurons in the locus ceruleus (LC) by MOR activation reduces wakefulness, blood pressure, and respiration [87]. However, the deleterious respiratory effects of opioids are primarily attributed to the high expression of MOR in the pre-Bötzinger complex, a region of the ventral medulla [88,89], which is thought to generate respiratory rhythm [90].
MOR desensitization is focal in cases of opioid tolerance, addiction, and withdrawal. This involves adaptation of dopamine release from the VTA and norepinephrine from the LC, producing hyperresponsivesness to pain and elevated alertness [34,87]. Mechanistically, MOR typically functions first through Gi/o to signal with temporal cessation after β-arrestin recruitment to the receptor [34,91]. With tolerance, signaling becomes β-arrestin-biased, reducing antinociception and producing enhanced respiratory depression [34,87,92,93]. Research into biased MOR signaling is ongoing, with the potential to severely reduce the negative central effects of opioids [94,95,96].
Whereas opioids primarily compete for endogenous opioid peptide binding sites on μ, κ, and δ receptors [97], vitamin C has broader activity in neurotransmission. Neurons and glia both rely heavily on a tightly regulated surplus of vitamin C (millimolar levels) via uptake through the glucose transport system and sodium-coupled active transport [79]. GLUT-1 transporters are primarily responsible for vitamin C delivery across the blood–brain barrier in the form of DHA [98]. Once DHA has been recycled to retained AA, subsequent release can be triggered by glutamate uptake in astrocytes, which has been proposed to cause extracellular swelling and allows diffusion of AA through activation of volume-regulated anion channels [99,100].
Vitamin C is known to modulate T-type Ca2+ channels, thus modulating neural excitability [9]. Recently, studies have suggested a pivotal role of these channels, particularly CaV3.2 T-type channels, in the processing of pain signals [101]. These channels regulate excitatory neurotransmission, notably glutamate, in peripheral nerve endings of nociceptors. These calcium channels are sensitive to inhibition by divalent metals such as zinc and contain a Zn2+ binding histidine residue (His191) on domain I. AA selectively suppresses CaV3.2 T-type channels via simultaneous binding of Zn2+ and AA at His191 [101]. Inactivation of these channels to allow calcium efflux by vitamin C may be nullified by the action of MOR activation to increase intracellular Ca2+ concentrations [42], yet this has not been determined experimentally.
AA is not a direct neurotransmitter; however, extracellular AA may act as a modulator of neurotransmission by distinct mechanisms. It can attenuate the activity of NMDA receptors in the forebrain through its various redox reactions [9] as discussed later, and may also play a role in the release of biogenic amines such as dopamine and pituitary neuropeptides [102].
Vitamin C also plays a significant role in the biosynthesis of neurotransmitters and neuropeptides directly involved in opioid-induced analgesia and dependence. As a cofactor in dopamine β-hydroxylase and 4-hydroxyphenylpyruvate dioxygenase, vitamin C is required for the synthesis of dopamine and upstream shunting of tyrosine, a precursor of dopamine, to alternative catabolic pathways. Immediate biotransformation products of tyrosine metabolism, tyramine [103,104,105,106] and L-DOPA [104,105,106,107], as well as further downstream products epinephrine/norepinephrine [108] and homogentisic acid [109] can act as direct antioxidants. AA has been shown to elevate recycling of tetrahydrobiopterin (BH4) [102], which is critical in the synthesis of small neurotransmitters such as dopamine, epinephrine, norepinephrine (via tyrosine hydroxylase; TH), serotonin (via tryptophan hydroxylase; TPH), and nitric oxide (NO, via nitric oxide synthase; NOS) [8]. Morphine administration has been associated with increased TH protein levels in the ventral tegmental area, but without changes in total TH amount in the nucleus accumbens [110,111]; both regions have been implicated in reward-seeking behavior [112]. Morphine has been shown to increase TPH activity in the cerebral cortex, midbrain, and pons-medulla, but not the spinal cord in rats [113]. NOS activity is known to be increased by morphine administration in neuronal and peripheral tissues (discussed later with MAPK activation). Combined, the action of vitamin C (to increase BH4 levels) and opioids (to upregulate enzyme levels and activity) lead to catecholamine, serotonin, and NO synthesis that complicates the redox status of target cells; although these compounds can act as antioxidants, morphine is generally known to increase ROS and RNS [114,115,116].
Beyond additive activity, AA may indirectly antagonize opioid activity through receptor binding competition via endomorphin synthesis. This antagonism is possible due to the ability of AA to maintain stores of Cu+ by direct reduction of Cu2+. Cu+ is involved in the synthesis of numerous small neuropeptides (e.g., endomorphins) as a cofactor in peptidylglycine α-amidating monooxygenase. This enzyme can also catabolize glycyl-fatty acids to produce fatty acid amides [117] with direct and indirect activities on the aforementioned neurotransmitter systems, but also endocannabinoid [118] and vanilloid [119,120] signaling, both of which are implicated in redox balance.
5. Opioids, Vitamin C, and Direct Oxidative Stress Modulation
Under normal conditions, diatomic oxygen concentrations are modulated by a series of enzymes that have the capacity to suppress radical production and propagation. These antioxidant enzymes consume glutathione or NADPH stores to reduce downstream DNA, lipid, and protein oxidation, which ultimately prevents mitochondrial and whole-cell damage from ROS and RNS. SOD, in tandem with CAT, and glutathione peroxidase (GPx) are the major endogenous antioxidant defenses that have been studied for both opioids and vitamin C.
As discussed in “Opioid Activity and Metabolism” above, morphinoids can function as direct antioxidants upon oxidative activation [53,54,55]. Despite this activity, morphine and other morphinoid-signaling functions cause overall increases in the production of ROS and RNS, yet the underlying mechanisms causing this have not been fully elucidated. However, the major antioxidant defense enzymes have been studied in response to opioid exposure: SOD activity has been shown to be decreased in human erythrocytes [121], plasma [12], and sperm [122], as well as in rat cerebrum [123], hippocampus [124], and liver [125]; CAT has been shown to be decreased in human erythrocytes [121] and plasma [12], as well as rat cerebrum [123] and livers [125]; and GPx has been shown to be decreased in human plasma [12] and sperm [122], as well as rat whole brain [126], cerebrum [123], and hippocampus [124], after opioid exposure. Few studies have looked at alternate redox enzymes, but have revealed increases in thioredoxin [107] as well as decreases in peroxiredoxin [127] and myeloperoxidase [128].
The effect of opioids on the major antioxidant enzymes above are straightforward, unlike the role of vitamin C. During oxidative stress, vitamin C primarily acts as an antioxidant species (as ascorbyl radical) to directly quench reactive species such as peroxynitrite and singlet oxygen via single-electron reduction [69,129,130]. Changes in antioxidant enzymes and oxidation markers (e.g., SOD, glutathione, and malondialdehyde) are highly cell- and tissue-dependent with regard to vitamin C status. Despite the ability of vitamin C to reduce overall oxidant load, thus lessening the required activity of enzymes such as SOD, CAT, and GPx, studies have shown mixed results. Human red blood cells from healthy, non-smoking volunteers have been shown to have decreased SOD activity after vitamin C supplementation [131]. In rats with vitamin C supplementation, SOD activity has been shown to be decreased in astrocytes under normoxia [132], yet activity is unchanged in red blood cells and liver [133]. In the sera of spontaneous hypertensive rats given a high-salt diet with vitamin C supplementation, SOD activity has been shown to be increased [134]. While SOD activity was not shown to be significantly different from controls in vitamin C supplemented patient sera after an acute experience of repeated diving apnea [135], it was shown to be increased in sera from type 2 diabetics given vitamin C [136], suggesting a potential cumulative adaptive response in SOD from prolonged oxidative stress with vitamin C exposure. In contrast, a small cohort study showed no significant change in SOD activity in the saliva of chronic smokers [137] given vitamin C, yet supplemented rats exposed to chronic cigarette smoke had increased liver SOD activity [138].
The complex story associated with vitamin C and redox-specific enzymes continues with CAT activity, where human red blood cells have been shown to not be affected by vitamin C supplementation [131]. CAT also was not affected in red blood cells or livers of vitamin C supplemented rats in one study [133], yet liver activity was increased in another study of the effects of vitamin C on cigarette smoke-exposed rats [138]. CAT activity has been shown to be elevated in the sera of rats under chronic variable stress [139]. In humans, vitamin C supplementation increased CAT activity in the first 24 h of hydrogen peroxide treatment in lymphocytes of a post-exercise cohort [140], but showed no significant differences from 24 to 48 h. Human serum from individuals exposed to the same diving apnea study mentioned previously showed no significant changes in CAT activity with vitamin C supplementation [135].
The impact on GPx has not been investigated as frequently for its role in oxidant stress with vitamin C supplementation. In normoxic, healthy human red blood cells, vitamin C supplementation has no effect on GPx activity [131]. In normoxic rats, supplementation increased GPx activity in red blood cells, but not in livers [133]. In contrast, after supplementation, GPx activity was unchanged in rat livers following chronic cigarette smoke exposure [141]. It was increased in supplemented type 2 diabetic human sera, however [136]. Malondialdehyde levels, downstream of GPx, were not significantly different in supplemented rats under chronic stress [139], yet were decreased in supplemented humans after acute exercise and in type 2 diabetes [136].
Overall, direct comparison of previous literature associated with vitamin C and antioxidant enzymes (SOD, CAT, and GPx), which were either increased [134,136,138,139,140] or unchanged [135,139,140,141] may be confounded by the samples assayed. In humans, samples in previous research have been limited to saliva and blood. Although these may reflect whole-body oxidative enzyme status, organ-specific effects are critical to define, as conditions such as smoking and type 2 diabetes are designated to have target organs (e.g., lungs and pancreas). Despite two of the human studies involving acute oxidative stress (diving apnea and exercise) and two involving chronic conditions (smoking and type 2 diabetes), the researchers used different samples (i.e., isolated leukocytes and serum for acute condition, and saliva and serum for chronic). Future studies should be standardized to a single type of sample, such as a serum, and address all three antioxidant defense enzymes (SOD, CAT, and GPx).
Rat studies are complicated by differences in sampling as well, but also by the presence of an active rat Gulo gene, which may therefore have implications on the pharmacology, particularly transport and retention, of exogenous vitamin C. Across rat models, oxidative-stress-linked condition or disease has either increased SOD and CAT or had no change across SOD, CAT, and GPx. Animal models are critical for vitamin C supplementation research as they allow for direct organ and tissue sampling, yet standardization (e.g., using a specific strain) is critical. Gulo knockout mice or rats are better suited for translatable research toward the local effects of vitamin C supplementation.
6. MOR Activity and NOS
Individual mechanistic interactions between vitamin C and opioids have not been researched previously. Despite this, both AA and opioids have been shown to separately affect signaling pathways at overlapping and discrete nodes with crosstalk potential. These pharmacodynamic interactions may underlie the capacity of vitamin C to ameliorate oxidative stress induced by opioids beyond ablation of free radicals by vitamin C or indirect changes in antioxidant defense enzymes. Pharmacokinetic interactions, e.g., metabolic changes in opioid structures that change ligand affinity to MOR [47] or intracellular redox cofactor-sensitive recycling of DHA to AA [74] should be taken into consideration for experimental validation of potential crosstalk in oxidative stress. A summary of the potential pathway crosstalk described here is provided in Figure 3 and described in detail below.
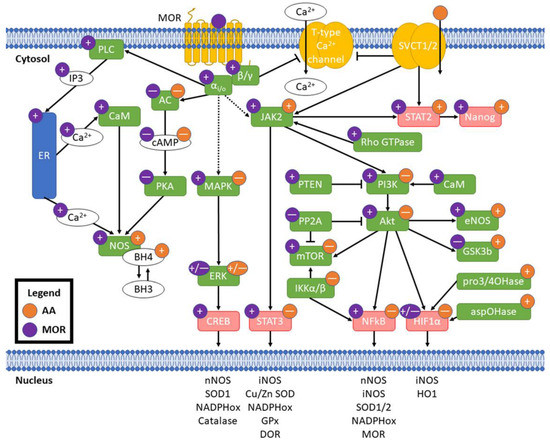
Figure 3.
Potential crosstalk between MOR activation and AA. Purple circles indicate effect of MOR activation and orange circles indicate effect of AA; “+” is an increase, “—” is a decrease, and “+/—” indicates mixed effects from literature. Pointed arrowheads indicate activation and flat arrowheads indicate decreased activity. Solid arrows indicate direct effects and dashed arrows indicate indirect/distal effects. Green boxes are enzymes and red are transcription factors. Transcription of example redox genes and proteins listed in the nucleus is affected by their linked transcription factors. MOR: μ opioid receptor; SVCT1/2: sodium-dependent vitamin C transporters 1/2; αi/o: Gαi/o subunit; β/γ: Gβ/γ subunit; PLC: phospholipase C; IP3: inositol 3-phosphate; ER: endoplasmic reticulum; CaM: calmodulin; NOS: nitric oxide synthases; BH3: trihydrobiopterin; BH4: tetrahydrobiopterin; AC: adenylate cyclase; cAMP: cyclic adenosine monophosphate; PKA: protein kinase A; pro3/4OHase: prolyl 3- and prolyl 4-hydroxylases; aspOHase: asparaginyl hydroxylase.
AA has mixed effects on the inhibitory G protein cascades initiated by MOR. Both MOR and AA lead to inhibition of AC and reduced cAMP levels [26,142,143,144]. MAPK signaling, induced directly from MOR activation, is linked to ROS generation in an ERK/JNK-dependent manner [145]. While AA has been shown to decrease MAPK signaling [146,147], ERK has been shown to be both activated and inhibited by both MOR [148,149] and AA [150,151,152]. Accumulation of β-arrestin at MOR decreases signaling post-activation [91], ceasing this activity. Increased cytosolic Ca2+ concentrations, another product of MOR activation [42], leads to NOS activation from direct calmodulin binding to the enzyme [153]. MOR activation has been shown to cause increased endothelial NOS (eNOS) activity and NO release in vascular tissue, potentially via μ3 opioid receptors [154], and has been linked to increased nNOS activity and NO production in neuronal tissue [155]. Morphine agonism on mitochondrial opioid receptors has also been shown to be coupled to calcium-dependent NO production from constitutive cNOS [156].
BH4 is required as a cofactor for many NOS enzymes which are upregulated by opioids. Overall, NOS activity has been shown to be increased by vitamin C [157] via increased BH4 levels. This phenomenon is most likely explained by the ability of vitamin C to recycle BH3, the reduced form of BH4 [158], and thus prevent BH3 from further catabolism. The additive effect of opioids and AA to promote NOS activity confers elevated NO, which can outcompete superoxide at SOD in a concentration-dependent manner [159]. NO can also combine with superoxide to form peroxynitrite (ONOO-), which has a longer half-life than superoxide [160]. Increased NOS activity in tandem with decreased SOD activity (and therefore elevated free superoxide) from morphine is likely to cause increased ONOO- production, but this has not yet been experimentally determined. Although ONOO- is difficult to measure directly in biological systems, its predominant marker, nitrotyrosine, has been shown to be elevated after morphine treatment [161]. A general scheme for nitrotyrosine formation is given in the section “Opioid Activity and Metabolism”, Figure 2B, where “Y” is either nitrite radical or ONOO-. This modified amino acid can disrupt protein–protein signaling at tyrosine residues directly and impact local binding environment due to pKa shift at the phenol moiety [162]. In contrast, vitamin C reduces the formation of ONOO- via mechanisms such as reductions in NADPH oxidase activity [163]. The degree to which the combination of vitamin C and morphine co-administration affects ONOO- levels has not yet been explored.
7. Vitamin C and MOR: Potential for Crosstalk
JAK/STAT signaling is crucial in cytokine-induced transcriptional regulation resulting from many cellular processes, particularly those involved in immunostasis, and can be activated by ROS generation [164]. The canonical pathway begins with ligand binding to a cytokine receptor, causing receptor dimerization and phosphorylation of the receptor by JAK1, JAK2, or JAK3. These phosphorylation sites recruit STAT proteins, which, after dimerization and phosphorylation, translocate to the nucleus to alter transcription. Opioid receptors have not been shown to bind latent JAK proteins. However, morphine has been shown to induce JAK2 phosphorylation with no change in JAK1 in reperfusion-ischemia-injured rat hearts [165]. The ability of MOR activation to induce JAK phosphorylation may be attributable to downstream rho GTPase activity [164]. Uptake of vitamin C through SVCT2 has been shown to be induced by phosphorylation of the C-terminus of SVCT2 by JAK2 [166], therefore suggesting a localized additive effect between morphine and vitamin C; both compounds have also been shown to cause downstream STAT2 phosphorylation [165,166] resulting in upregulation of Nanog, a transcription factor noted in pluripotency and cancer. JAK2 is also capable of phosphorylating STAT3; while studies on morphine have shown increased activation [165,167], vitamin C abrogates STAT3 activation [168,169] potentially by quenching activation from ROS. Furthermore, activated STAT3 has been shown to regulate iNOS [170,171], NADPH oxidases, GPx, SOD3, and OPRD1 [172], which directly encodes for the δ opioid receptor (DOR).
JAK2 activation by morphine has also been described to upregulate the phosphoinositol-3-kinase (PI3K)/Akt (protein kinase B) pathway [165,173]. This cycle-regulating pathway is induced by a variety of growth signals such as insulin, EGF, and calmodulin (upregulated directly by MOR activation), whereby activated PI3K phosphorylates and activates Akt, which can then activate factors such as mTOR [174] and eNOS [175]. Vitamin C has been shown to antagonize these pathways in cancer cells [146,176]. Interactions between opioids and vitamin C on this pathway are further complicated in the ability of morphine to upregulate PTEN [177], a PI3K blocker, while downregulating PP2A phosphorylation, thereby increasing the activity of Akt. Phosphorylated Akt promotes NO and superoxide production from the mitochondrion via dysregulated NADPH oxidases [178,179], leading to further Akt phosphorylation. Generation of free radicals, known to be induced by MOR, can also induce NF-κB and HIF1α translocation to the nucleus [180]. Directly downstream of Akt, mTOR is activated by morphine [181,182,183], yet is repressed by vitamin C; mTOR is central to growth signaling, pathway integration, and numerous degenerative diseases. Inactivation of PP2A by MOR has been shown to increase mTOR signaling [184]. Similar to opposing actions of morphine and vitamin C on mTOR, GSK3β, a negative regulator of glucose metabolism (and therefore oxygen consumption) by Akt, is inactivated (phosphorylated) in the presence of morphine [165], but is activated (via reduced phosphorylation) by vitamin C [185].
Elevated ROS generation, a product of MOR activity [186], can cause NF-κB translocation to the nucleus via modulation of upstream kinases [187], but NF-κB translocation has been shown to be blocked by AA [188]. This transcription factor regulates the expression of a myriad of pro-inflammatory and redox-mediating factors and enzymes; some of these include SOD1 [189], SOD2 [190], Cu/Zn SOD [191], iNOS [192], nNOS [193], NAD(P)H quinone oxidoreductases [194], and even MOR itself [195]. As mentioned previously, Akt can activate NF-κB; Akt activity is linked to morphine signaling [196], but blocked by vitamin C [76]. Translocation of NF-κB is canonically controlled by IKKα/IKKβ heterodimers (with or without a/an IKKγ subunit), which sequester NF-κB into the cytoplasm. Degradation of this IKKα/IKKβ complex via phosphorylation is inhibited by DHA but not AA [197]. The IKKα/IKKβ complex can phosphorylate mTOR [198], further activating it, yet mTOR is repressed by AA. Directly downstream of the IKK complex, IκBα has been shown to be inhibited by AA, as well as NF-κB itself [188]. Synthesizing the known references above leads to the hypothesis that competition of AA:DHA between mTOR and the IKK complex may act as a redox-sensitive switch to control gene regulation by NF-κB, yet this requires further exploration.
HIF1α is a major oxygen sensor and regulator of redox status whose activity is induced by ROS and Akt and inhibited by AA [199], yet has been shown to have mixed effects from opioids [200,201,202,203,204]. Its transcriptional activity leads to the production of antioxidant genes such as iNOS and heme oxygenase-1 [205]. As an upstream cofactor, vitamin C is required for prolyl 3-hydroxylase, prolyl 4-hydroxylases, and asparaginyl hydroxylases to act on HIF1α in order to deactivate its transcriptional activity and ubiquitinate it for degradation [206,207]. HIF1α is also established as a regulator of mitochondrial fatty acid metabolism [208], which is further impacted by aberrant redox signaling due to MOR activation. Both trimethyllysine hydroxylase and γ-butyrobetaine hydroxylase, which utilize vitamin C as a cofactor, are necessary for the synthesis of carnitine that facilitates fatty acid import into the mitochondrion. Morphine has been shown to increase triglyceride content in rat serum, brain [209], and cultured cardiac cells, potentially via triglyceride lipase inhibition [210], yet its effects on individual fatty acids remain to be explored. By combining the known decreases in free fatty acids due to opioid exposure, along with increased concentrations of carnitine from vitamin C, there may be a potential metabolic shift toward oxidative stress in the mitochondria, yet this has not been explored experimentally.
8. Conclusions and Future Directions
Opioids are the leading class of drugs of abuse worldwide and continue to rise in popularity while retaining vital roles for analgesia in the clinic. Vitamin C is a well-tolerated exogenous antioxidant with great potential to ameliorate some of the side effects associated with acute and chronic opioid use, but more research must be done to understand the potentially beneficial interactions. While previous research in humans has predominantly focused on integrated physiological responses, such as reductions in pain scores, opioid consumption, and withdrawal symptoms, the underlying mechanisms are poorly understood. The direct antioxidant capacity of vitamin C is one explanation for its effectiveness, but further research is required to optimize dosing and recommended regimens.
Previous studies of vitamin C co-administration have not addressed changes in respiratory parameters, such as blood oxygenation and tidal volume, known to be severely altered by opioids [211,212,213,214]; the impact of vitamin C on potentially fatal OIRD is unknown. These respiratory data are facile and non-invasive to acquire from humans, particularly in clinical settings where they are commonly monitored [215,216]. In animal models, blood oxygenation (SpO2) monitoring is commonplace using pulse oximetry [94,217,218], while plethysmography measurements require more complex instrumentation [219,220,221]. Altered lung function conferred from OIRD may also result in local signaling alterations, particularly in chronic opioid use; animal models are ideal for measuring these tissue changes, as lung biopsy is often not suitable for individuals already undergoing surgery or recovering from chronic opioid use. Illicit use of opioids often involves vaporizing or smoking as well, further implying the need for further investigation into local lung effects.
Further considerations should be made within a local tissue context with regard to metabolism. While many common opioids are prodrugs of active compounds with higher MOR affinity, their metabolites may also have MOR activity, yet these metabolites may be sequestered for excretion before they reach peripheral organs in a sufficient quantity for appreciable activity. For example, morphine-3- and morphine-6-gluconurides, but not morphine, have been implicated in kidney failure [222,223], but excess AA is contraindicated in individuals with kidney disease due to production of oxalate from AA [71]. Hydrophilic metabolites of opioids generated from first-pass metabolism may not sufficiently pass through the blood–brain barrier, thus the route of administration should also be explored as well and given significant consideration, particularly due to the well-established roles of vitamin C in neurotransmission.
The effects of opioids on endogenous antioxidant enzymes have been established, but the role of vitamin C has not been researched extensively. In other oxidative-stress-related conditions and diseases, AA has been shown to have mixed effects on SOD, CAT, and GPx, but these results are difficult to compare due to the types of samples assayed. Animal models are necessary in this context for exploration of tissue and organ-specific effects. Cell-based models may be suitable for these studies, as direct measurements of oxidative stress such as markers of mitochondrial stress (e.g., oxygen consumption and hydrogen ion generation), as well as generation of radical species, can be kinetically monitored.
Individual signaling mechanisms, such as those involving STAT3 and NF-κB, require validation for opioid and AA co-administration. While opioids have been shown to increase, and AA has been shown to decrease, each of these transcription factors’ activities (binding to target genes) is dependent upon upstream signaling from opioids and AA. Therefore, the dose and period of administration, in other words the strength and persistence of upstream signaling, are critical in validating changes in these signaling pathways. Animal models are implicit to define translatable effects of vitamin C and opioid co-administration, yet the presence or absence of Gulo and standardization of strain/species used must be taken into account.
Overall vitamin C is safe, relatively non-toxic, inexpensive, and widely available, which justifies it as a potential facile therapeutic or adjuvant for recovery after opioid intoxication. However, much further research is necessary to qualify previous observations of its capacity to ameliorate opioid side effects and define the underlying signaling interactions.
Author Contributions
Conceptualization, M.N. and J.B.; methodology, M.N.; investigation, M.N. and H.C.; data curation, M.N. and H.C.; writing—original draft preparation, M.N. and H.C.; writing—review and editing, J.B.; visualization, M.N.; supervision, J.B.; project administration, M.N. and J.B.; funding acquisition, J.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Defense Threat Reduction Agency grant number HDTRA1-20-1-0008.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article.
Acknowledgments
The authors would like to acknowledge Suzanne Danley for administrative and technical support.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AA | ascorbic acid |
| AC | adenylate cyclase |
| Akt | protein kinase B |
| aspOHase | asparaginyl hydroxylase |
| BH3 | trihydrobiopterin |
| BH4 | tetrahydrobiopterin |
| CaM | calmodulin |
| cAMP | cyclic adenosine monophosphate |
| CAT | catalase |
| cNOS | constitutive nitric oxide synthase |
| CREB | cAMP response element-binding protein |
| DHA | dehydroascorbic acid |
| DOR | δ opioid receptor |
| eNOS | endothelial nitric oxide synthase |
| ER | endoplasmic reticulum |
| ERK | extracellular-regulated signaling kinase |
| GPCR | G-protein-coupled receptor |
| GPx | glutathione peroxidase |
| GSK3β | glycogen synthase kinase 3β |
| Gulo | L-gulonolactone oxidase |
| HIF1α | hypoxia-induced factor 1α |
| IKK | IκB kinase |
| IP3 | inositol-3-phosphate |
| JAK | janus kinase |
| LC | locus ceruleus |
| MAPK | mitogen-activated protein kinase |
| MOR | μ opioid receptor |
| mTOR | mammalian target of rapamycin |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| nNOS | neuronal nitric oxide synthase |
| NO | nitric oxide |
| OIRD | opioid-induced respiratory disorder |
| ONOO- | peroxynitrite |
| PI3K | phosphoinositol-3-kinase |
| PKA | protein kinase A |
| PLC | phospholipase C |
| PP2A | protein phosphatase 2A |
| pro3/4OHase | prolyl 3- and prolyl 4-hydroxylases |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| SOD | superoxide dismutase |
| STAT1/2/3 | signal transducer and activator of transcription 1/2/3 |
| SVCT1/2 | sodium-dependent vitamin C transporters 1/2 |
| TH | tyrosine hydroxylase |
| TPH | tryptophan hydroxylase |
| VTA | ventral tegmental area |
| αi/o | Gαi/o subunit |
| β/γ | Gβ/γ subunit |
References
- Swegle, J.M.; Logemann, C. Management of common opioid-induced adverse effects. Am. Fam. Physician 2006, 74, 1347–1354. [Google Scholar] [PubMed]
- Benyamin, R.; Trescot, A.M.; Datta, S.; Buenaventura, R.; Adlaka, R.; Sehgal, N.; Glaser, S.E.; Vallejo, R. Opioid complications and side effects. Pain Physician 2008, 11, S105–S120. [Google Scholar] [CrossRef] [PubMed]
- Porreca, F.; Ossipov, M.H. Nausea and Vomiting Side Effects with Opioid Analgesics during Treatment of Chronic Pain: Mechanisms, Implications, and Management Options. Pain Med. 2009, 10, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Vowles, K.E.; McEntee, M.L.; Julnes, P.S.; Frohe, T.; Ney, J.P.; van der Goes, D.N. Rates of opioid misuse, abuse, and addiction in chronic pain. Pain 2015, 156, 569–576. [Google Scholar] [CrossRef]
- Boom, M.; Niesters, M.; Sarton, E.; Aarts, L.; Smith, T.W.; Dahan, A. Non-Analgesic Effects of Opioids: Opioid-induced Respiratory Depression. Curr. Pharm. Des. 2012, 18, 5994–6004. [Google Scholar] [CrossRef]
- Talwar, D.; McConnachie, A.; Welsh, P.; Upton, M.; O'Reilly, D.; Smith, G.D.; Watt, G.; Sattar, N. Which Circulating Antioxidant Vitamins Are Confounded by Socioeconomic Deprivation? The MIDSPAN Family Study. PLoS ONE 2010, 5, e11312. [Google Scholar] [CrossRef]
- Bloodbook Blood Test Results—Normal Ranges. Available online: www.bloodbook.com/ranges.html (accessed on 4 June 2022).
- Bhagavan, N.V.; Ha, C.-E. Protein and Amino Acid Metabolism. In Essentials of Medical Biochemistry; Elsevier: Amsterdam, The Netherlands, 2015; pp. 227–268. [Google Scholar]
- Majewska, M.D.; Bell, J.A.; London, E.D. Regulation of the NMDA receptor by redox phenomena: Inhibitory role of ascorbate. Brain Res. 1990, 537, 328–332. [Google Scholar] [CrossRef]
- Moores, J. Vitamin C: A wound healing perspective. Br. J. Community Nurs. 2013, 18, S6–S11. [Google Scholar] [CrossRef]
- Cerullo, G.; Negro, M.; Parimbelli, M.; Pecoraro, M.; Perna, S.; Liguori, G.; Rondanelli, M.; Cena, H.; D’Antona, G. The Long History of Vitamin C: From Prevention of the Common Cold to Potential Aid in the Treatment of COVID-19. Front. Immunol. 2020, 11, 574029. [Google Scholar] [CrossRef]
- Zhou, J.; Si, P.; Ruan, Z.; Ma, S.; Yan, X.; Sun, L.; Peng, F.; Yuan, H.; Cai, D.; Ding, D.; et al. Primary studies on heroin abuse and injury induced by oxidation and lipoperoxidation. Chin. Med. J. 2001, 114, 297–302. [Google Scholar]
- Zhou, J.F.; Yan, X.F.; Ruan, Z.R.; Peng, F.Y.; Cai, D.; Yuan, H.; Sun, L.; Ding, D.Y.; Xu, S.S. Heroin abuse and nitric oxide, oxidation, peroxidation, lipoperoxidation. Biomed. Environ. Sci. 2000, 13, 131–139. [Google Scholar] [PubMed]
- Johnston, P.; Chahl, L.A. Chronic treatment with ascorbic acid inhibits the morphine withdrawal response in guinea-pigs. Neurosci. Lett. 1992, 135, 23–27. [Google Scholar] [CrossRef]
- Alaei, H.A.; Ramshini, E.; Talkhooncheh, M.; Shahidani, S. The effect of vitamin C on morphine self-administration in rats. Adv. Biomed. Res. 2014, 3, 178. [Google Scholar] [CrossRef]
- Evangelou, A.; Kalfakakou, V.; Georgakas, P.; Koutras, V.; Vezyraki, P.; Iliopoulou, L.; Vadalouka, A. Ascorbic acid (vitamin C) effects on withdrawal syndrome of heroin abusers. In Vivo 2000, 14, 363–366. [Google Scholar]
- Zelfand, E. Vitamin C, Pain and Opioid Use Disorder. Integr. Med. 2020, 19, 18–29. [Google Scholar]
- Scott, J.; Winfield, A.; Kennedy, E.; Bond, C. Laboratory study of the effects of citric and ascorbic acids on injections prepared with brown heroin. Int. J. Drug Policy 2000, 11, 417–422. [Google Scholar] [CrossRef]
- Andersen, J.M.; Bogen, I.L.; Karinen, R.; Brochmann, G.W.; Mørland, J.; Vindenes, V.; Boix, F. Does the preparation for intravenous administration affect the composition of heroin injections? A controlled laboratory study. Addiction 2021, 116, 3104–3112. [Google Scholar] [CrossRef]
- Kanazi, G.E.; El-Khatib, M.F.; Karam, V.G.Y.; Hanna, J.E.; Masri, B.; Aouad, M.T. Effect of vitamin C on morphine use after laparoscopic cholecystectomy: A randomized controlled trial. Can. J. Anaesth. 2012, 59, 538–543. [Google Scholar] [CrossRef]
- Chen, S.; Roffey, D.; Dion, C.-A.; Arab, A.; Wai, E.K. Effect of Perioperative Vitamin C Supplementation on Postoperative Pain and the Incidence of Chronic Regional Pain Syndrome. Clin. J. Pain 2016, 32, 179–185. [Google Scholar] [CrossRef]
- Tunay, D.L.; Ilgınel, M.T.; Ünlügenç, H.; Tunay, M.; Karacaer, F.; Biricik, E. Comparison of the effects of preoperative melatonin or vitamin C administration on postoperative analgesia. Bosn. J. Basic Med. Sci. 2020, 20, 117–124. [Google Scholar] [CrossRef]
- Chaitanya, N.C.; Muthukrishnan, A.; Krishnaprasad, C.; Sanjuprasanna, G.; Pillay, P.; Mounika, B. An Insight and Update on the Analgesic Properties of Vitamin C. J. Pharm. Bioallied Sci. 2018, 10, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.C.; McCall, C. The role of vitamin C in the treatment of pain: New insights. J. Transl. Med. 2017, 15, 77. [Google Scholar] [CrossRef] [PubMed]
- E Willette, R.; Thomas, B.L.; Barnett, G. Inhibition of morphine analgesia by ascorbate. Res. Commun. Chem. Pathol. Pharmacol. 1983, 42, 485–491. [Google Scholar] [CrossRef]
- Khanna, N.; Sharma, S.K. Megadoses of vitamin C prevent the development of tolerance and physical dependence on morphine in mice. Life Sci. 1983, 33, 401–404. [Google Scholar] [CrossRef]
- Alaei, H.; Esmaeili, M.; Nasimi, A.; Pourshanazari, A. Ascorbic acid decreases morphine self-administration and withdrawal symptoms in rats. Pathophysiology 2005, 12, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Enrico, P.; A Mura, M.; Esposito, G.; Serra, P.; Migheli, R.; De Natale, G.; Desole, M.S.; Miele, M.; Miele, E. Effect of naloxone on morphine-induced changes in striatal dopamine metabolism and glutamate, ascorbic acid and uric acid release in freely moving rats. Brain Res. 1998, 797, 94–102. [Google Scholar] [CrossRef]
- Desole, M.S.; Esposito, G.; Fresu, L.; Migheli, R.; Enrico, P.; Mura, M.A.; De Natale, G.; Miele, E.; Miele, M. Effects of morphine treatment and withdrawal on striatal and limbic monoaminergic activity and ascorbic acid oxidation in the rat. Brain Res. 1996, 723, 154–161. [Google Scholar] [CrossRef]
- Enrico, P.; Esposito, G.; Mura, M.A.; Fresu, L.; De Natale, G.; Miele, E.; Desole, M.S.; Miele, M. Effect of morphine on striatal dopamine metabolism and ascorbic acid and uric acid release in freely moving rats. Brain Res. 1997, 745, 173–182. [Google Scholar] [CrossRef]
- Ordan, B.A.; Cvejic, S.; Devi, L.A. Opioids and Their Complicated Receptor Complexes. Neuropsychopharmacology 2000, 23, S5–S18. [Google Scholar] [CrossRef]
- Ballantyne, J.C.; Sullivan, M.D. Discovery of endogenous opioid systems: What it has meant for the clinician's understanding of pain and its treatment. Pain 2017, 158, 2290–2300. [Google Scholar] [CrossRef]
- Pasternak, G.; Pan, Y.-X. Mu opioid receptors in pain management. Acta Anaesthesiol. Taiwanica 2011, 49, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Valentino, R.J.; Volkow, N.D. Untangling the complexity of opioid receptor function. Neuropsychopharmacology 2018, 43, 2514–2520. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef] [PubMed]
- Human Protein Atlas. Available online: https://www.proteinatlas.org/ (accessed on 15 May 2022).
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, N.; Smith, H.S.; Manchikanti, L. Peripherally acting opioids and clinical implications for pain control. Pain Physician 2011, 14, 249–258. [Google Scholar] [CrossRef]
- Connor, M.; Christie, M. Opioid receptor signalling mechanisms. Clin. Exp. Pharmacol. Physiol. 1999, 26, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Gonzalez-Zulueta, M.; Huang, H.; Herring, W.J.; Ahn, S.; Ginty, D.D.; Dawson, V.L.; Dawson, T.M. Dynamic regulation of neuronal NO synthase transcription by calcium influx through a CREB family transcription factor-dependent mechanism. Proc. Natl. Acad. Sci. USA 2000, 97, 8617–8622. [Google Scholar] [CrossRef]
- Zhang, X.; Odom, D.T.; Koo, S.-H.; Conkright, M.D.; Canettieri, G.; Best, J.; Chen, H.; Jenner, R.; Herbolsheimer, E.; Jacobsen, E.; et al. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 4459–4464. [Google Scholar] [CrossRef]
- Listos, J.; Łupina, M.; Talarek, S.; Mazur, A.; Orzelska-Górka, J.; Kotlińska, J. The Mechanisms Involved in Morphine Addiction: An Overview. Int. J. Mol. Sci. 2019, 20, 4302. [Google Scholar] [CrossRef]
- Smith, S.H. Opioid metabolism. In Mayo Clinic Proceedings; Elsevier: Amsterdam, The Netherlands, 2009; pp. 613–624. [Google Scholar]
- Stone, A.N.; Mackenzie, P.; Galetin, A.; Houston, J.B.; Miners, J.O. Isoform selectivity and kinetics of morphine 3- and 6-glucuronidation by human udp-glucuronosyltransferases: Evidence for atypical glucuronidation kinetics by ugt2b7. Drug Metab. Dispos. 2003, 31, 1086–1089. [Google Scholar] [CrossRef]
- Veith, A.; Moorthy, B. Role of cytochrome P450s in the generation and metabolism of reactive oxygen species. Curr. Opin. Toxicol. 2018, 7, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Feltrin, C.; Farias, I.V.; Sandjo, L.P.; Reginatto, F.H.; Simões, C.M.O. Effects of Standardized Medicinal Plant Extracts on Drug Metabolism Mediated by CYP3A4 and CYP2D6 Enzymes. Chem. Res. Toxicol. 2020, 33, 2408–2419. [Google Scholar] [CrossRef] [PubMed]
- Klimas, R.; Mikus, G. Morphine-6-glucuronide is responsible for the analgesic effect after morphine administration: A quantitative review of morphine, morphine-6-glucuronide, and morphine-3-glucuronide. Br. J. Anaesth. 2014, 113, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Kang, Y.S.; Bickel, U.; Pardridge, W.M. Blood-brain barrier permeability to morphine-6-glucuronide is markedly reduced compared with morphine. Drug Metab. Dispos. 1997, 25, 768–771. [Google Scholar]
- Samer, C.F.; Daali, Y.; Wagner, M.; Hopfgartner, G.; Eap, C.B.; Rebsamen, M.C.; Rossier, M.F.; Hochstrasser, D.; Dayer, P.; Desmeules, J.A. The effects of CYP2D6 and CYP3A activities on the pharmacokinetics of immediate release oxycodone. Br. J. Pharmacol. 2010, 160, 907–918. [Google Scholar] [CrossRef]
- Balyan, R.; Mecoli, M.; Venkatasubramanian, R.; Chidambaran, V.; Kamos, N.; Clay, S.; Moore, D.L.; Mavi, J.; Glover, C.D.; Szmuk, P.; et al. CYP2D6 pharmacogenetic and oxycodone pharmacokinetic association study in pediatric surgical patients. Pharmacogenomics 2017, 18, 337–348. [Google Scholar] [CrossRef]
- Owen, J.A.; Nakatsu, K. Diacetylmorphine (heroin) hydrolases in human blood. Can. J. Physiol. Pharmacol. 1983, 61, 870–875. [Google Scholar] [CrossRef]
- Lockridge, O.; Mottershaw-Jackson, N.; Eckerson, H.W.; La Du, B.N. Hydrolysis of diacetylmorphine (heroin) by human serum cholinesterase. J. Pharmacol. Exp. Ther. 1980, 215, 1–8. [Google Scholar]
- Garbuz, O.; Gulea, A.; Dyniewicz, J.; Zablocka, B.; Lipkowski, A.W. The non-opioid receptor, antioxidant properties of morphine and the opioid peptide analog biphalin. Peptides 2015, 63, 1–3. [Google Scholar] [CrossRef]
- Gülçin, I.; Beydemir, S.; Alici, H.A.; Elmastaş, M.; Büyükokuroğlu, M.E. In vitro antioxidant properties of morphine. Pharmacol. Res. 2004, 49, 59–66. [Google Scholar] [CrossRef]
- Nilsen-Moe, A.; Reinhardt, C.R.; Glover, S.D.; Liang, L.; Hammes-Schiffer, S.; Hammarström, L.; Tommos, C. Proton-Coupled Electron Transfer from Tyrosine in the Interior of a de novo Protein: Mechanisms and Primary Proton Acceptor. J. Am. Chem. Soc. 2020, 142, 11550–11559. [Google Scholar] [CrossRef] [PubMed]
- Radi, R. Protein Tyrosine Nitration: Biochemical Mechanisms and Structural Basis of Functional Effects. Acc. Chem. Res. 2013, 46, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Tyndale, R.F.; Droll, K.P.; Sellers, E. Genetically deficient CYP2D6 metabolism provides protection against oral opiate dependence. Pharmacogenetics 1997, 7, 375–379. [Google Scholar] [CrossRef]
- Samer, C.; Daali, Y.; Wagner, M.; Hopfgartner, G.; Eap, C.; Rebsamen, M.; Rossier, M.; Hochstrasser, D.; Dayer, P.; Desmeules, J. Genetic polymorphisms and drug interactions modulating CYP2D6 and CYP3A activities have a major effect on oxycodone analgesic efficacy and safety. J. Cereb. Blood Flow Metab. 2010, 160, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Toki, S.; Yamano, S. Production of Morphinone as a Metabolite of Morphine and Its Physiological Role. YAKUGAKU ZASSHI 1999, 119, 249–267. [Google Scholar] [CrossRef][Green Version]
- Yamano, S.; Takahashi, A.; Todaka, T.; Toki, S. In vivo and in vitro formation of morphinone from morphine in rat. Xenobiotica 1997, 27, 645–656. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Amatore, C.; Arbault, S.; Ferreira, D.C.M.; Tapsoba, I.; Verchier, Y. Vitamin C stimulates or attenuates reactive oxygen and nitrogen species (ROS, RNS) production depending on cell state: Quantitative amperometric measurements of oxidative bursts at PLB-985 and RAW 264.7 cells at the single cell level. J. Electroanal. Chem. 2008, 615, 34–44. [Google Scholar] [CrossRef]
- Cobley, J.N.; McHardy, H.; Morton, J.P.; Nikolaidis, M.G.; Close, G.L. Influence of vitamin C and vitamin E on redox signaling: Implications for exercise adaptations. Free Radic. Biol. Med. 2015, 84, 65–76. [Google Scholar] [CrossRef]
- Paciolla, C.; Fortunato, S.; Dipierro, N.; Paradiso, A.; De Leonardis, S.; Mastropasqua, L.; de Pinto, M.C. Vitamin C in Plants: From Functions to Biofortification. Antioxidants 2019, 8, 519. [Google Scholar] [CrossRef]
- Yang, H. Conserved or Lost: Molecular Evolution of the Key Gene GULO in Vertebrate Vitamin C Biosynthesis. Biochem. Genet. 2013, 51, 413–425. [Google Scholar] [CrossRef]
- Jiao, Y.; Zhang, J.; Yan, J.; Stuart, J.; Gibson, G.; Lu, L.; Willaims, R.; Wang, Y.J.; Gu, W. Differential gene expression between wild-type and Gulo-deficient mice supplied with vitamin C. Genet. Mol. Biol. 2011, 34, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Curran, C.P.; Nebert, D.W.; Patel, K.V.; Williams, M.T.; Vorhees, C.V. Effect of vitamin C deficiency during postnatal development on adult behavior: Functional phenotype ofGulo(−/−)knockout mice. Genes Brain Behav. 2012, 11, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Aumailley, L.; Warren, A.; Garand, C.; Dubois, M.J.; Paquet, E.R.; Le Couteur, D.G.; Marette, A.; Cogger, V.C.; Lebel, M. Vitamin C modulates the metabolic and cytokine profiles, alleviates hepatic endoplasmic reticulum stress, and increases the life span of Gulo−/− mice. Aging 2016, 8, 458–483. [Google Scholar] [CrossRef] [PubMed]
- Shyu, K.-G.; Chang, C.-C.; Yeh, Y.-C.; Sheu, J.-R.; Chou, D.-S. Mechanisms of Ascorbyl Radical Formation in Human Platelet-Rich Plasma. BioMed Res. Int. 2014, 2014, 614506. [Google Scholar] [CrossRef] [PubMed]
- ScienceLab.com Material Safety Data Sheet Ascorbic Acid MSDS 2022. Available online: https://sciencelab.com/ (accessed on 12 April 2022).
- Levine, M.; Rumsey, S.C.; Daruwala, R.; Park, J.B.; Wang, Y. Criteria and Recommendations for Vitamin C Intake. JAMA 1999, 281, 1415–1423. [Google Scholar] [CrossRef]
- Mandl, J.; Szarka, A.; Bánhegyi, G. Vitamin C: Update on physiology and pharmacology. J. Cereb. Blood Flow Metab. 2009, 157, 1097–1110. [Google Scholar] [CrossRef]
- Buettner, G.R.; Jurkiewicz, B.A. Catalytic metals, ascorbate and free radicals: Combinations to avoid. Radiat. Res. 1996, 145, 532–541. [Google Scholar] [CrossRef]
- Li, X.; Cobb, C.E.; Hill, K.E.; Burk, R.F.; May, J.M. Mitochondrial Uptake and Recycling of Ascorbic Acid. Arch. Biochem. Biophys. 2001, 387, 143–153. [Google Scholar] [CrossRef]
- Lykkesfeldt, J.; Tveden-Nyborg, P. The Pharmacokinetics of Vitamin C. Nutrients 2019, 11, 2412. [Google Scholar] [CrossRef]
- Li, Y.; Schellhorn, H.E. New Developments and Novel Therapeutic Perspectives for Vitamin C. J. Nutr. 2007, 137, 2171–2184. [Google Scholar] [CrossRef] [PubMed]
- Rock, C.L.; Jacob, R.A.; Bowen, P. Update on the Biological Characteristics of the Antioxidant Micronutrients: Vitamin C, Vitamin E, and the Carotenoids. J. Am. Diet. Assoc. 1996, 96, 693–702. [Google Scholar] [CrossRef]
- Lane, D.J.; Richardson, D.R. The active role of vitamin C in mammalian iron metabolism: Much more than just enhanced iron absorption! Free Radic. Biol. Med. 2014, 75, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Ballaz, S.J.; Rebec, G.V. Neurobiology of vitamin C: Expanding the focus from antioxidant to endogenous neuromodulator. Pharmacol. Res. 2019, 146, 104321. [Google Scholar] [CrossRef] [PubMed]
- Jacob, R.A.; Sotoudeh, G. Vitamin C Function and Status in Chronic Disease. Nutr. Clin. Care 2002, 5, 66–74. [Google Scholar] [CrossRef]
- Figueroa-Méndez, R.; Rivas-Arancibia, S. Vitamin C in Health and Disease: Its Role in the Metabolism of Cells and Redox State in the Brain. Front. Physiol. 2015, 6, 397. [Google Scholar] [CrossRef]
- Mou, L.; Lankford-Turner, P.; Leander, M.V.; Bissonnette, R.P.; Donahoe, R.M.; Royal, W. RXR-induced TNF-α suppression is reversed by morphine in activated U937 cells. J. Neuroimmunol. 2004, 147, 99–105. [Google Scholar] [CrossRef]
- Gu, P.F.; Wu, C.F.; Yang, J.Y.; Shang, Y.; Hou, Y.; Bi, X.L.; Dai, F. Differential effects of drug-induced ascorbic acid release in the striatum and nucleus accumbens of freely moving rats. Neurosci. Lett. 2006, 399, 79–84. [Google Scholar] [CrossRef]
- Cisternas, P.; Silva-Alvarez, C.; Martínez, F.; Fernández, E.; Ferrada, L.; Oyarce, K.; Salazar, K.; Bolanos, J.P.; Nualart, F. The oxidized form of vitamin C, dehydroascorbic acid, regulates neuronal energy metabolism. J. Neurochem. 2014, 129, 663–671. [Google Scholar] [CrossRef]
- Johnston, C.S.; Corte, C.; Swan, P.D. Marginal vitamin C status is associated with reduced fat oxidation during submaximal exercise in young adults. Nutr. Metab. 2006, 3, 35. [Google Scholar] [CrossRef]
- Chambial, S.; Dwivedi, S.; Shukla, K.K.; John, P.J.; Sharma, P. Vitamin C in Disease Prevention and Cure: An Overview. Indian J. Clin. Biochem. 2013, 28, 314–328. [Google Scholar] [CrossRef] [PubMed]
- Kosten, T.R.; George, T.P. The Neurobiology of Opioid Dependence: Implications for Treatment. Sci. Pract. Perspect. 2002, 1, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Bubier, J.A.; He, H.; Philip, V.M.; Roy, T.; Hernandez, C.M.; Bernat, R.; Donohue, K.D.; O’Hara, B.F.; Chesler, E.J. Genetic variation regulates opioid-induced respiratory depression in mice. Sci. Rep. 2020, 10, 14970. [Google Scholar] [CrossRef]
- Hayes, J.A.; Kottick, A.; Picardo, M.C.D.; Halleran, A.D.; Smith, R.D.; Smith, G.D.; Saha, M.S.; Del Negro, C.A. Transcriptome of neonatal preBötzinger complex neurones in Dbx1 reporter mice. Sci. Rep. 2017, 7, 8669. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.C.; Ellenberger, H.H.; Ballanyi, K.; Richter, D.W.; Feldman, J.L. Pre-Bötzinger Complex: A Brainstem Region that May Generate Respiratory Rhythm in Mammals. Science 1991, 254, 726–729. [Google Scholar] [CrossRef]
- Raehal, K.M.; Bohn, L.M. β-Arrestins: Regulatory Role and Therapeutic Potential in Opioid and Cannabinoid Receptor-Mediated Analgesia. Arrestins-Pharmacol. Ther. Potential 2014, 219, 427–443. [Google Scholar] [CrossRef]
- Raehal, K.M.; Walker, J.K.L.; Bohn, L. Morphine Side Effects in β-Arrestin 2 Knockout Mice. J. Pharmacol. Exp. Ther. 2005, 314, 1195–1201. [Google Scholar] [CrossRef]
- Bohn, L.M.; Gainetdinov, R.; Lin, F.-T.; Lefkowitz, R.J.; Caron, M.G. μ-Opioid receptor desensitization by β-arrestin-2 determines morphine tolerance but not dependence. Nature 2000, 408, 720–723. [Google Scholar] [CrossRef]
- Stahl, E.L.; Schmid, C.L.; Acevedo-Canabal, A.; Read, C.; Grim, T.W.; Kennedy, N.M.; Bannister, T.D.; Bohn, L.M. G protein signaling–biased mu opioid receptor agonists that produce sustained G protein activation are noncompetitive agonists. Proc. Natl. Acad. Sci. USA 2021, 118, e2102178118. [Google Scholar] [CrossRef]
- Gillis, A.; Kliewer, A.; Kelly, E.; Henderson, G.; Christie, M.J.; Schulz, S.; Canals, M. Critical Assessment of G Protein-Biased Agonism at the μ-Opioid Receptor. Trends Pharmacol. Sci. 2020, 41, 947–959. [Google Scholar] [CrossRef]
- Stanczyk, M.A.; Kandasamy, R. Biased agonism: The quest for the analgesic holy grail. PAIN Rep. 2018, 3, e650. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, G.W.; Pan, Y.-X. Mu Opioids and Their Receptors: Evolution of a Concept. Pharmacol. Rev. 2013, 65, 1257–1317. [Google Scholar] [CrossRef] [PubMed]
- Agus, D.B.; Gambhir, S.S.; Pardridge, W.M.; Spielholz, C.; Baselga, J.; Vera, J.C.; Golde, D.W. Vitamin C crosses the blood-brain barrier in the oxidized form through the glucose transporters. J. Clin. Investig. 1997, 100, 2842–2848. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J.R.; Lawen, A. The Glutamate Aspartate Transporter (GLAST) Mediates l-Glutamate-Stimulated Ascorbate-Release via Swelling-Activated Anion Channels in Cultured Neonatal Rodent Astrocytes. Cell Biophys. 2013, 65, 107–119. [Google Scholar] [CrossRef]
- Lane, D.J.R.; Lawen, A. Ascorbate and plasma membrane electron transport—Enzymes vs efflux. Free Radic. Biol. Med. 2009, 47, 485–495. [Google Scholar] [CrossRef]
- Sekiguchi, F.; Kawabata, A. T-type Calcium Channels: Functional Regulation and Implication in Pain Signaling. J. Pharmacol. Sci. 2013, 122, 244–250. [Google Scholar] [CrossRef]
- Ward, M.S.; Lamb, J.; May, J.M.; Harrison, F.E. Behavioral and monoamine changes following severe vitamin C deficiency. J. Neurochem. 2013, 124, 363–375. [Google Scholar] [CrossRef]
- Yen, G.-C.; Hsieh, C.-L. Antioxidant Effects of Dopamine and Related Compounds. Biosci. Biotechnol. Biochem. 1997, 61, 1646–1649. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef] [PubMed]
- Colamartino, M.; Santoro, M.; Duranti, G.; Sabatini, S.; Ceci, R.; Testa, A.; Padua, L.; Cozzi, R. Evaluation of Levodopa and Carbidopa Antioxidant Activity in Normal Human Lymphocytes In Vitro: Implication for Oxidative Stress in Parkinson’s Disease. Neurotox. Res. 2015, 27, 106–117. [Google Scholar] [CrossRef]
- Álvarez-Diduk, R.; Galano, A. Adrenaline and Noradrenaline: Protectors against Oxidative Stress or Molecular Targets? J. Phys. Chem. B 2015, 119, 3479–3491. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.A.; Chae, S.; Lee, K.H.; Zhang, R.; Jung, M.S.; You, H.J.; Kim, J.S.; Hyun, J.W. Antioxidant effect of homogenetisic acid on hydrogen peroxide induced oxidative stress in human lung fibroblast cells. Biotechnol. Bioprocess Eng. 2005, 10, 556–563. [Google Scholar] [CrossRef]
- Boundy, V.A.; Gold, S.J.; Messer, C.J.; Chen, J.; Son, J.H.; Joh, T.H.; Nestler, E.J. Regulation of Tyrosine Hydroxylase Promoter Activity by Chronic Morphine in TH9.0-LacZ Transgenic Mice. J. Neurosci. 1998, 18, 9989–9995. [Google Scholar] [CrossRef]
- Beitner-Johnson, D.; Nestler, E.J. Morphine and Cocaine Exert Common Chronic Actions on Tyrosine Hydroxylase in Dopaminergic Brain Reward Regions. J. Neurochem. 1991, 57, 344–347. [Google Scholar] [CrossRef]
- Bariselli, S.; Glangetas, C.; Tzanoulinou, S.; Bellone, C. Ventral tegmental area subcircuits process rewarding and aversive experiences. J. Neurochem. 2016, 139, 1071–1080. [Google Scholar] [CrossRef]
- Boadle-Bibe, M.C.; Johannessen, J.N.; Narasimhachari, N.; Phan, T.-H. Activation of cortical tryptophan hydroxylase by acute morphine treatment: Blockade by 6-hydroxydopamine. Eur. J. Pharmacol. 1987, 139, 193–204. [Google Scholar] [CrossRef]
- Bhat, R.S.; Bhaskaran, M.; Mongia, A.; Hitosugi, N.; Singhal, P.C. Morphine-induced macrophage apoptosis: Oxidative stress and strategies for modulation. J. Leukoc. Biol. 2004, 75, 1131–1138. [Google Scholar] [CrossRef]
- Cai, Y.; Yang, L.; Hu, G.; Chen, X.; Niu, F.; Yuan, L.; Liu, H.; Xiong, H.; Arikkath, J.; Buch, S. Regulation of morphine-induced synaptic alterations: Role of oxidative stress, ER stress, and autophagy. J. Cell Biol. 2016, 215, 245–258. [Google Scholar] [CrossRef]
- Skrabalova, J.; Drastichova, Z.; Novotny, J. Morphine as a Potential Oxidative Stress-Causing Agent. Mini-Rev. Org. Chem. 2013, 10, 367–372. [Google Scholar] [CrossRef]
- Wilcox, B.J.; Ritenour-Rodgers, K.J.; Asser, A.S.; Baumgart, L.E.; Baumgart, M.A.; Boger, D.L.; DeBlassio, J.L.; Delong, M.A.; Glufke, U.; Henz, M.E.; et al. N-Acylglycine Amidation: Implications for the Biosynthesis of Fatty Acid Primary Amides. Biochemistry 1999, 38, 3235–3245. [Google Scholar] [CrossRef] [PubMed]
- Lipina, C.; Hundal, H.S. Modulation of cellular redox homeostasis by the endocannabinoid system. Open Biol. 2016, 6, 150276. [Google Scholar] [CrossRef]
- Al-Azzam, N.; Teegala, L.R.; Pokhrel, S.; Ghebreigziabher, S.; Chachkovskyy, T.; Thodeti, S.; Gavilanes, I.; Covington, K.; Thodeti, C.K.; Paruchuri, S. Transient Receptor Potential Vanilloid channel regulates fibroblast differentiation and airway remodeling by modulating redox signals through NADPH Oxidase 4. Sci. Rep. 2020, 10, 9827. [Google Scholar] [CrossRef] [PubMed]
- Farfariello, V.; Amantini, C.; Santoni, G. Transient receptor potential vanilloid 1 activation induces autophagy in thymocytes through ROS-regulated AMPK and Atg4C pathways. J. Leukoc. Biol. 2012, 92, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Salarian, A.; Kadkhodaee, M.; Zahmatkesh, M.; Seifi, B.; Bakhshi, E.; Akhondzadeh, S.; Adeli, S.; Askari, H.; Arbabi, M. Opioid Use Disorder Induces Oxidative Stress and Inflammation: The Attenuating Effect of Methadone Maintenance Treatment. Iran. J. Psychiatry 2018, 13, 46–54. [Google Scholar]
- Ajayi, A.F.; Akhigbe, R.E. Codeine-induced sperm DNA damage is mediated predominantly by oxidative stress rather than apoptosis. Redox Rep. 2020, 25, 33–40. [Google Scholar] [CrossRef]
- Mohamed, H.M.; Mahmoud, A.M. Chronic exposure to the opioid tramadol induces oxidative damage, inflammation and apoptosis, and alters cerebral monoamine neurotransmitters in rats. Biomed. Pharmacother. 2019, 110, 239–247. [Google Scholar] [CrossRef]
- Motaghinejad, M.; Karimian, M.; Motaghinejad, O.; Shabab, B.; Yazdani, I.; Fatima, S. Protective effects of various dosage of Curcumin against morphine induced apoptosis and oxidative stress in rat isolated hippocampus. Pharmacol. Rep. 2015, 67, 230–235. [Google Scholar] [CrossRef]
- Samarghandian, S.; Afshari, R.; Farkhondeh, T. Effect of long-term treatment of morphine on enzymes, oxidative stress indices and antioxidant status in male rat liver. Int. J. Clin. Exp. Med. 2014, 7, 1449–1453. [Google Scholar]
- Abdel-Zaher, A.O.; Mostafa, M.G.; Farghaly, H.S.; Hamdy, M.M.; Abdel-Hady, R.H. Role of oxidative stress and inducible nitric oxide synthase in morphine-induced tolerance and dependence in mice. Effect of alpha-lipoic acid. Behav. Brain Res. 2013, 247, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Bu, Q.; Yang, Y.; Yan, G.; Hu, Z.; Hu, C.; Duan, J.; Lv, L.; Zhou, J.; Zhao, J.; Shao, X.; et al. Proteomic analysis of the nucleus accumbens in rhesus monkeys of morphine dependence and withdrawal intervention. J. Proteom. 2012, 75, 1330–1342. [Google Scholar] [CrossRef] [PubMed]
- Nyssen, P.; Mouithys-Mickalad, A.; Minguet, G.; Sauvage, E.; Wouters, J.; Franck, T.; Hoebeke, M. Morphine, a potential inhibitor of myeloperoxidase activity. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2236–2244. [Google Scholar] [CrossRef] [PubMed]
- Kramarenko, G.G.; Hummel, S.G.; Martin, S.M.; Buettner, G.R. Ascorbate Reacts with Singlet Oxygen to Produce Hydrogen Peroxide. Photochem. Photobiol. 2006, 82, 1634–1637. [Google Scholar] [CrossRef]
- Buettner, G.R.; Jurkiewicz, B.A. Ascorbate Radical: A Valuable Marker of Oxidative Stress. In Analysis of Free Radicals in Biological Systems; Birkhäuser Basel: Basel, Switherland, 1995; pp. 145–164. [Google Scholar]
- Brennan, L.A.; Morris, G.M.; Wasson, G.R.; Hannigan, B.M.; Barnett, Y.A. The effect of vitamin C or vitamin E supplementation on basal and H2O2-induced DNA damage in human lymphocytes. Br. J. Nutr. 2000, 84, 195–202. [Google Scholar] [CrossRef]
- Kao, P.-F.; Lee, W.-S.; Liu, J.-C.; Chan, P.; Tsai, J.-C.; Hsu, Y.-H.; Chang, W.-Y.; Cheng, T.-H.; Liao, S.-S. Downregulation of superoxide dismutase activity and gene expression in cultured rat brain astrocytes after incubation with vitamin C. Pharmacology 2003, 69, 1–6. [Google Scholar] [CrossRef]
- Chen, L.H.; Thacker, R.R. An increase in glutathione peroxidase activity induced by high supplementation of vitamin C in rats. Nutr. Res. 1984, 4, 657–664. [Google Scholar] [CrossRef]
- Chen, X.; Touyz, R.M.; Park, J.B.; Schiffrin, E.L. Antioxidant Effects of Vitamins C and E Are Associated With Altered Activation of Vascular NADPH Oxidase and Superoxide Dismutase in Stroke-Prone SHR. Hypertension 2001, 38, 606–611. [Google Scholar] [CrossRef]
- Sureda, A.; Batle, J.M.; Tauler, P.; Ferrer, M.D.; Tur, J.A.; Pons, A. Vitamin C supplementation influences the antioxidant response and nitric oxide handling of erythrocytes and lymphocytes to diving apnea. Eur. J. Clin. Nutr. 2006, 60, 838–846. [Google Scholar] [CrossRef]
- Mahmoudabadi, M.M.S.; Rahbar, A.R. Effect of EPA and Vitamin C on Superoxide Dismutase, Glutathione Peroxidase, Total Antioxidant Capacity and Malondialdehyde in Type 2 Diabetic Patients. Oman Med. J. 2014, 29, 39–45. [Google Scholar] [CrossRef]
- Sedighe, B.; Maryam, B.; Fahimeh, A.; Somayyeh, A. Bigom T Effect of Vitamin C on Salivary Superoxide Dismutase Ac-tivity in Smokers. Aftrican J. Biotechnol. 2011, 10, 7267–7270. [Google Scholar]
- Helen, A.; Vijayammal, P.L. Vitamin C Supplementation on Hepatic Oxidative Stress Induced by Cigarette Smoke. J. Appl. Toxicol. 1997, 17, 289–295. [Google Scholar] [CrossRef]
- Salami, S.A.; Salahdeen, H.M.; Moronkola, O.T.; Murtala, B.A.; Raji, Y. Vitamin C supplementation during chronic variable stress exposure modulates contractile functions of testicular artery and sperm parameters in male Wistar rats. Middle East Fertil. Soc. J. 2020, 25, 8. [Google Scholar] [CrossRef]
- Khassaf, M.; McArdle, A.; Esanu, C.; Vasilaki, A.; Griffiths, R.D.; Brodie, D.A.; Jackson, M.J. Effect of Vitamin C Supplements on Antioxidant Defence and Stress Proteins in Human Lymphocytes and Skeletal Muscle. J. Physiol. 2003, 549, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Ueta, E.; Tadokoro, Y.; Yamamoto, T.; Yamane, C.; Suzuki, E.; Nanba, E.; Otsuka, Y.; Kurata, T. The Effect of Cigarette Smoke Exposure and Ascorbic Acid Intake on Gene Expression of Antioxidant Enzymes and Other Related Enzymes in the Livers and Lungs of Shionogi Rats with Osteogenic Disorders. Toxicol. Sci. 2003, 73, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Kaya, F.; Belin, S.; Diamantidis, G.; Fontes, M. Ascorbic acid is a regulator of the intracellular cAMP concentration: Old molecule, new functions? FEBS Lett. 2008, 582, 3614–3618. [Google Scholar] [CrossRef]
- Rahman, F.; Al Frouh, F.; Bordignon, B.; Fraterno, M.; Landrier, J.-F.; Peiretti, F.; Fontes, M. Ascorbic acid is a dose-dependent inhibitor of adipocyte differentiation, probably by reducing cAMP pool. Front. Cell Dev. Biol. 2014, 2, 29. [Google Scholar] [CrossRef]
- Doulas, N.L.; Constantopoulos, A.; Litsios, B. Effect of Ascorbic Acid on Guinea Pig Adrenal Adenylate Cyclase Activity and Plasma Cortisol. J. Nutr. 1987, 117, 1108–1114. [Google Scholar] [CrossRef]
- Schattauer, S.S.; Bedini, A.; Summers, F.; Reilly-Treat, A.; Andrews, M.M.; Land, B.B.; Chavkin, C. Reactive oxygen species (ROS) generation is stimulated by κ opioid receptor activation through phosphorylated c-Jun N-terminal kinase and inhibited by p38 mitogen-activated protein kinase (MAPK) activation. J. Biol. Chem. 2019, 294, 16884–16896. [Google Scholar] [CrossRef]
- Su, X.; Shen, Z.; Yang, Q.; Sui, F.; Pu, J.; Ma, J.; Ma, S.; Yao, D.; Ji, M.; Hou, P. Vitamin C kills thyroid cancer cells through ROS-dependent inhibition of MAPK/ERK and PI3K/AKT pathways via distinct mechanisms. Theranostics 2019, 9, 4461–4473. [Google Scholar] [CrossRef]
- Su, X.; Li, P.; Han, B.; Jia, H.; Liang, Q.; Wang, H.; Gu, M.; Cai, J.; Li, S.; Zhou, Y.; et al. Vitamin C sensitizes BRAFV600E thyroid cancer to PLX4032 via inhibiting the feedback activation of MAPK/ERK signal by PLX4032. J. Exp. Clin. Cancer Res. 2021, 40, 34. [Google Scholar] [CrossRef]
- Rosas, M.; Porru, S.; Sabariego, M.; Piludu, M.A.; Giorgi, O.; Corda, M.G.; Acquas, E. Effects of morphine on place conditioning and ERK1/2 phosphorylation in the nucleus accumbens of psychogenetically selected Roman low- and high-avoidance rats. Psychopharmacology 2018, 235, 59–69. [Google Scholar] [CrossRef]
- De Freitas, B.G.; Pereira, L.M.; Santa-Cecília, F.V.; Hösch, N.G.; Picolo, G.; Cury, Y.; Zambelli, V.O. Mitogen-Activated Protein Kinase Signaling Mediates Morphine Induced-Delayed Hyperalgesia. Front. Neurosci. 2019, 13, 1018. [Google Scholar] [CrossRef]
- Ferrada, L.; Magdalena, R.; Barahona, M.; Ramírez, E.; Sanzana, C.; Gutiérrez, J.; Nualart, F. Two Distinct Faces of Vitamin C: AA vs. DHA. Antioxidants 2021, 10, 215. [Google Scholar] [CrossRef]
- Park, H.J.; Ock, S.M.; Kim, H.J.; Park, H.J.; Lee, Y.B.; Choi, J.M.; Cho, C.S.; Lee, J.Y.; Cho, B.K.; Cho, D.H. Vitamin C attenuates ERK signalling to inhibit the regulation of collagen production by LL-37 in human dermal fibroblasts. Exp. Dermatol. 2010, 19, e258–e264. [Google Scholar] [CrossRef]
- Ulrich-Merzenich, G.; Zeitler, H.; Panek, D.; Bokemeyer, D.; Vetter, H. Vitamin C promotes human endothelial cell growth via the ERK-signaling pathway. Eur. J. Nutr. 2007, 46, 87–94. [Google Scholar] [CrossRef]
- Su, Z.; Blazing, M.A.; Fan, D.; George, S.E. The Calmodulin-Nitric Oxide Synthase Interaction. J. Biol. Chem. 1995, 270, 29117–29122. [Google Scholar] [CrossRef]
- Cadet, P.; Bilfinger, T.V.; Fimiani, C.; Peter, D.; Stefano, G.B. Human Vascular and Cardiac Endothelia Express Mu Opiate Receptor Transcripts. Endothelium 2000, 7, 185–191. [Google Scholar] [CrossRef]
- Toda, N.; Kishioka, S.; Hatano, Y.; Toda, H. Interactions between morphine and nitric oxide in various organs. J. Anesthesia 2009, 23, 554–568. [Google Scholar] [CrossRef]
- Stefano, G.B.; Mantione, K.; Capellan, L.; Casares, F.M.; Challenger, S.; Ramin, R.; Samuel, J.M.; Snyder, C.; Kream, R.M. Morphine stimulates nitric oxide release in human mitochondria. J. Bioenerg. Biomembr. 2015, 47, 409–417. [Google Scholar] [CrossRef]
- D’Uscio, L.V.; Milstien, S.; Richardson, D.; Smith, L.; Katusic, Z.S. Long-Term Vitamin C Treatment Increases Vascular Tetrahydrobiopterin Levels and Nitric Oxide Synthase Activity. Circ. Res. 2003, 92, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Kuzkaya, N.; Weissmann, N.; Harrison, D.G.; Dikalov, S. Interactions of Peroxynitrite, Tetrahydrobiopterin, Ascorbic Acid, and Thiols. J. Biol. Chem. 2003, 278, 22546–22554. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Koppenol, W.H. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and ugly. Am. J. Physiol. Cell Physiol. 1996, 271, C1424–C1437. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric Oxide and Peroxynitrite in Health and Disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef]
- Muscoli, C.; Cuzzocrea, S.; Ndengele, M.M.; Mollace, V.; Porreca, F.; Fabrizi, F.; Esposito, E.; Masini, E.; Matuschak, G.M.; Salvemini, D. Therapeutic manipulation of peroxynitrite attenuates the development of opiate-induced antinociceptive tolerance in mice. J. Clin. Investig. 2007, 117, 3530–3539. [Google Scholar] [CrossRef] [PubMed]
- Batthyány, C.; Bartesaghi, S.; Mastrogiovanni, M.; Lima, A.; Demicheli, V.; Radi, R. Tyrosine-Nitrated Proteins: Proteomic and Bioanalytical Aspects. Antioxid. Redox Signal. 2017, 26, 313–328. [Google Scholar] [CrossRef]
- Straaten, H.M.O.-V.; Man, A.M.S.-D.; De Waard, M.C. Vitamin C revisited. Crit. Care 2014, 18, 460. [Google Scholar] [CrossRef]
- Pelletier, S.; Duhamel, F.; Coulombe, P.; Popoff, M.R.; Meloche, S. Rho Family GTPases Are Required for Activation of Jak/STAT Signaling by G Protein-Coupled Receptors. Mol. Cell. Biol. 2003, 23, 1316–1333. [Google Scholar] [CrossRef]
- Gross, E.R.; Hsu, A.K.; Gross, G.J. The JAK/STAT pathway is essential for opioid-induced cardioprotection: JAK2 as a mediator of STAT3, Akt, and GSK-3β. Am. J. Physiol. Circ. Physiol. 2006, 291, H827–H834. [Google Scholar] [CrossRef]
- Han, Z.; Zhang, Z.; Guan, Y.; Chen, B.; Yu, M.; Zhang, L.; Fang, J.; Gao, Y.; Guo, Z. New insights into Vitamin C function: Vitamin C induces JAK2 activation through its receptor-like transporter SVCT2. Int. J. Biol. Macromol. 2021, 173, 379–398. [Google Scholar] [CrossRef]
- Chen, J.-X.; Huang, K.-M.; Liu, M.; Jiang, J.-X.; Liu, J.-P.; Zhang, Y.-X.; Yang, C.; Xin, W.-J.; Zhang, X.-Q. Activation of TLR4/STAT3 signaling in VTA contributes to the acquisition and maintenance of morphine-induced conditioned place preference. Behav. Brain Res. 2017, 335, 151–157. [Google Scholar] [CrossRef]
- Sun, X.; Wu, S.; Xing, D. The reactive oxygen species-Src-Stat3 pathway provokes negative feedback inhibition of apoptosis induced by high-fluence low-power laser irradiation. FEBS J. 2010, 277, 4789–4802. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Xu, J.; Ji, W.; Wang, L.; Wang, A. Upregulation of TMEFF2 is involved in the antiproliferative effects of vitamin C and tyrphostin AG490 on GES-1 and AGS cells. Oncol. Lett. 2018, 17, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Puram, S.V.; Yeung, C.M.; Jahani-Asl, A.; Lin, C.; De La Iglesia, N.; Konopka, G.; Jackson-Grusby, L.; Bonni, A. STAT3-iNOS Signaling Mediates EGFRvIII-Induced Glial Proliferation and Transformation. J. Neurosci. 2012, 32, 7806–7818. [Google Scholar] [CrossRef] [PubMed]
- Queval, C.J.; Song, O.-R.; Deboosere, N.; Delorme, V.; Debrie, A.-S.; Iantomasi, R.; Veyron-Churlet, R.; Jouny, S.; Redhage, K.; Deloison, G.; et al. STAT3 Represses Nitric Oxide Synthesis in Human Macrophages upon Mycobacterium tuberculosis Infection. Sci. Rep. 2016, 6, 29297. [Google Scholar] [CrossRef]
- Rouillard, A.D.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermott, M.G.; Ma’Ayan, A. The harmonizome: A collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016, 2016, baw100. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Jiang, L.; Wang, J.; Chai, Z.; Xiong, W. Morphine promotes angiogenesis by activating PI3K/Akt/HIF-1α pathway and upregulating VEGF in hepatocellular carcinoma. J. Gastrointest. Oncol. 2021, 12, 1761–1772. [Google Scholar] [CrossRef]
- Hahn-Windgassen, A.; Nogueira, V.; Chen, C.-C.; Skeen, J.E.; Sonenberg, N.; Hay, N. Akt Activates the Mammalian Target of Rapamycin by Regulating Cellular ATP Level and AMPK Activity. J. Biol. Chem. 2005, 280, 32081–32089. [Google Scholar] [CrossRef]
- Li, C.-J.; Elsasser, T.H.; Kahl, S. AKT/eNOS signaling module functions as a potential feedback loop in the growth hormone signaling pathway. J. Mol. Signal. 2009, 4, 1. [Google Scholar] [CrossRef][Green Version]
- Fang, Q.; Naidu, K.A.; A Naidu, K.; Zhao, H.; Sun, M.; Dan, H.C.; Nasir, A.; E Kaiser, H.; Cheng, J.Q.; Nicosia, S.V.; et al. Ascorbyl stearate inhibits cell proliferation and tumor growth in human ovarian carcinoma cells by targeting the PI3K/AKT pathway. Anticancer Res. 2006, 26, 203–209. [Google Scholar]
- Yang, L.; Wang, S.; Sung, B.; Lim, G.; Mao, J. Morphine Induces Ubiquitin-Proteasome Activity and Glutamate Transporter Degradation. J. Biol. Chem. 2008, 283, 21703–21713. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Browning, E.A.; Hong, N.; DeBolt, K.; Sorokina, E.M.; Liu, W.; Birnbaum, M.; Fisher, A.B. Membrane depolarization is the trigger for PI3K/Akt activation and leads to the generation of ROS. Am. J. Physiol. Circ. Physiol. 2012, 302, H105–H114. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Koundouros, N.; Poulogiannis, G. Phosphoinositide 3-Kinase/Akt Signaling and Redox Metabolism in Cancer. Front. Oncol. 2018, 8, 160. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, J.; Yang, C.; Huang, X.; Han, M.; Kang, F.; Li, J. Morphine promotes the malignant biological behavior of non-small cell lung cancer cells through the MOR/Src/mTOR pathway. Cancer Cell Int. 2021, 21, 622. [Google Scholar] [CrossRef]
- Xu, J.-T.; Zhao, J.-Y.; Zhao, X.; Ligons, D.; Tiwari, V.; Atianjoh, F.E.; Lee, C.-Y.; Liang, L.; Zang, W.; Njoku, D.; et al. Opioid receptor–triggered spinal mTORC1 activation contributes to morphine tolerance and hyperalgesia. J. Clin. Investig. 2014, 124, 592–603. [Google Scholar] [CrossRef]
- Mazei-Robison, M.S.; Koo, J.W.; Friedman, A.K.; Lansink, C.S.; Robison, A.J.; Vinish, M.; Krishnan, V.; Kim, S.; Siuta, M.A.; Galli, A.; et al. Role for mTOR Signaling and Neuronal Activity in Morphine-Induced Adaptations in Ventral Tegmental Area Dopamine Neurons. Neuron 2011, 72, 977–990. [Google Scholar] [CrossRef]
- Delgoffe, G.M. PP2A's restraint of mTOR is critical for T(reg) cell activity. Nat. Immunol. 2016, 17, 478–479. [Google Scholar] [CrossRef]
- Ali, M.A.; Eid, R.M.; Hanafi, M.Y. Vitamin C and E chronic supplementation differentially affect hepatic insulin signaling in rats. Life Sci. 2018, 194, 196–204. [Google Scholar] [CrossRef]
- Zahmatkesh, M.; Kadkhodaee, M.; Salarian, A.; Seifi, B.; Adeli, S. Impact of opioids on oxidative status and related signaling pathways: An integrated view. J. Opioid Manag. 2017, 13, 241–251. [Google Scholar] [CrossRef]
- Lingappan, K. NF-κB in oxidative stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Cárcamo, J.M.; Pedraza, A.; Bórquez-Ojeda, A.O.; Golde, D.W. Vitamin C Suppresses TNFα-Induced NFκB Activation by Inhibiting IκBα Phosphorylation. Biochemistry 2002, 41, 12995–13002. [Google Scholar] [CrossRef] [PubMed]
- Das, K.C.; Lewis-Molock, Y.; White, C.W. Activation of NF-kappa B and elevation of MnSOD gene expression by thiol reducing agents in lung adenocarcinoma (A549) cells. Am. J. Physiol. Cell. Mol. Physiol. 1995, 269, L588–L602. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Fang, F.; Clair, D.K.S.; Josson, S.; Sompol, P.; Spasojevic, I.; Clair, W.H.S. Suppression of RelB-mediated manganese superoxide dismutase expression reveals a primary mechanism for radiosensitization effect of 1α,25-dihydroxyvitamin D3 in prostate cancer cells. Mol. Cancer Ther. 2007, 6, 2048–2056. [Google Scholar] [CrossRef] [PubMed]
- I Rojo, A.; Salinas, M.; Martín, D.; Perona, R.; Cuadrado, A. Regulation of Cu/Zn-Superoxide Dismutase Expression via the Phosphatidylinositol 3 Kinase/Akt Pathway and Nuclear Factor- B. J. Neurosci. 2004, 24, 7324–7334. [Google Scholar] [CrossRef]
- Guo, G.; Bhat, N.R. Hypoxia/Reoxygenation Differentially Modulates NF-κB Activation and iNOS Expression in Astrocytes and Microglia. Antioxid. Redox Signal. 2006, 8, 911–918. [Google Scholar] [CrossRef]
- Nakata, S.; Tsutsui, M.; Shimokawa, H.; Yamashita, T.; Tanimoto, A.; Tasaki, H.; Ozumi, K.; Sabanai, K.; Morishita, T.; Suda, O.; et al. Statin Treatment Upregulates Vascular Neuronal Nitric Oxide Synthase Through Akt/NF-κB Pathway. Arter. Thromb. Vasc. Biol. 2007, 27, 92–98. [Google Scholar] [CrossRef]
- Yao, K.-S.; O'Dwyer, P.J. Involvement of NF-κB in the induction of NAD(P)H: Quinone oxidoreductase (DT-diaphorase) by hypoxia, oltipraz and mitomycin C. Biochem. Pharmacol. 1995, 49, 275–282. [Google Scholar] [CrossRef]
- Kraus, J.; Börner, C.; Giannini, E.; Höllt, V. The Role of Nuclear Factor κB in Tumor Necrosis Factor-Regulated Transcription of the Human μ-Opioid Receptor Gene. Mol. Pharmacol. 2003, 64, 876–884. [Google Scholar] [CrossRef]
- Nennig, S.; Schank, J. The Role of NFkB in Drug Addiction: Beyond Inflammation. Alcohol Alcohol. 2017, 52, 172–179. [Google Scholar] [CrossRef]
- Cárcamo, J.M.; Pedraza, A.; Bórquez-Ojeda, O.; Zhang, B.; Sanchez, R.; Golde, D.W. Vitamin C Is a Kinase Inhibitor: Dehydroascorbic Acid Inhibits IκBα Kinase β. Mol. Cell. Biol. 2004, 24, 6645–6652. [Google Scholar] [CrossRef] [PubMed]
- Dan, H.C.; Ebbs, A.; Pasparakis, M.; Van Dyke, T.; Bassères, D.; Baldwin, A.S. Akt-dependent Activation of mTORC1 Complex Involves Phosphorylation of mTOR (Mammalian Target of Rapamycin) by IκB Kinase α (IKKα). J. Biol. Chem. 2014, 289, 25227–25240. [Google Scholar] [CrossRef] [PubMed]
- Muellner, M.K.; Schreier, S.M.; Schmidbauer, B.; Moser, M.; Quehenberger, P.; Kapiotis, S.; Goldenberg, H.; Laggner, H. Vitamin C inhibits NO-induced stabilization of HIF-1α in HUVECs. Free Radic. Res. 2010, 44, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Balasubramanian, S.; Wang, J.; Chandrashekhar, Y.; Charboneau, R.; Barke, R. Morphine inhibits VEGF expression in myocardial ischemia. Surgery 2003, 134, 336–344. [Google Scholar] [CrossRef]
- Takabuchi, S.; Hirota, K.; Oda, S.; Nishi, K.; Oda, T.; Shingu, K.; Adachi, T.; Fukuda, K. Opioid receptor stimulation does not affect cellular hypoxia-induced gene responses mediated by hypoxia-inducible factor 1 in cultured cell lines. J. Anesthesia 2005, 19, 263–265. [Google Scholar] [CrossRef]
- Daijo, H.; Kai, S.; Tanaka, T.; Wakamatsu, T.; Kishimoto, S.; Suzuki, K.; Harada, H.; Takabuchi, S.; Adachi, T.; Fukuda, K.; et al. Fentanyl activates hypoxia-inducible factor 1 in neuronal SH-SY5Y cells and mice under non-hypoxic conditions in a μ-opioid receptor-dependent manner. Eur. J. Pharmacol. 2011, 667, 144–152. [Google Scholar] [CrossRef]
- Koodie, L.; Ramakrishnan, S.; Roy, S. Morphine Suppresses Tumor Angiogenesis through a HIF-1α/p38MAPK Pathway. Am. J. Pathol. 2010, 177, 984–997. [Google Scholar] [CrossRef]
- Tang, X.; Chen, Y.; Ran, H.; Jiang, Y.; He, B.; Wang, B.; Kong, S.; Wang, H. Systemic Morphine Treatment Derails Normal Uterine Receptivity, Leading to Embryo Implantation Failure in Mice1. Biol. Reprod. 2015, 92, 118. [Google Scholar] [CrossRef]
- Cheng, L.; Yu, H.; Yan, N.; Lai, K.; Xiang, M. Hypoxia-Inducible Factor-1α Target Genes Contribute to Retinal Neuroprotection. Front. Cell. Neurosci. 2017, 11, 20. [Google Scholar] [CrossRef]
- Masson, N.; Ratcliffe, P. HIF prolyl and asparaginyl hydroxylases in the biological response to intracellular O2 levels. J. Cell Sci. 2003, 116, 3041–3049. [Google Scholar] [CrossRef]
- Natarajan, R.; Salloum, F.N.; Fisher, B.J.; Kukreja, R.C.; FowlerIII, A.A. Hypoxia Inducible Factor-1 Activation by Prolyl 4-Hydroxylase-2 Gene Silencing Attenuates Myocardial Ischemia Reperfusion Injury. Circ. Res. 2006, 98, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Mylonis, I.; Simos, G.; Paraskeva, E. Hypoxia-Inducible Factors and the Regulation of Lipid Metabolism. Cells 2019, 8, 214. [Google Scholar] [CrossRef] [PubMed]
- al Sagair, O.A. Effect of morphine sulphate on total lipids and triglycerides contents in serum and brain regions of rat. Med. Islamic World Sci. 2005, 15, 117–125. [Google Scholar]
- Li, L.; Wang, J.; Li, D.; Zhang, H. Morphine increases myocardial triacylglycerol through regulating adipose triglyceride lipase S406 phosphorylation. Life Sci. 2021, 283, 119866. [Google Scholar] [CrossRef]
- Drummond, G.B.; Lafferty, B. Oxygen saturation decreases acutely when opioids are given during anaesthesia. Br. J. Anaesth. 2010, 104, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Kiyatkin, E.A. Respiratory depression and brain hypoxia induced by opioid drugs: Morphine, oxycodone, heroin, and fentanyl. Neuropharmacology 2019, 151, 219–226. [Google Scholar] [CrossRef]
- Pattinson, K.T.S. Opioids and the control of respiration. Br. J. Anaesth. 2008, 100, 747–758. [Google Scholar] [CrossRef]
- Bouillon, T.; Bruhn, J.; Roepcke, H.; Hoeft, A. Opioid-induced respiratory depression is associated with increased tidal volume variability. Eur. J. Anaesthesiol. 2003, 20, 127–133. [Google Scholar] [CrossRef]
- Dong, T.W.; MacLeod, D.B.; Santoro, A.; Augustine, Z.; Barth, S.; Cooter, M.; E Moon, R. A Methodology to Explore Ventilatory Chemosensitivity and Opioid-Induced Respiratory Depression Risk. J. Appl. Physiol. 2020, 129, 500–507. [Google Scholar] [CrossRef]
- Kor, J.J.; Sprung, J.; Khanna, A.K.; Weingarten, T.N. Continuous Monitoring Detected Respiratory Depressive Episodes in Proximity to Adverse Respiratory Events During the PRODIGY Trial. J. Patient Saf. 2022, 10–97. [Google Scholar] [CrossRef]
- Stone, L.S.; German, J.P.; Kitto, K.F.; Fairbanks, C.A.; Wilcox, G.L. Morphine and Clonidine Combination Therapy Improves Therapeutic Window in Mice: Synergy in Antinociceptive but Not in Sedative or Cardiovascular Effects. PLoS ONE 2014, 9, e109903. [Google Scholar] [CrossRef] [PubMed]
- Baehr, C.A.; Kelcher, A.H.; Khaimraj, A.; Reed, D.E.; Pandit, S.G.; Aucoin, D.; Averick, S.; Pravetoni, M. Monoclonal Antibodies Counteract Opioid-Induced Behavioral and Toxic Effects in Mice and Rats. J. Pharmacol. Exp. Ther. 2020, 375, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Ding, X.; Funk, G.; Greer, J.J. Ampakine CX717 Protects against Fentanyl-induced Respiratory Depression and Lethal Apnea in Rats. Anesthesiology 2009, 110, 1364–1370. [Google Scholar] [CrossRef] [PubMed]
- Baby, S.M.; Gruber, R.B.; Young, A.P.; MacFarlane, P.M.; Teppema, L.J.; Lewis, S.J. Bilateral carotid sinus nerve transection exacerbates morphine-induced respiratory depression. Eur. J. Pharmacol. 2018, 834, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Kim, D.-I.; Oh, T.G.; Pao, G.M.; Kim, J.-H.; Palmiter, R.D.; Banghart, M.R.; Lee, K.-F.; Evans, R.M.; Han, S. Neural basis of opioid-induced respiratory depression and its rescue. Proc. Natl. Acad. Sci. USA 2021, 118, e2022134118. [Google Scholar] [CrossRef]
- Osborne, R.; Joel, S.; Grebenik, K.; Trew, D.; Slevin, M. The pharmacokinetics of morphine and morphine glucuronides in kidney failure. Clin. Pharmacol. Ther. 1993, 54, 158–167. [Google Scholar] [CrossRef]
- Osborne, R.J.; Joel, S.P.; Slevin, M.L. Morphine intoxication in renal failure: The role of morphine-6-glucuronide. BMJ 1986, 292, 1548–1549. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).