
Next-Generation Sequencing Advances the Genetic Diagnosis of Cerebral Cavernous Malformation (CCM)

 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Clinical Assessment
2.2. Molecular Genetic Analyses
2.3. Quantitative Real-Time PCR
2.4. RNA Isolation, cDNA Synthesis and KRIT1 Transcript Analysis
3. Results
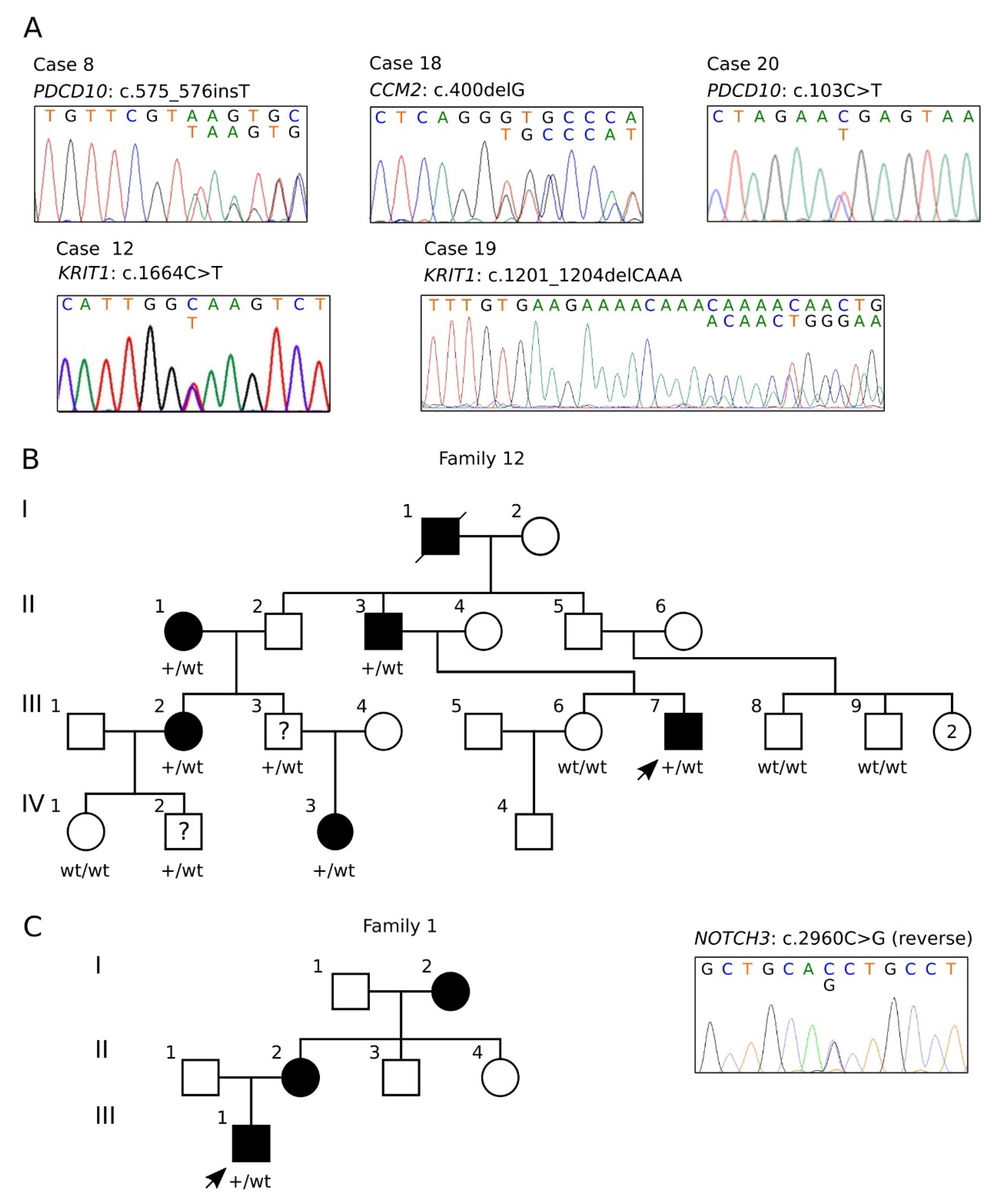
3.1. NGS-Based Genetic Screening for CCM Disease
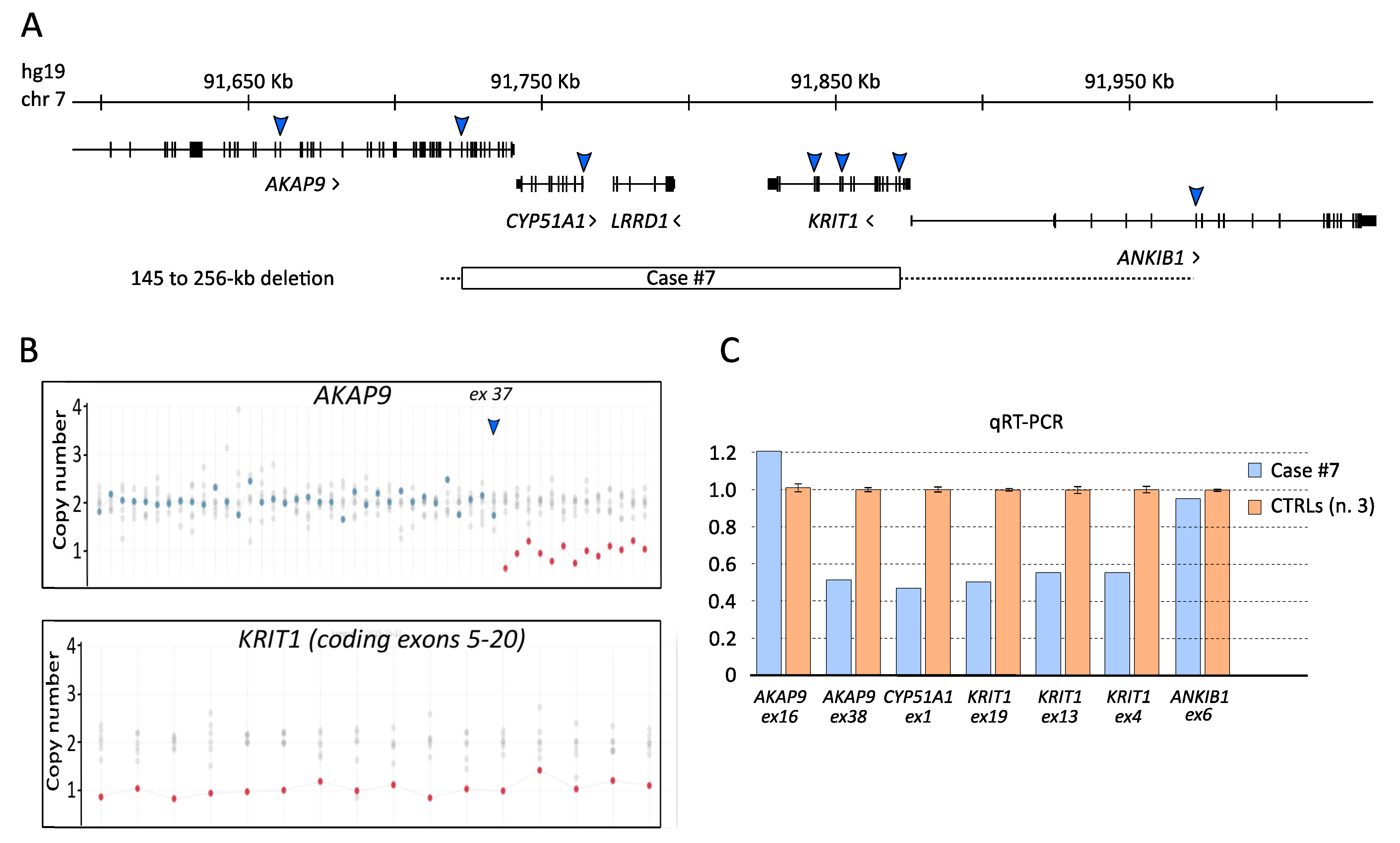
3.2. Characterization of the KRTI1 Genomic Deletion Identified in Case #7
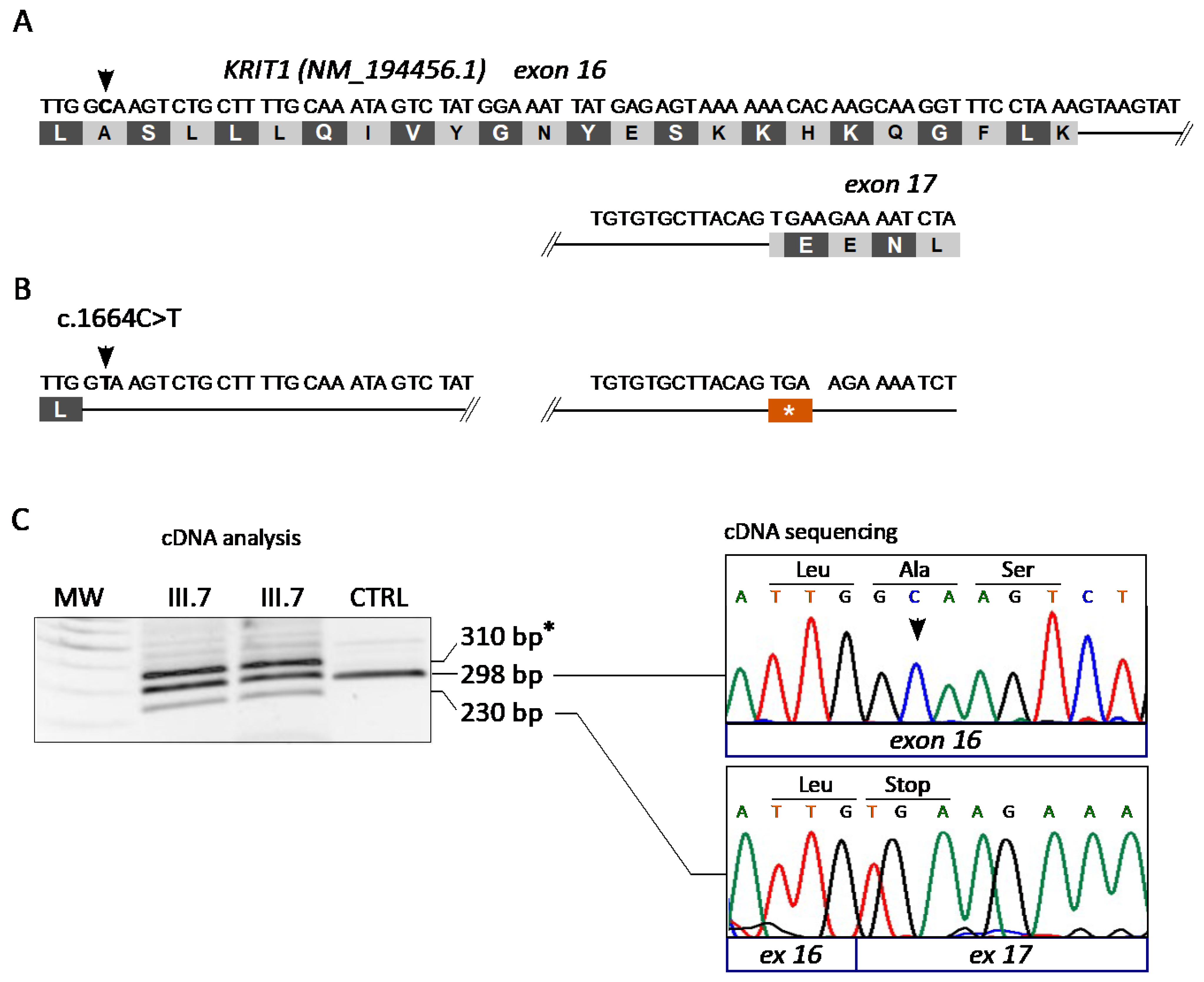
3.3. The KRIT1 c.1664C>T Variant Leads to Aberrant Exon Skipping and Segregates with CCM Disease
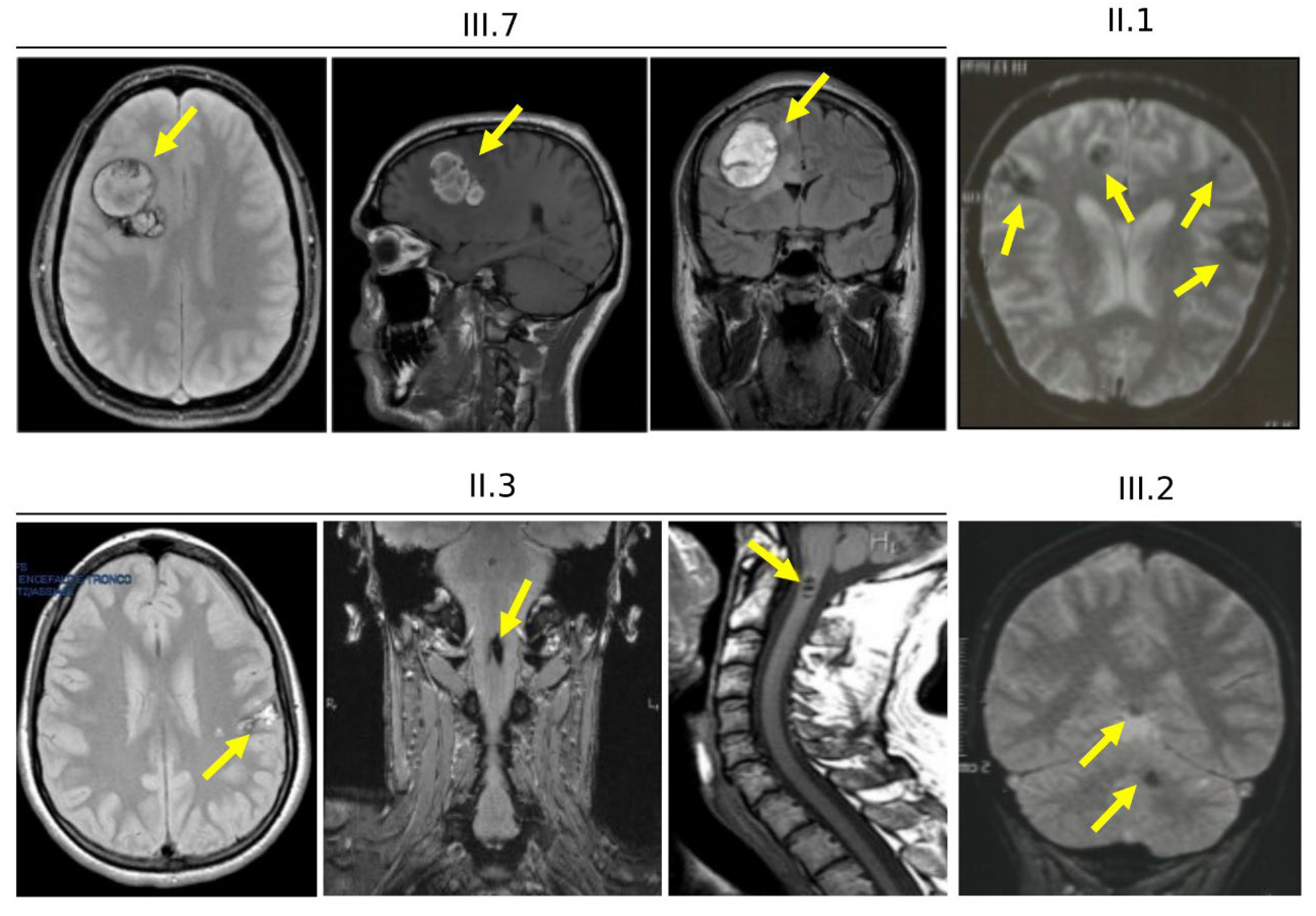
3.4. Clinical Features of Cases Carrying the KRIT1 c.1664C>T Variant (Family 12)
3.5. Characterization of the NOTCH3 c.2960G>C Variant Identified in Case #1
3.6. Clinical Features and Family History of Case #1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Batra, S.; Lin, D.; Recinos, P.F.; Zhang, J.; Rigamonti, D. Cavernous malformations: Natural history, diagnosis and treatment. Nat. Rev. Neurol. 2009, 5, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Fontanella, M. Cerebral Cavernous Malformations (CCM); Minerva Medica: Rome, Italy, 2015. [Google Scholar]
- Rigamonti, D. Cavernous Malformations of the Nervous System; Cambridge University Press: Cambridge, UK, 2011; p. 195. [Google Scholar]
- Flemming, K.D. Incidence, Prevalence, and Clinical Presentation of Cerebral Cavernous Malformations. Methods Mol. Biol. 2020, 2152, 27–33. [Google Scholar] [PubMed]
- Mabray, M.; Hart, B. Clinical Imaging of Cerebral Cavernous Malformations: Computed Tomography and Magnetic Resonance Imaging. Methods Mol. Biol. 2020, 2152, 85–96. [Google Scholar] [PubMed]
- Fontanella, M.; Panciani, P.P.; Spena, G.; Roca, E.; Migliorati, K.; Ambrosi, C.; Sturiale, C.L.; Retta, F.S. Professional athletes and cerebral cavernomas: An obstacle to overcome. J. Sports Med. Phys. Fit. 2015, 55, 1046–1047. [Google Scholar]
- Fontanella, M.M.; Zanin, L.; Fiorindi, A.; Spena, G.; Nicolosi, F.; Belotti, F.; Panciani, P.; Cornali, C.; Doglietto, F. Surgical Management of Brain Cavernous Malformations. Methods Mol. Biol. 2020, 2152, 109–128. [Google Scholar] [PubMed]
- Couteulx, S.L.-L.; Jung, H.H.; Labauge, P.; Houtteville, J.-P.; Lescoat, C.; Cecillon, M.; Marechal, E.; Joutel, A.; Bach, J.-F.; Tournier-Lasserve, E. Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat. Genet. 1999, 23, 189–193. [Google Scholar] [CrossRef]
- Sahoo, T.; Johnson, E.W.; Thomas, J.W.; Kuehl, P.M.; Jones, T.L.; Dokken, C.G.; Touchman, J.W.; Gallione, C.J.; Lee-Lin, S.-Q.; Kosofsky, B.; et al. Mutations in the gene encoding KRIT1, a Krev-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1). Hum. Mol. Genet. 1999, 8, 2325–2333. [Google Scholar] [CrossRef] [Green Version]
- Liquori, C.L.; Berg, M.J.; Siegel, A.M.; Huang, E.; Zawistowski, J.S.; Stoffer, T.; Verlaan, D.; Balogun, F.; Hughes, L.; Leedom, T.P.; et al. Mutations in a gene encoding a novel protein containing a phosphotyrosine-binding domain cause type 2 cerebral cavernous malformations. Am. J. Hum. Genet. 2003, 73, 1459–1464. [Google Scholar] [CrossRef] [Green Version]
- Bergametti, F.; Denier, C.; Labauge, P.; Arnoult, M.; Boetto, S.; Clanet, M.; Coubes, P.; Echenne, B.; Ibrahim, R.; Irthum, B.; et al. Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am. J. Hum. Genet. 2005, 76, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Choquet, H.; Pawlikowska, L.; Lawton, M.T.; Kim, H. Genetics of cerebral cavernous malformations: Current status and future prospects. J. Neurosurg. Sci. 2015, 59, 211–220. [Google Scholar]
- Tournier-Lasserve, E. Molecular Genetic Screening of CCM Patients: An Overview. Methods Mol. Biol. 2020, 2152, 49–57. [Google Scholar] [PubMed]
- Peyre, M.; Miyagishima, D.; Bielle, F.; Chapon, F.; Sierant, M.; Venot, Q.; Lerond, J.; Marijon, P.; Abi-Jaoude, S.; Le Van, T.; et al. Somatic PIK3CA Mutations in Sporadic Cerebral Cavernous Malformations. N. Engl. J. Med. 2021, 385, 996. [Google Scholar] [CrossRef] [PubMed]
- Weng, J.; Yang, Y.; Song, D.; Huo, R.; Li, H.; Chen, Y.; Nam, Y.; Zhou, Q.; Jiao, Y.; Fu, W.; et al. Somatic MAP3K3 mutation defines a subclass of cerebral cavernous malformation. Am. J. Hum. Genet. 2021, 108, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Hong, T.; Xiao, X.; Ren, J.; Cui, B.; Zong, Y.; Zou, J.; Kou, Z.; Jiang, N.; Meng, G.; Zeng, G.; et al. Somatic MAP3K3 and PIK3CA mutations in sporadic cerebral and spinal cord cavernous malformations. Brain 2021, 144, 2648–2658. [Google Scholar] [CrossRef]
- Retta, S.F.; Avolio, M.; Francalanci, F.; Procida, S.; Balzac, F.; Degani, S.; Tarone, G.; Silengo, L. Identification of Krit1B: A novel alternative splicing isoform of cerebral cavernous malformation gene-1. Gene 2004, 325, 63–78. [Google Scholar] [CrossRef]
- Jiang, X.; Padarti, A.; Qu, Y.; Sheng, S.; Abou-Fadel, J.; Badr, A.; Zhang, J. Alternatively spliced isoforms reveal a novel type of PTB domain in CCM2 protein. Sci. Rep. 2019, 9, 15808. [Google Scholar] [CrossRef]
- Spiegler, S.; Rath, M.; Paperlein, C.; Felbor, U. Cerebral Cavernous Malformations: An Update on Prevalence, Molecular Genetic Analyses, and Genetic Counselling. Mol. Syndr. 2018, 9, 60–69. [Google Scholar] [CrossRef]
- Trapani, E.; Retta, S.F. Cerebral cavernous malformation (CCM) disease: From monogenic forms to genetic susceptibility factors. J. Neurosurg. Sci. 2015, 59, 201–209. [Google Scholar]
- Choquet, H.; Trapani, E.; Goitre, L.; Trabalzini, L.; Akers, A.; Fontanella, M.; Hart, B.L.; Morrison, L.A.; Pawlikowska, L.; Kim, H.; et al. Cytochrome P450 and matrix metalloproteinase genetic modifiers of disease severity in Cerebral Cavernous Malformation type 1. Free Radic. Biol. Med. 2016, 92, 100–109. [Google Scholar] [CrossRef] [Green Version]
- Retta, S.F.; Glading, A.J. Oxidative stress and inflammation in cerebral cavernous malformation disease pathogenesis: Two sides of the same coin. Int. J. Biochem. Cell Biol. 2016, 81 Pt B, 254–270. [Google Scholar] [CrossRef] [Green Version]
- Choquet, H.; Kim, H. Genome-wide Genotyping of Cerebral Cavernous Malformation Type 1 Individuals to Identify Genetic Modifiers of Disease Severity. Methods Mol. Biol. 2020, 2152, 77–84. [Google Scholar] [PubMed]
- Retta, S.F.; Perrelli, A.; Trabalzini, L.; Finetti, F. From Genes and Mechanisms to Molecular-Targeted Therapies: The Long Climb to the Cure of Cerebral Cavernous Malformation (CCM) Disease. Methods Mol. Biol. 2020, 2152, 3–25. [Google Scholar] [PubMed]
- Goitre, L.; Balzac, F.; Degani, S.; Degan, P.; Marchi, S.; Pinton, P.; Retta, S.F. KRIT1 regulates the homeostasis of intracellular reactive oxygen species. PLoS ONE 2010, 5, e11786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goitre, L.; De Luca, E.; Braggion, S.; Trapani, E.; Guglielmotto, M.; Biasi, F.; Forni, M.; Moglia, A.; Trabalzini, L.; Retta, S.F. KRIT1 loss of function causes a ROS-dependent upregulation of c-Jun. Free Radic. Biol. Med. 2014, 68, 134–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchi, S.; Corricelli, M.; Trapani, E.; Bravi, L.; Pittaro, A.; Monache, S.D.; Ferroni, L.; Patergnani, S.; Missiroli, S.; Goitre, L.; et al. Defective autophagy is a key feature of cerebral cavernous malformations. EMBO Mol. Med. 2015, 7, 1403–1417. [Google Scholar] [CrossRef]
- Marchi, S.; Retta, S.F.; Pinton, P. Cellular processes underlying cerebral cavernous malformations: Autophagy as another point of view. Autophagy 2016, 12, 424–425. [Google Scholar] [CrossRef]
- Marchi, S.; Trapani, E.; Corricelli, M.; Goitre, L.; Pinton, P.; Retta, S.F. Beyond multiple mechanisms and a unique drug: Defective autophagy as pivotal player in cerebral cavernous malformation pathogenesis and implications for targeted therapies. Rare Dis. 2016, 4, e1142640. [Google Scholar] [CrossRef] [Green Version]
- Goitre, L.; DiStefano, P.V.; Moglia, A.; Nobiletti, N.; Baldini, E.; Trabalzini, L.; Keubel, J.; Trapani, E.; Shuvaev, V.V.; Muzykantov, V.R.; et al. Up-regulation of NADPH oxidase-mediated redox signaling contributes to the loss of barrier function in KRIT1 deficient endothelium. Sci. Rep. 2017, 7, 8296. [Google Scholar] [CrossRef] [Green Version]
- Antognelli, C.; Trapani, E.; Delle Monache, S.; Perrelli, A.; Fornelli, C.; Retta, F.; Cassoni, P.; Talesa, V.N.; Retta, S.F. Data in support of sustained upregulation of adaptive redox homeostasis mechanisms caused by KRIT1 loss-of-function. Data Brief 2018, 16, 929–938. [Google Scholar] [CrossRef]
- Antognelli, C.; Trapani, E.; Delle Monache, S.; Perrelli, A.; Daga, M.; Pizzimenti, S.; Barrera, G.; Cassoni, P.; Angelucci, A.; Trabalzini, L.; et al. KRIT1 loss-of-function induces a chronic Nrf2-mediated adaptive homeostasis that sensitizes cells to oxidative stress: Implication for Cerebral Cavernous Malformation disease. Free Radic Biol Med 2018, 115, 202–218. [Google Scholar] [CrossRef]
- Cianfruglia, L.; Perrelli, A.; Fornelli, C.; Magini, A.; Gorbi, S.; Salzano, A.M.; Antognelli, C.; Retta, F.; Benedetti, V.; Cassoni, P.; et al. KRIT1 Loss-Of-Function Associated with Cerebral Cavernous Malformation Disease Leads to Enhanced. Antioxidants 2019, 8, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antognelli, C.; Perrelli, A.; Armeni, T.; Nicola Talesa, V.; Retta, S.F. Dicarbonyl Stress and S-Glutathionylation in Cerebrovascular Diseases: A Focus on Cerebral Cavernous Malformations. Antioxidants 2020, 9, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrelli, A.; Retta, S.F. Polymorphisms in genes related to oxidative stress and inflammation: Emerging links with the pathogenesis and severity of Cerebral Cavernous Malformation disease. Free Radic. Biol. Med. 2021, 172, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Fontanella, M.M.; Bacigaluppi, S.; Doglietto, F.; Zanin, L.; Agosti, E.; Panciani, P.; Belotti, F.; Saraceno, G.; Spena, G.; Draghi, R.; et al. An international call for a new grading system for cerebral and cerebellar cavernomas. J. Neurosurg. Sci. 2021, 65, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Vercelli, G.G.; Cofano, F.; Santonio, F.V.; Vincitorio, F.; Zenga, F.; Garbossa, D. Natural History, Clinical, and Surgical Management of Cavernous Malformations. Methods Mol. Biol. 2020, 2152, 35–46. [Google Scholar]
- Cerrato, P.; Bergui, M.; Imperiale, D.; Baima, C.; Grasso, M.; Giraudo, M.; Lentini, A.; Lopiano, L.; Bradac, G.B.; Bergamasco, B. Occipital neuralgia as isolated symptom of an upper cervical cavernous angioma. J. Neurol. 2002, 249, 1464–1465. [Google Scholar] [CrossRef]
- Benedetti, V.; Pellegrino, E.; Brusco, A.; Piva, R.; Retta, S.F. Next Generation Sequencing (NGS) Strategies for Genetic Testing of Cerebral Cavernous Malformation (CCM) Disease. Methods Mol. Biol. 2020, 2152, 59–75. [Google Scholar]
- Rashkin, S.; Jun, G.; Chen, S.; Genetics and Epidemiology of Colorectal Cancer Consortium (GECCO); Abecasis, G. R. Optimal sequencing strategies for identifying disease-associated singletons. PLoS Genet. 2017, 13, e1006811. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Khezri, N.; Eckert, S.; Weinstock-Guttman, B.; Sawyer, R.N. A novel genetic variant of NOTCH3 gene associated with familial CADASIL-like disease. Int. J. Stroke 2018, 13, 242. Available online: https://journals.sagepub.com/doi/10.1177/1747493018802481#_i426) (accessed on 8 October 2021).
- Ricci, C.; Cerase, A.; Riolo, G.; Manasse, G.; Battistini, S. KRIT1 Gene in Patients with Cerebral Cavernous Malformations: Clinical Features and Molecular Characterization of Novel Variants. J. Mol. Neurosci. 2021, 71, 1876–1883. [Google Scholar] [CrossRef] [PubMed]
- Ren, A.A.; Snellings, D.A.; Su, Y.S.; Hong, C.C.; Castro, M.; Tang, A.T.; Detter, M.R.; Hobson, N.; Girard, R.; Romanos, S.; et al. PIK3CA and CCM mutations fuel cavernomas through a cancer-like mechanism. Nature 2021, 594, 271–276. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, R.; Marini, V.; Rinaldi, C.; Origone, P.; Dorcaratto, A.; Avolio, M.; Goitre, L.; Forni, M.; Capra, V.; Alafaci, C.; et al. Mutation analysis of CCM1, CCM2 and CCM3 genes in a cohort of Italian patients with cerebral cavernous malformation. Brain Pathol. 2011, 21, 215–224. [Google Scholar] [CrossRef]
- Ku, C.S.; Naidoo, N.; Pawitan, Y. Revisiting Mendelian disorders through exome sequencing. Hum. Genet. 2011, 129, 351–370. [Google Scholar] [CrossRef] [PubMed]
- Bamshad, M.J.; Ng, S.B.; Bigham, A.W.; Tabor, H.K.; Emond, M.J.; Nickerson, D.A.; Shendure, J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 2011, 12, 745–755. [Google Scholar] [CrossRef]
- Zhang, X. Exome sequencing greatly expedites the progressive research of Mendelian diseases. Front. Med. 2014, 8, 42–57. [Google Scholar] [CrossRef]
- Xue, Y.; Ankala, A.; Wilcox, W.R.; Hegde, M.R. Solving the molecular diagnostic testing conundrum for Mendelian disorders in the era of next-generation sequencing: Single-gene, gene panel, or exome/genome sequencing. Genet. Med. 2015, 17, 444–451. [Google Scholar] [CrossRef] [Green Version]
- Guazzi, P.; Goitre, L.; Ferro, E.; Cutano, V.; Martino, C.; Trabalzini, L.; Retta, S.F. Identification of the Kelch family protein Nd1-L as a novel molecular interactor of KRIT1. PLoS ONE 2012, 7, e44705. [Google Scholar] [CrossRef] [Green Version]
- Tang, A.T.; Choi, J.P.; Kotzin, J.J.; Yang, Y.; Hong, C.; Hobson, N.; Girard, R.; Zeineddine, H.A.; Lightle, R.; Moore, T.; et al. Endothelial TLR4 and the microbiome drive cerebral cavernous malformations. Nature 2017, 545, 305–310. [Google Scholar] [CrossRef] [Green Version]
- De Luca, E.; Perrelli, A.; Swamy, H.; Nitti, M.; Passalacqua, M.; Furfaro, A.L.; Salzano, A.M.; Scaloni, A.; Glading, A.J.; Retta, S.F. Protein kinase Cα regulates the nucleocytoplasmic shuttling of KRIT1. J. Cell Sci. 2021, 134, jcs250217. [Google Scholar] [CrossRef]
- Vieceli Dalla Sega, F.; Mastrocola, R.; Aquila, G.; Fortini, F.; Fornelli, C.; Zotta, A.; Cento, A.S.; Perrelli, A.; Boda, E.; Pannuti, A.; et al. KRIT1 Deficiency Promotes Aortic Endothelial Dysfunction. Int. J. Mol. Sci. 2019, 20, 4930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Li, Y.; Zou, J.; Polster, S.P.; Lightle, R.; Moore, T.; Dimaano, M.; He, T.-C.; Weber, C.R.; Awad, I.A.; et al. The cerebral cavernous malformation disease causing gene KRIT1 participates in intestinal epithelial barrier maintenance and regulation. FASEB J. 2019, 33, 2132–2143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girard, R.; Zeineddine, H.A.; Fam, M.D.; Mayampurath, A.; Cao, Y.; Shi, C.; Shenkar, R.; Polster, S.; Jesselson, M.; Duggan, R.; et al. Plasma Biomarkers of Inflammation Reflect Seizures and Hemorrhagic Activity of Cerebral Cavernous Malformations. Transl. Stroke Res. 2018, 9, 34–43. [Google Scholar] [CrossRef]
- Flemming, K.D.; Kumar, S.; Brown, R.D., Jr.; Singh, R.J.; Whitehead, K.; McCreath, L.; Lanzino, G. Cavernous Malformation Hemorrhagic Presentation at Diagnosis Associated with Low 25-Hydroxy-Vitamin D Level. Cerebrovasc. Dis. 2020, 49, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Gibson, C.C.; Zhu, W.; Davis, C.T.; Bowman-Kirigin, J.A.; Chan, A.; Ling, J.; Walker, A.; Goitre, L.; Monache, S.D.; Retta, S.F.; et al. Strategy for identifying repurposed drugs for the treatment of cerebral cavernous malformation. Circulation 2015, 131, 289–299. [Google Scholar] [CrossRef]
- Moglia, A.; Goitre, L.; Gianoglio, S.; Baldini, E.; Trapani, E.; Genre, A.; Scattina, A.; Dondo, G.; Trabalzini, L.; Beekwilder, J.; et al. Evaluation of the bioactive properties of avenanthramide analogs produced in recombinant yeast. Biofactors 2015, 41, 15–27. [Google Scholar] [CrossRef]
- Moglianetti, M.; De Luca, E.; Pedone, D.; Marotta, R.; Catelani, T.; Sartori, B.; Amenitsch, H.; Retta, S.F.; Pompa, P.P. Platinum nanozymes recover cellular ROS homeostasis in an oxidative stress-mediated disease model. Nanoscale 2016, 8, 3739–3752. [Google Scholar] [CrossRef]
- Perrelli, A.; Goitre, L.; Salzano, A.M.; Moglia, A.; Scaloni, A.; Retta, S.F. Biological Activities, Health Benefits, and Therapeutic Properties of Avenanthramides: From Skin Protection to Prevention and Treatment of Cerebrovascular Diseases. Oxid. Med. Cell. Longev. 2018, 2018, 6015351. [Google Scholar] [CrossRef]
- De Luca, E.; Pedone, D.; Moglianetti, M.; Pulcini, D.; Perrelli, A.; Retta, S.F.; Pompa, P.P. Multifunctional Platinum@BSA-Rapamycin Nanocarriers for the Combinatorial Therapy of Cerebral Cavernous Malformation. ACS Omega 2018, 3, 15389–15398. [Google Scholar] [CrossRef]
- Moglianetti, M.; Pedone, D.; Udayan, G.; Retta, S.F.; Debellis, D.; Marotta, R.; Turco, A.; Rella, S.; Malitesta, C.; Bonacucina, G.; et al. Intracellular Antioxidant Activity of Biocompatible Citrate-Capped Palladium Nanozymes. Nanomaterials 2020, 10, 99. [Google Scholar] [CrossRef] [Green Version]
- Perrelli, A.; Fatehbasharzad, P.; Benedetti, V.; Ferraris, C.; Fontanella, M.; De Luca, E.; Moglianetti, M.; Battaglia, L.; Retta, S.F. Towards precision nanomedicine for cerebrovascular diseases with emphasis on Cerebral Cavernous Malformation (CCM). Expert Opin. Drug Deliv. 2021, 7, 849–876. [Google Scholar] [CrossRef] [PubMed]
- Sehrawat, S.; Ernandez, T.; Cullere, X.; Takahashi, M.; Ono, Y.; Komarova, Y.; Mayadas, T.N. AKAP9 regulation of microtubule dynamics promotes Epac1-induced endothelial barrier properties. Blood 2011, 117, 708–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Zhao, J. Novel oxidative stress-related prognostic biomarkers for melanoma associated with tumor metastasis. Medicine 2021, 100, e24866. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Kan, H.; Sakurai, K.; Arai, N.; Inui, S.; Kobayashi, S.; Kato, D.; Ueki, Y.; Matsukawa, N. Iron leakage owing to blood-brain barrier disruption in small vessel disease CADASIL. Neurology 2020, 95, e1188–e1198. [Google Scholar] [CrossRef]
- Rutten, J.W.; Hack, R.J.; Duering, M.; Gravesteijn, G.; Dauwerse, J.G.; Overzier, M.; van den Akker, E.B.; Slagboom, E.; Holstege, H.; Nho, K.; et al. Broad phenotype of cysteine-altering NOTCH3 variants in UK Biobank: CADASIL to nonpenetrance. Neurology 2020, 95, e1835–e1843. [Google Scholar] [CrossRef]
- Ruchoux, M.M.; RKalaria, N.; Román, G.C. The pericyte: A critical cell in the pathogenesis of CADASIL. Cereb. Circ. Cogn. Behav. 2021, 2, 100031. [Google Scholar] [CrossRef]
- Østergaard, L.; Engedal, T.S.; Moreton, F.; Hansen, M.B.; Wardlaw, J.M.; Dalkara, T.; Markus, H.S.; Muir, K.W. Cerebral small vessel disease: Capillary pathways to stroke and cognitive decline. J. Cereb. Blood Flow Metab. 2016, 36, 302–325. [Google Scholar] [CrossRef]
- Wang, K.; Zhang, H.; He, Y.; Jiang, Q.; Tanaka, Y.; Park, I.-H.; Pober, J.S.; Min, W.; Zhou, H.J. Mural Cell-Specific Deletion of Cerebral Cavernous Malformation 3 in the Brain Induces Cerebral Cavernous Malformations. Arter. Thromb. Vasc. Biol. 2020, 40, 2171–2186. [Google Scholar] [CrossRef]
- Clatterbuck, R.E.; Eberhart, C.G.; Crain, B.J.; Rigamonti, D. Ultrastructural and immunocytochemical evidence that an incompetent blood-brain barrier is related to the pathophysiology of cavernous malformations. J. Neurol. Neurosurg. Psychiatry 2001, 71, 188–192. [Google Scholar] [CrossRef] [Green Version]
- Pedapati, R.; Vishnu, V.Y.; Singh, M.B.; Goyal, V.; Garg, A.; Srivastava, P. CADASIL and Cavernomas: A Common Mechanism. Ann. Indian Acad. Neurol. 2020, 23, 570–572. [Google Scholar]
- Schulz, G.B.; Wieland, E.; Wüstehube-Lausch, J.; Boulday, G.; Moll, I.; Tournier-Lasserve, E.; Fischer, A. Cerebral Cavernous Malformation-1 Protein Controls DLL4-Notch3 Signaling Between the Endothelium and Pericytes. Stroke 2015, 46, 1337–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, B.P.; Nannoni, S.; Harshfield, E.L.; Tozer, D.; Gräf, S.; Bell, S.; Markus, H.S. variants are more common than expected in the general population and associated with stroke and vascular dementia: An analysis of 200,000 participants. J. Neurol. Neurosurg. Psychiatry 2021, 92, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Bordes, C.; Sargurupremraj, M.; Mishra, A.; Debette, S. Genetics of common cerebral small vessel disease. Nat. Rev. Neurol. 2022, 18, 84–101. [Google Scholar] [CrossRef]
- Li, Z.; Xiao, J.; Liu, M.; Cui, J.; Lian, B.; Sun, Y.; Li, C. Notch3 regulates ferroptosis via ROS-induced lipid peroxidation in NSCLC cells. FEBS Open Bio 2022, 6, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Neves, K.B.; Harvey, A.P.; Moreton, F.; Montezano, A.C.; Rios, F.J.; Alves-Lopes, R.; Cat, A.N.D.; Rocchicciolli, P.; Delles, C.; Joutel, A.; et al. ER stress and Rho kinase activation underlie the vasculopathy of CADASIL. JCI Insight 2019, 4, e131344. [Google Scholar] [CrossRef]
- Whitehead, K.J.; Chan, A.C.; Navankasattusas, S.; Koh, W.; London, N.R.; Ling, J.; Mayo, A.H.; Drakos, S.G.; Jones, C.A.; Zhu, W.; et al. The cerebral cavernous malformation signaling pathway promotes vascular integrity via Rho GTPases. Nat. Med. 2009, 15, 177–184. [Google Scholar] [CrossRef] [Green Version]
- Stockton, R.A.; Shenkar, R.; Awad, I.A.; Ginsberg, M.H. Cerebral cavernous malformations proteins inhibit Rho kinase to stabilize vascular integrity. J. Exp. Med. 2010, 207, 881–896. [Google Scholar] [CrossRef] [Green Version]
- Richardson, B.T.; Dibble, C.F.; Borikova, A.L.; Johnson, G.L. Cerebral cavernous malformation is a vascular disease associated with activated RhoA signaling. Biol. Chem. 2013, 394, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Adachi, K.; Yoshizaki, K.; Kunimoto, S.; Kalaria, R.N.; Watanabe, A. Mutations in NOTCH3 cause the formation and retention of aggregates in the endoplasmic reticulum, leading to impaired cell proliferation. Hum. Mol. Genet. 2010, 19, 79–89. [Google Scholar] [CrossRef] [Green Version]
- Wüstehube, J.; Bartol, A.; Liebler, S.S.; Brütsch, R.; Zhu, Y.; Felbor, U.; Sure, U.; Augustin, H.G.; Fischer, A. Cerebral cavernous malformation protein CCM1 inhibits sprouting angiogenesis by activating DELTA-NOTCH signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 12640–12645. [Google Scholar] [CrossRef] [Green Version]
- You, C.; Sandalcioglu, I.E.; Dammann, P.; Felbor, U.; Sure, U.; Zhu, Y. Loss of CCM3 impairs DLL4-Notch signalling: Implication in endothelial angiogenesis and in inherited cerebral cavernous malformations. J. Cell. Mol. Med. 2013, 17, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Hasan, S.S.; Fischer, A. Notch Signaling in Familial Cerebral Cavernous Malformations and Immunohistochemical Detection of Cleaved Notch1 Intracellular Domain. Methods Mol. Biol. 2020, 2152, 427–435. [Google Scholar] [PubMed]
- Wu, K.L.; Wu, C.-A.; Wu, C.-W.; Chan, S.H.; Chang, A.Y.; Chan, J.Y. Redox-sensitive oxidation and phosphorylation of PTEN contribute to enhanced activation of PI3K/Akt signaling in rostral ventrolateral medulla and neurogenic hypertension in spontaneously hypertensive rats. Antioxid. Redox. Signal. 2013, 18, 36–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Park, J.; Han, S.-J.; Yang, S.Y.; Yoon, H.J.; Park, I.; Woo, H.A.; Lee, S.-R. Redox regulation of tumor suppressor PTEN in cell signaling. Redox Biol. 2020, 34, 101553. [Google Scholar] [CrossRef] [PubMed]
- Huu, T.N.; Park, J.; Zhang, Y.; Park, I.; Yoon, H.; Woo, H.; Lee, S.-R. Redox Regulation of PTEN by Peroxiredoxins. Antioxidants 2021, 10, 302. [Google Scholar]
- Kwon, J.; Lee, S.-R.; Yang, K.-S.; Ahn, Y.; Kim, Y.J.; Stadtman, E.R.; Rhee, S.G. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc. Natl. Acad. Sci. USA 2004, 101, 16419–16424. [Google Scholar] [CrossRef] [Green Version]
- Connor, K.; Subbaram, S.; Regan, K.J.; Nelson, K.K.; Mazurkiewicz, J.E.; Bartholomew, P.J.; Aplin, A.E.; Tai, Y.-T.; Aguirre-Ghiso, J.; Flores, S.C.; et al. Mitochondrial H2O2 regulates the angiogenic phenotype via PTEN oxidation. J. Biol. Chem. 2005, 280, 16916–16924. [Google Scholar] [CrossRef] [Green Version]
- Nogueira, V.; Park, Y.; Chen, C.-C.; Xu, P.-Z.; Chen, M.-L.; Tonic, I.; Unterman, T.; Hay, N. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell 2008, 14, 458–470. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Browning, E.A.; Hong, N.; DeBolt, K.; Sorokina, E.M.; Liu, W.; Birnbaum, M.; Fisher, A.B. Membrane depolarization is the trigger for PI3K/Akt activation and leads to the generation of ROS. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H105–H114. [Google Scholar] [CrossRef] [Green Version]
- Koundouros, N.; Poulogiannis, G. Phosphoinositide 3-Kinase/Akt Signaling and Redox Metabolism in Cancer. Front. Oncol. 2018, 8, 160. [Google Scholar] [CrossRef]
- Ushio-Fukai, M.; Frey, R.S.; Fukai, T.; Malik, A.B. Reactive oxygen species and endothelial permeability. Curr. Top. Membr. 2008, 61, 147–189. [Google Scholar]
- Bäumer, A.T.; Freyhaus, H.T.; Sauer, H.; Wartenberg, M.; Kappert, K.; Schnabel, P.; Konkol, C.; Hescheler, J.; Vantler, M.; Rosenkranz, S. Phosphatidylinositol 3-kinase-dependent membrane recruitment of Rac-1 and p47phox is critical for alpha-platelet-derived growth factor receptor-induced production of reactive oxygen species. J. Biol. Chem. 2008, 283, 7864–7876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kma, L.; Baruah, T.J. The interplay of ROS and the PI3K/Akt pathway in autophagy regulation. Biotechnol. Appl. Biochem. 2022, 69, 248–264. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.-C.; Liu, P.-F.; Chang, C.-H.; Lin, Y.-C.; Chen, Y.-J.; Shu, C.-W. The interplay of autophagy and oxidative stress in the pathogenesis and therapy of retinal degenerative diseases. Cell Biosci. 2022, 12, 1. [Google Scholar] [CrossRef]
- Zhang, Y.-Q.; Liang, Y.-K.; Wu, Y.; Chen, M.; Chen, W.-L.; Li, R.-H.; Zeng, Y.-Z.; Huang, W.-H.; Wu, J.-D.; Zeng, D.; et al. Notch3 inhibits cell proliferation and tumorigenesis and predicts better prognosis in breast cancer through transactivating PTEN. Cell Death Dis. 2021, 12, 502. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | MIM | Disease |
|---|---|---|
| ABCC6 | 603234 | Pseudoxanthoma elasticum and arterial calcification generalized of infancy 2 (GACI2) |
| ACTA2 | 102620 | Multisystemic smooth muscle dysfunction syndrome (MSMDS) and aortic aneurysm familial thoracic 6 (AAT6) |
| ACVRL1 | 601284 | Telangiectasia hereditary hemorrhagic type 2 (HHT2) and pulmonary hypertension primary 1 (PPH1) |
| APP | 104760 | Cerebral amyloid angiopathy app-related (CAA-APP) and Alzheimer’s disease (AD) |
| BRCC3 | 300617 | Moyamoya disease 4 with short stature hypergonadotropic hypogonadism and facial dysmorphism (MYMY4) and T-Cell prolymphocytic leukemia (T-PLL) |
| CCM2 | 607929 | Cerebral cavernous malformations 2 (CCM2) and cerebral cavernous malformations (CCM) |
| COL3A1 | 120180 | Ehlers–Danlos syndrome vascular type (EDSVASC) and polymicrogyria with or without vascular-type Ehlers–Danlos syndrome (PMGEDSV) |
| COL4A1 | 120130 | Retinal arteries tortuosity of (RATOR) and angiopathy hereditary with nephropathy aneurysms and muscle cramps (HANAC) |
| COL4A2 | 120090 | Brain small vessel disease 2 (BSVD2) and intracerebral hemorrhage (ICH) |
| ENG | 131195 | Telangiectasia hereditary hemorrhagic type 1 (HHT1) and hereditary hemorrhagic telangiectasia (HHT) |
| FBN1 | 134797 | Marfan syndrome (MFS) and stiff skin syndrome (SSKS) |
| GLA | 300644 | Fabry disease (FD) and rare cardiomyopathy |
| HTRA1 | 602194 | Cerebral arteriopathy autosomal recessive with subcortical infarcts and leukoencephalopathy (CARASIL) and cerebral arteriopathy autosomal dominant with subcortical infarcts and leukoencephalopathy type 2 (CADASIL2) |
| JAM3 | 606871 | Hemorrhagic destruction of the brain subependymal calcification and cataracts (HDBSCC) and Jacobsen syndrome (JBS) |
| KRIT1 | 604214 | Cerebral Cavernous Malformations (CCM) and Cerebral Cavernous Malformations type 1 (CCM1) |
| MYH11 | 160745 | Aortic aneurysm familial thoracic 4 (AAT4) and familial thoracic aortic aneurysm and aortic dissection (FAA) |
| MYLK | 600922 | Aortic aneurysm familial thoracic 7 (AAT7) and megacystis-microcolon-intestinal hypoperistalsis syndrome (MMIHS) |
| NOTCH3 | 600276 | Cerebral arteriopathy autosomal dominant with subcortical infarcts and leukoencephalopathy type 1 (CADASIL1) and lateral meningocele syndrome (LMNS) |
| PDCD10 | 609118 | Cerebral Cavernous Malformations 3 (CCM3) and Cerebral Cavernous Malformations (CCM) |
| SAMHD1 | 606754 | Aicardi–Goutieres syndrome 5 (AGS5) and Chilblain Lupus 2 (CHBL2) |
| TGFBR1 | 190181 | Multiple Self-Healing Squamous Epithelioma (MSSE) and Loeys–Dietz Syndrome 1 (LDS1) |
| TGFBR2 | 190182 | Loeys–Dietz syndrome 2 (LDS2) and colorectal cancer hereditary nonpolyposis type 6 (HNPCC6) |
| TREX1 | 606609 | Vasculopathy retinal with cerebral leukodystrophy (RVCL) and Aicardi–Goutieres syndrome 1 (AGS1) |
| ID | Sex | Gene | Ref Seq a | cDNA/ Genomic DNA | Protein | Variant Type | GnomAd v.2.1 Frequency | ACMG Classification | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | M | NOTCH3 | NM_000435.2 (LRG n.a.) | c.2960C>G | p.(Thr987Ser) | Missense | 14/215,582 | Benign—PM1, PP2, BS1, BP4 | [42] |
| 2 | M | ND | |||||||
| 3 | F | ND | |||||||
| 4 b | M | PTEN | NM_000314.8 (LRG_311) | c.959dup | p.(Leu320PhefsTer5) | Frameshift | Not found | Pathogenic—PVS1, PM2, PP3 | This report |
| 5 | M | ND | |||||||
| 6 | M | ND | |||||||
| 7 | F | KRIT1 | NC_000007.13 | g.(91715729_91718698)_(91864237_91972337)del | Large genomic deletion | Not found | Pathogenic | [39] | |
| 8 | F | PDCD10 | NM_007217.4 (LRG_651) | c.575_576insT | p.(Ser193LysfsTer36) | Frameshift | Not found | Pathogenic—PVS1, PM2, PP3 | See ClinVar SCV000808927.1 |
| 9 | F | ND | |||||||
| 10 | M | ND | |||||||
| 11 | F | ND | |||||||
| 12 | M | KRIT1 | NM_194456.1 (LRG_650) | c.1664C>T | p.(Ala555Ter) | Splicing | Not found | Pathogenic—PM2, PP1, PP5, PS3, PS4 | [43] |
| 13 | F | ND | |||||||
| 14 | F | ND | |||||||
| 15 | F | ND | |||||||
| 16 | F | ND | |||||||
| 17 | M | ND | |||||||
| 18 | M | CCM2 | NM_031443.4 (LRG_664t1) | c.400delG | p.(Val134CysfsTer22) | Frameshift | Not found | Likely Pathogenic—PVS1, PM2 | This report |
| 19 | F | KRIT1 | NM_194456.1 (LRG_650) | c.1057_1060del | p.(Gly353AsnfsTer17) | Frameshift | Not found | Pathogenic—PVS1, PP3, PM2 | This report |
| 20 | F | PDCD10 | NM_007217.4 (LRG_651) | c.103C>T | p.(Arg35Ter) | Nonsense | Not found | Pathogenic—PVS1, PP5, PM2, PP3 | See ClinVar VCV000372445.5 |
| 21 | F | ND |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benedetti, V.; Canzoneri, R.; Perrelli, A.; Arduino, C.; Zonta, A.; Brusco, A.; Retta, S.F. Next-Generation Sequencing Advances the Genetic Diagnosis of Cerebral Cavernous Malformation (CCM). Antioxidants 2022, 11, 1294. https://doi.org/10.3390/antiox11071294
Benedetti V, Canzoneri R, Perrelli A, Arduino C, Zonta A, Brusco A, Retta SF. Next-Generation Sequencing Advances the Genetic Diagnosis of Cerebral Cavernous Malformation (CCM). Antioxidants. 2022; 11(7):1294. https://doi.org/10.3390/antiox11071294
Chicago/Turabian StyleBenedetti, Valerio, Rosalia Canzoneri, Andrea Perrelli, Carlo Arduino, Andrea Zonta, Alfredo Brusco, and Saverio Francesco Retta. 2022. "Next-Generation Sequencing Advances the Genetic Diagnosis of Cerebral Cavernous Malformation (CCM)" Antioxidants 11, no. 7: 1294. https://doi.org/10.3390/antiox11071294
APA StyleBenedetti, V., Canzoneri, R., Perrelli, A., Arduino, C., Zonta, A., Brusco, A., & Retta, S. F. (2022). Next-Generation Sequencing Advances the Genetic Diagnosis of Cerebral Cavernous Malformation (CCM). Antioxidants, 11(7), 1294. https://doi.org/10.3390/antiox11071294