
Allopurinol Lowers Serum Urate but Does Not Reduce Oxidative Stress in CKD

 , and
, and
Abstract
1. Introduction
2. Methods
2.1. Study Subjects
2.2. Xanthine Oxidase Activity
2.3. Endothelial Protein Expression
2.4. Endothelium-Dependent and Independent Dilation
2.5. Carotid Intima-Media Thickness (CIMT)
2.6. Metabolomics Analysis
2.7. Statistical Analysis
3. Results
3.1. Clinical Characteristics of the Participants
3.2. Correlation of Baseline XO Activity and Expression with Makers of Vascular Function and Oxidative Stress
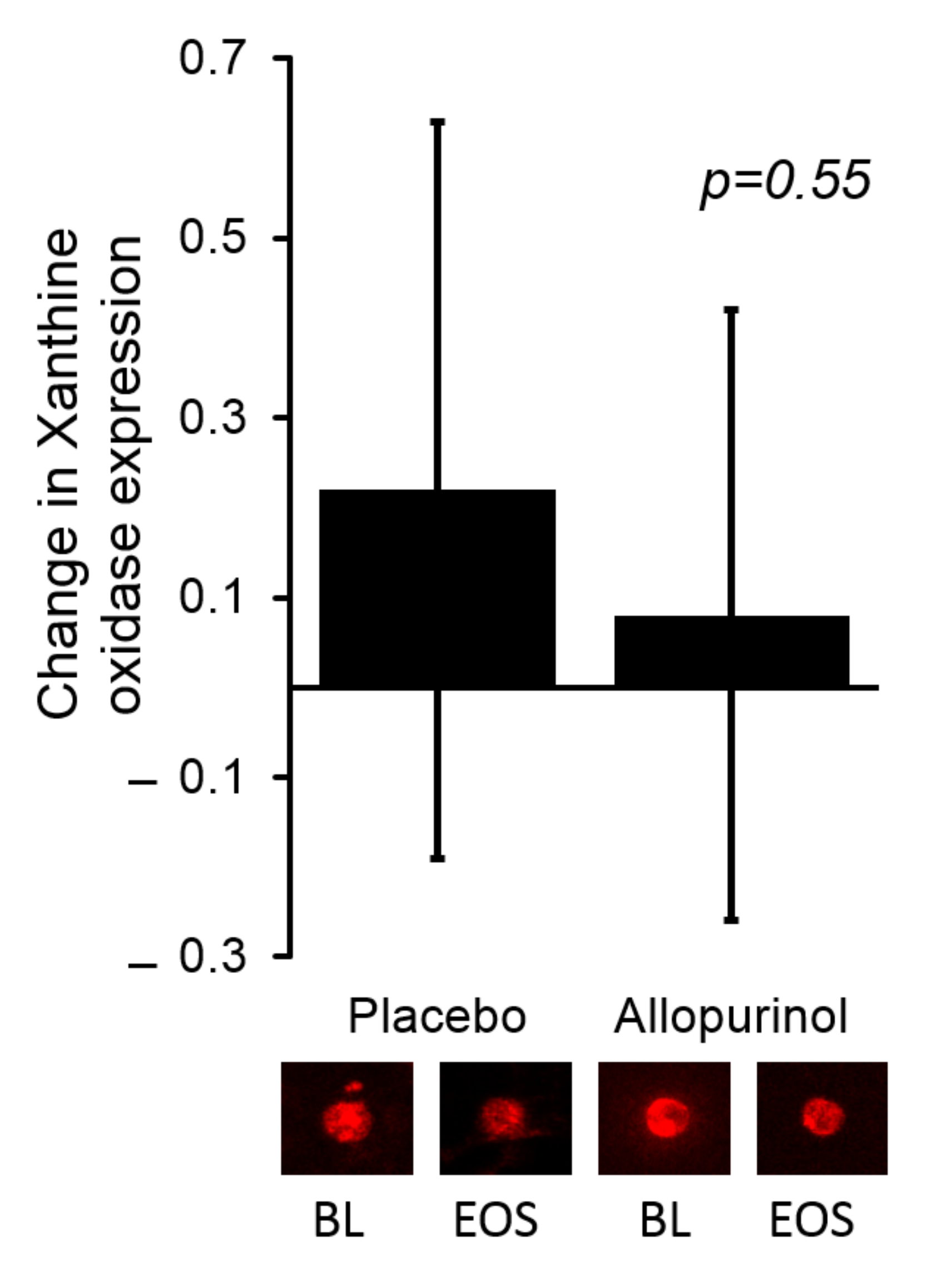
3.3. The Effects of Allopurinol on Serum Urate and XO Activity
3.4. Correlation of Change in Serum XO Activity with Makers of Vascular and Kidney Function and Markers of Oxidative Stress
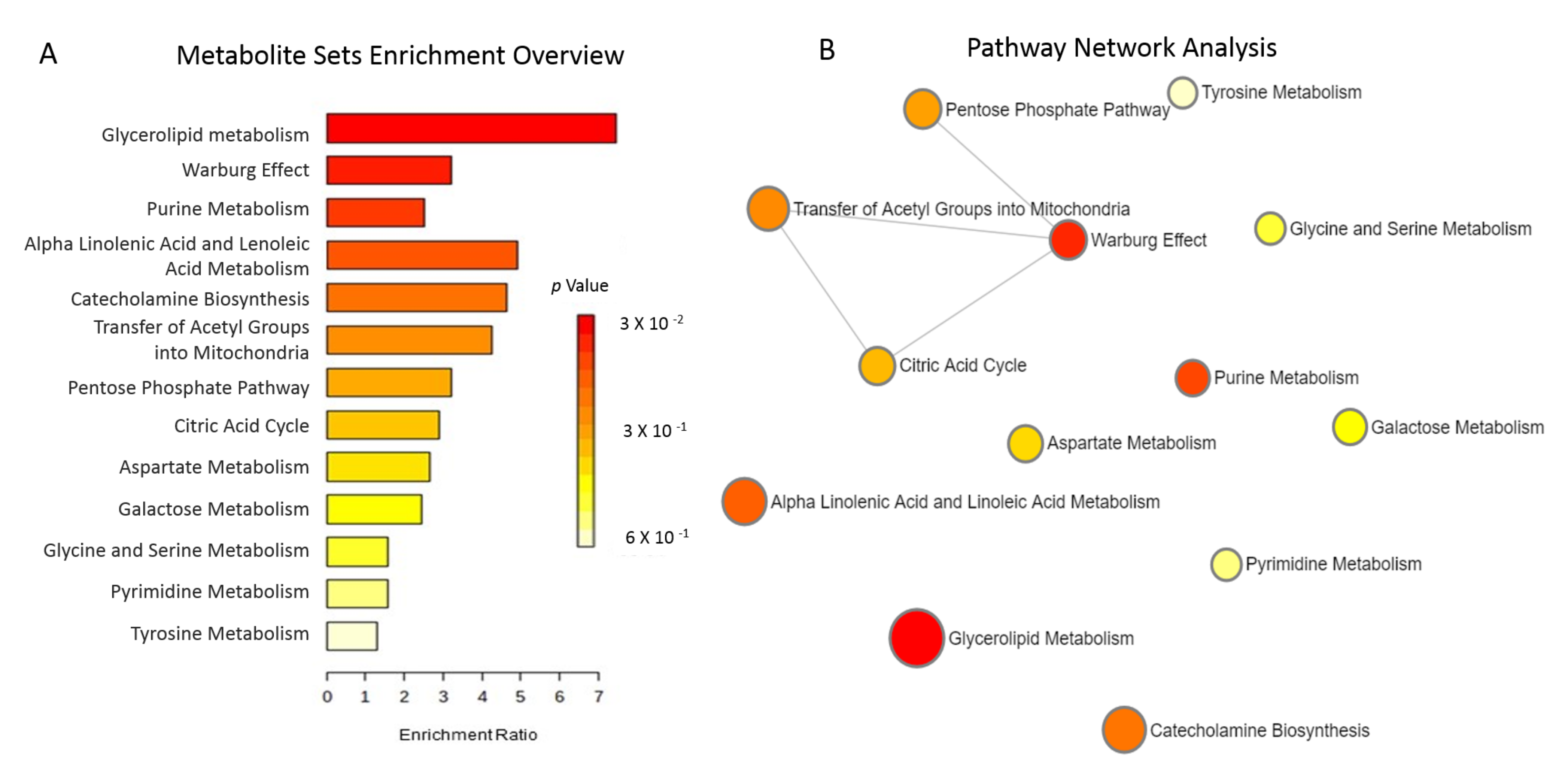
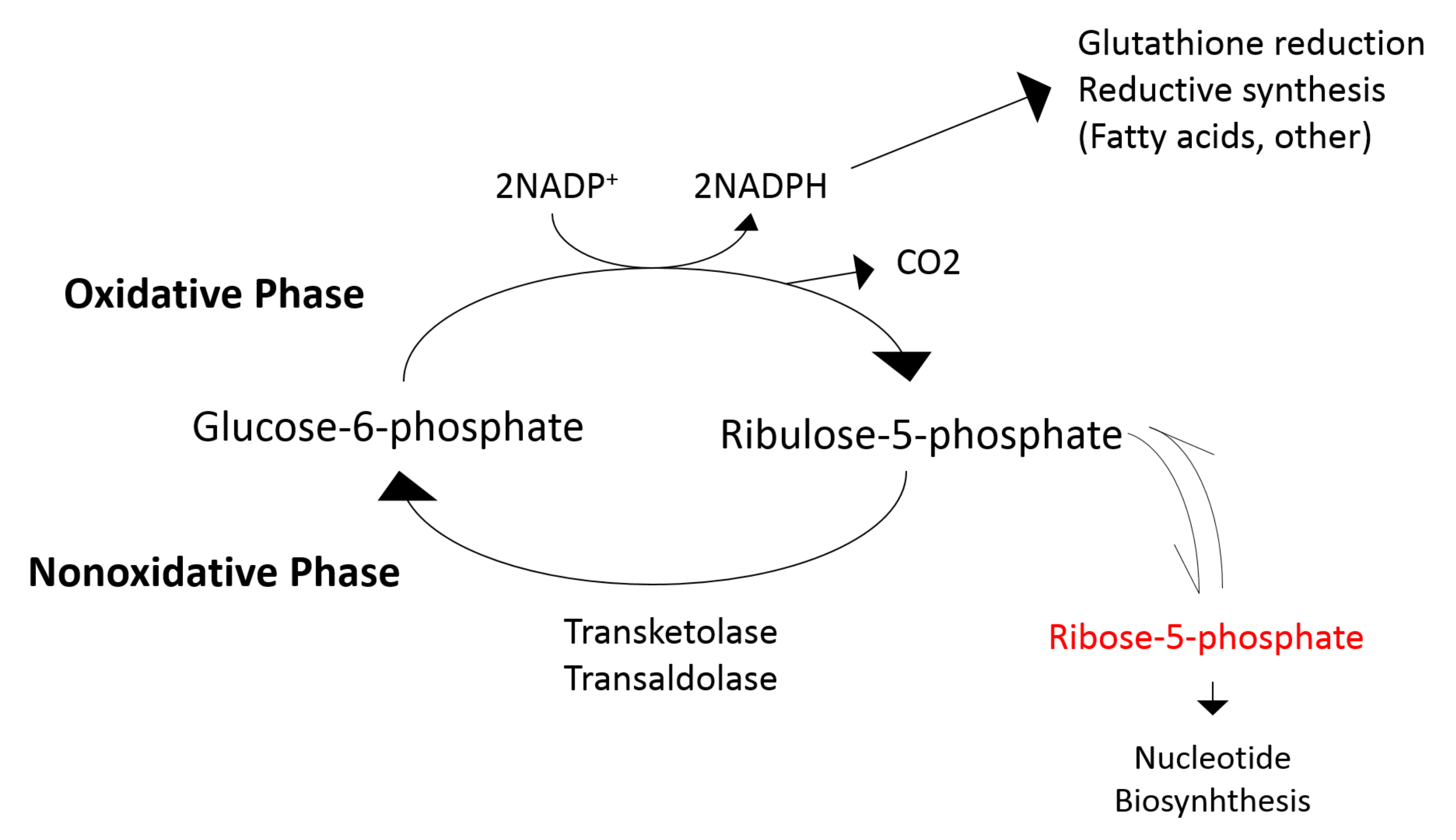
3.5. The Effects of Allopurinol on the Metabolome in Patients with CKD
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhu, Y.; Pandya, B.J.; Choi, H.K. Comorbidities of gout and hyperuricemia in the US general population: NHANES 2007–2008. Am. J. Med. 2012, 125, 679–687.e1. [Google Scholar] [CrossRef] [PubMed]
- BBrantsma, A.H.; Bakker, S.J.L.; Hillege, H.L.; de Zeeuw, D.; de Jong, P.E.; Gansevoort, R.T. Cardiovascular and renal outcome in subjects with K/DOQI stage 1-3 chronic kidney disease: The importance of urinary albumin excretion. Nephrol. Dial. Transplant. 2008, 23, 3851–3858. [Google Scholar] [CrossRef] [PubMed]
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- McCullough, P.A.; Jurkovitz, C.T.; Pergola, P.E.; McGill, J.B.; Brown, W.W.; Collins, A.J.; Chen, S.C.; Li, S.; Singh, A.; Norris, K.C.; et al. Independent components of chronic kidney disease as a cardiovascular risk state: Results from the Kidney Early Evaluation Program (KEEP). Arch. Intern. Med. 2007, 167, 1122–1129. [Google Scholar] [CrossRef]
- McCullough, P.A.; Li, S.; Jurkovitz, C.T.; Stevens, L.; Collins, A.J.; Chen, S.-C.; Norris, K.C.; McFarlane, S.; Johnson, B.; Shlipak, M.G.; et al. Chronic kidney disease, prevalence of premature cardiovascular disease, and relationship to short-term mortality. Am. Heart J. 2008, 156, 277–283. [Google Scholar] [CrossRef]
- Weiner, D.E.; Tighiouart, H.; Stark, P.C.; Amin, M.G.; MacLeod, B.; Griffith, J.L.; Salem, D.N.; Levey, A.S.; Sarnak, M.J. Kidney disease as a risk factor for recurrent cardiovascular disease and mortality. Am. J. Kidney Dis. 2004, 44, 198–206. [Google Scholar] [CrossRef]
- Madero, M.; Sarnak, M.J.; Wang, X.; Greene, T.; Beck, G.J.; Kusek, J.W.; Collins, A.J.; Levey, A.S.; Menon, V. Uric acid and long-term outcomes in CKD. Am. J. Kidney Dis. 2009, 53, 796–803. [Google Scholar] [CrossRef]
- Battelli, M.G.; Polito, L.; Bortolotti, M.; Bolognesi, A. Xanthine oxidoreductase-derived reactive species: Physiological and pathological effects. Oxid Med. Cell Longev. 2016, 2016, 3527579. [Google Scholar] [CrossRef]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox. Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef]
- Ohara, Y.; Peterson, T.E.; Harrison, D.G. Hypercholesterolemia increases endothelial superoxide anion production. J. Clin. Investig. 1993, 91, 2546–2551. [Google Scholar] [CrossRef]
- El Boueiz, A.; Damarla, M.; Hassoun, P.M. Xanthine oxidoreductase in respiratory and cardiovascular disorders. Am. J. Physiol Lung Cell Mol. Physiol. 2008, 294, L830–L840. [Google Scholar] [CrossRef] [PubMed]
- Omizo, H.; Tamura, Y.; Morimoto, C.; Ueno, M.; Hayama, Y.; Kuribayashi-Okuma, E.; Uchida, S.; Shibata, S. Cardio-renal protective effect of the xanthine oxidase inhibitor febuxostat in the 5/6 nephrectomy model with hyperuricemia. Sci. Rep. 2020, 10, 9326. [Google Scholar] [CrossRef] [PubMed]
- Jalal, D.I.; Chonchol, M.; Chen, W.; Targher, G. Uric acid as a target of therapy in CKD. Am. J. Kidney Dis. 2013, 61, 134–146. [Google Scholar] [CrossRef]
- Goicoechea, M.; de Vinuesa, S.G.; Verdalles, U.; Verde, E.; Macias, N.; Santos, A.; de Jose, A.P.; Cedeño, S.; Linares, T.; Luño, J. Allopurinol and progression of CKD and cardiovascular events: Long-term follow-up of a randomized clinical trial. Am. J. Kidney Dis. 2015, 65, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Goicoechea, M.; De Vinuesa, S.G.; Verdalles, U.; Ruiz-Caro, C.; Ampuero, J.; Rincón, A.; Arroyo, D.; Luño, J. Effect of allopurinol in chronic kidney disease progression and cardiovascular risk. Clin. J. Am. Soc. Nephrol. 2010, 5, 1388–1393. [Google Scholar] [CrossRef] [PubMed]
- Badve, S.V.; Pascoe, E.M.; Tiku, A.; Boudville, N.; Brown, F.G.; Cass, A.; Clarke, P.; Dalbeth, N.; Day, R.O.; de Zoysa, J.R.; et al. Effects of Allopurinol on the Progression of Chronic Kidney Disease. N. Engl. J. Med. 2020, 382, 2504–2513. [Google Scholar] [CrossRef] [PubMed]
- Doria, A.; Galecki, A.T.; Spino, C.; Pop-Busui, R.; Cherney, D.Z.; Lingvay, I.; Parsa, A.; Rossing, P.; Sigal, R.J.; Afkarian, M.; et al. Serum urate lowering with allopurinol and kidney function in type 1 diabetes. N. Engl. J. Med. 2020, 382, 2493–2503. [Google Scholar] [CrossRef]
- Singh, J.A.; Cleveland, J.D. Hypersensitivity reactions with allopurinol and febuxostat: A study using the Medicare claims data. Ann. Rheum Dis. 2020, 79, 529–535. [Google Scholar] [CrossRef]
- Mohammed, E.; Browne, L.D.; Kumar AU, A.; Adeeb, F.; Fraser, A.D.; Stack, A.G. Prevalence and treatment of gout among patients with chronic kidney disease in the Irish health system: A national study. PLoS ONE 2019, 14, e0210487. [Google Scholar] [CrossRef]
- Zhang, X.-W.; Li, Q.-H.; Xu, Z.-D.; Dou, J.-J. Mass spectrometry-based metabolomics in health and medical science: A systematic review. RSC Adv. 2020, 10, 3092–3104. [Google Scholar] [CrossRef]
- Jalal, D.I.; Decker, E.; Perrenoud, L.; Nowak, K.; Bispham, N.; Mehta, T.; Smits, G.; You, Z.; Seals, D.; Chonchol, M.; et al. Vascular function and uric acid-lowering in stage 3 CKD. J. Am. Soc. Nephrol. 2017, 28, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Levey, A.S.; Bosch, J.P.; Lewis, J.B.; Greene, T.; Rogers, N.; Roth, D. A more accurate method to estimate glomerular filtration rate from serum creatinine: A new prediction equation. Modification of Diet in Renal Disease Study Group. Ann. Intern. Med. 1999, 130, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Meijer, B.; Seinen, M.L.; Hosman, T.; Linskens, R.K.; Kneppelhout, J.-K.; Peters, G.J.; Mulder, C.J.; A Van Bodegraven, A.; De Boer, N.K. High inter-individual variability of serum xanthine oxidoreductase activity in IBD patients. Nucleosides Nucleotides Nucleic Acids 2018, 37, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Ni, Z.; Oveisi, F.; Liang, K.; Pandian, R. Enhanced nitric oxide inactivation and protein nitration by reactive oxygen species in renal insufficiency. Hypertension 2002, 39, 135–141. [Google Scholar] [CrossRef]
- Celermajer, D.S.; Sorensen, K.; Ryalls, M.; Robinson, J.; Thomas, O.; Leonard, J.V.; Deanfield, J.E. Impaired endothelial function occurs in the systemic arteries of children with homozygous homocystinuria but not in their heterozygous parents. J. Am. Coll Cardiol. 1993, 22, 854–858. [Google Scholar] [CrossRef][Green Version]
- Jablonski, K.L.; Decker, E.; Perrenoud, L.; Kendrick, J.; Chonchol, M.; Seals, U.R.; Jalal, D. Assessment of vascular function in patients with chronic kidney disease. J. Vis. Exp. 2014, 88, e51478. [Google Scholar] [CrossRef]
- Andrews, E.S.; Perrenoud, L.; Nowak, K.L.; You, Z.; Pasch, A.; Chonchol, M.; Kendrick, J.; Jalal, D. Examining the effects of uric acid-lowering on markers vascular of calcification and CKD-MBD; A post-hoc analysis of a randomized clinical trial. PLoS ONE 2018, 13, e0205831. [Google Scholar] [CrossRef]
- Tompkins, S.C.; Sheldon, R.; Rauckhorst, A.J.; Noterman, F.M.; Solst, S.R.; Buchanan, J.L.; Mapuskar, K.A.; Pewa, A.D.; Gray, L.R.; Oonthonpan, L.; et al. Disrupting mitochondrial pyruvate uptake directs glutamine into the TCA cycle away from glutathione synthesis and impairs hepatocellular tumorigenesis. Cell Rep. 2019, 28, 2608–2619.e6. [Google Scholar] [CrossRef]
- Metabolomics Core Facility. Available online: https://medicine.uiowa.edu/diabetes/metabolomics-core-facility (accessed on 1 February 2021).
- Li, B.; Tang, J.; Yang, Q.; Li, S.; Cui, X.; Li, Y.H.; Chen, Y.Z.; Xue, W.; Li, X.; Zhu, F. NOREVA: Normalization and evaluation of MS-based metabolomics data. Nucleic Acids Res. 2017, 45, W162–W170. [Google Scholar] [CrossRef]
- Levey, A.S.; Stevens, L.A.; Schmid, C.H.; Zhang, Y.L.; Castro, A.F., III; Feldman, H.I.; Kusek, J.W.; Eggers, P.; Van Lente, F.; Greene, T. A new equation to estimate glomerular filtration rate. Ann. Intern. Med. 2009, 150, 604–612. [Google Scholar] [CrossRef]
- Fanelli, G.; Gevi, F.; Belardo, A.; Zolla, L. Metabolic patterns in insulin-sensitive male hypogonadism. Cell Death Dis. 2018, 9, 653. [Google Scholar] [CrossRef] [PubMed]
- Houston, M.; Estevez, A.; Chumley, P.; Aslan, M.; Marklund, S.; Parks, D.A.; Freeman, B.A. Binding of xanthine oxidase to vascular endothelium. Kinetic characterization and oxidative impairment of nitric oxide-dependent signaling. J. Biol. Chem. 1999, 274, 4985–4994. [Google Scholar] [CrossRef] [PubMed]
- Nomura, J.; Busso, N.; Ives, A.; Matsui, C.; Tsujimoto, S.; Shirakura, T.; Tamura, M.; Kobayashi, T.; So, A.; Yamanaka, Y. Xanthine oxidase inhibition by febuxostat attenuates experimental atherosclerosis in mice. Sci. Rep. 2014, 4, 4554. [Google Scholar] [CrossRef] [PubMed]
- Elion, G.B. Nobel Lecture. The purine path to chemotherapy. Biosci. Rep. 1989, 9, 509–529. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Nivorozhkin, A.; Szabó, C. Therapeutic effects of xanthine oxidase inhibitors: Renaissance half a century after the discovery of allopurinol. Pharmacol. Rev. 2006, 58, 87–114. [Google Scholar] [CrossRef]
- Landmesser, U.; Drexler, H. Allopurinol and endothelial function in heart failure: Future or fantasy? Circulation 2002, 106, 173–175. [Google Scholar] [CrossRef][Green Version]
- Hellsten-Westing, Y. Immunohistochemical localization of xanthine oxidase in human cardiac and skeletal muscle. Histochemistry 1993, 100, 215–222. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Beckman, J.S.; Beckman, T.K.; Wheat, J.K.; Cash, T.G.; Freeman, B.A.; Parks, D.A. Circulating xanthine oxidase: Potential mediator of ischemic injury. Am. J. Physiol. 1990, 258 Pt 1, G564–G570. [Google Scholar] [CrossRef]
- Farquharson, C.A.J.; Butler, R.; Hill, A.; Belch, J.J.F.; Struthers, A.D. Allopurinol improves endothelial dysfunction in chronic heart failure. Circulation 2002, 106, 221–226. [Google Scholar] [CrossRef]
- Doehner, W.; Schoene, N.; Rauchhaus, M.; Leyva-Leon, F.; Pavitt, D.V.; Reaveley, D.A.; Schuler, G.; Coats, A.J.; Anker, S.D.; Hambrecht, R. Effects of xanthine oxidase inhibition with allopurinol on endothelial function and peripheral blood flow in hyperuricemic patients with chronic heart failure: Results from 2 placebo-controlled studies. Circulation 2002, 105, 2619–2624. [Google Scholar] [CrossRef]
- Sarnesto, A.; Linder, N.; O Raivio, K. Organ distribution and molecular forms of human xanthine dehydrogenase/xanthine oxidase protein. Lab. Investig. 1996, 74, 48–56. [Google Scholar] [PubMed]
- Day, R.O.; Graham, G.G.; Hicks, M.; McLachlan, A.J.; Stocker, S.; Williams, K.M. Clinical pharmacokinetics and pharmacodynamics of allopurinol and oxypurinol. Clin. Pharmacokinet. 2007, 46, 623–644. [Google Scholar] [CrossRef] [PubMed]
- Aranda, R.; Doménech, E.; Rus, A.D.; Real, J.T.; Sastre, J.; Vina, J.; Pallardó, F.V. Age-related increase in xanthine oxidase activity in human plasma and rat tissues. Free Radic. Res. 2007, 41, 1195–1200. [Google Scholar] [CrossRef] [PubMed]
- Newaz, M.A.; Adeeb, N.N. Detection of xanthine oxidase in human plasma. Med. J. Malaysia 1998, 53, 70–75. [Google Scholar]
- Tan, S.; Radi, R.; Gaudier, F.L.; A Evans, R.; Rivera, A.; A Kirk, K.; A Parks, D. Physiologic levels of uric acid inhibit xanthine oxidase in human plasma. Pediatr. Res. 1993, 34, 303–307. [Google Scholar] [CrossRef]
- Radi, R.; Tan, S.; Prodanov, E.; Evans, R.A.; Parks, D.A. Inhibition of xanthine oxidase by uric acid and its influence on superoxide radical production. Biochim. Biophys. Acta 1992, 1122, 178–182. [Google Scholar] [CrossRef]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.C.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Grüning, N.-M.; Krüger, A.; Tauqeer Alam, M.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 2015, 90, 927–963. [Google Scholar] [CrossRef]
- Nobrega-Pereira, S.; Fernandez-Marcos, P.J.; Brioche, T.; Gomez-Cabrera, M.C.; Salvador-Pascual, A.; Flores, J.M.; Viña, M.C.G.-C.A.S.-P.J.; Serrano, M. G6PD protects from oxidative damage and improves healthspan in mice. Nat. Commun. 2016, 7, 10894. [Google Scholar] [CrossRef]
- Pilz, R.B.; Willis, R.C.; Boss, G.R. The influence of ribose 5-phosphate availability on purine synthesis of cultured human lymphoblasts and mitogen-stimulated lymphocytes. J. Biol. Chem. 1984, 259, 2927–2935. [Google Scholar] [CrossRef]
- Beardmore, T.D.; Kelley, W.N. Mechanism of allopurinol-mediated inhibition of pyrimidine biosynthesis. J. Lab. Clin. Med. 1971, 78, 696–704. [Google Scholar]
- Hauser, E.R.; Finkelstein, J.E.; Valle, D.; Brusilow, S.W. Allopurinol-induced orotidinuria. A test for mutations at the ornithine carbamoyltransferase locus in women. N. Engl. J. Med. 1990, 322, 1641–1645. [Google Scholar] [CrossRef] [PubMed]
- Tani, T.; Okamoto, K.; Fujiwara, M.; Katayama, A.; Tsuruoka, S. Metabolomics analysis elucidates unique influences on purine/pyrimidine metabolism by xanthine oxidoreductase inhibitors in a rat model of renal ischemia-reperfusion injury. Mol. Med. 2019, 25, 40. [Google Scholar] [CrossRef] [PubMed]
- Kopple, J.D. Phenylalanine and tyrosine metabolism in chronic kidney failure. J. Nutr. 2007, 137 (Suppl. 1), 1586S–1590S. [Google Scholar] [CrossRef] [PubMed]
- Hornykiewicz, O. The discovery of dopamine deficiency in the parkinsonian brain. J. Neural. Transm. Suppl. 2006, 70, 9–15. [Google Scholar]
- Kachroo, A.; Schwarzschild, M.A. Allopurinol reduces levels of urate and dopamine but not dopaminergic neurons in a dual pesticide model of Parkinson’s disease. Brain Res. 2014, 1563, 103–109. [Google Scholar] [CrossRef]
- Crotty, G.F.; Ascherio, A.; Schwarzschild, M.A. Targeting urate to reduce oxidative stress in Parkinson disease. Exp. Neurol. 2017, 298 Pt B, 210–224. [Google Scholar] [CrossRef]
- Prentki, M.; Madiraju, S.R.M. Glycerolipid metabolism and signaling in health and disease. Endocr Rev. 2008, 29, 647–676. [Google Scholar] [CrossRef]
- Eyer, F.; Zilker, T. Bench-to-bedside review: Mechanisms and management of hyperthermia due to toxicity. Critical Care. 2007, 11, 236. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Characteristics | Allopurinol (n = 14) | Placebo (n = 14) | p Value |
|---|---|---|---|
| Age (years) | 61 ± 13 | 59 ± 8 | 0.27 |
| Male sex (n (%)) | 10 (71) | 11 (79) | >0.99 |
| Race (n (%)) | |||
| Caucasian | 9 (64) | 12 (86) | 0.38 |
| African American | 2 (14) | 1 (7) | |
| Other | 3 (22) | 1 (7) | |
| Baseline diabetes | 8 (57) | 9 (64) | 0.70 |
| Baseline cardiovascular disease | 1 (7) | 1 (7) | >0.99 |
| Systolic BP (mmHg) | 126 ± 15 | 127 ± 15 | 0.96 |
| Diastolic BP (mmHg) | 76 ± 11 | 74 ± 10 | 0.5 |
| BMI (kg/m2) | 32.0 ± 4.2 | 35.1 ± 5.0 | 0.17 |
| Hemoglobin A1C (%) | 6.6 ± 1.8 | 6.3 ± 1.3 | 0.81 |
| Creatinine (mg/dL) | 1.80 ± 0.4 | 1.75 ± 0.4 | 0.91 |
| CKD- EPI eGFR (mL/min/1.73 m2) | 39.9 ± 10.8 | 42.4 ± 11.3 | 0.58 |
| ACR (mg/g) | 160 ± 213 | 379 ± 583 | 0.89 |
| BA- FMD (% change) | 4.1 ± 5.4 | 6.2 ± 6.3 | 0.35 |
| NMD (% change) | 19.7 ± 8.4 | 16.3 ± 9.9 | 0.23 |
| CIMT (mm) | 0.75 ± 0.18 | 0.78 ± 0.19 | 0.93 |
| OxLDL | 43.2 ± 10.4 | 52.5 ± 17.9 | 0.08 |
| Endothelial NT * | 0.79 ± 0.14 | 0.87 ± 0.21 | 0.52 |
| Serum urate (mg/dL) | 8.4 ± 1.5 | 8.4 ± 1.3 | 0.86 |
| Serum XO activity mU/mL | 0.80 ± 0.51 | 0.78 ± 0.30 | 0.37 |
| Endothelial XO * | 1.00 | 1.00 | >0.99 |
| Measures of Vascular Function | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Variable | BA-FMD | NMD | CIMT | oxLDL | Endothelial NT * | |||||
| r | p Value | r | p Value | r | p Value | r | p Value | r | p Value | |
| Serum urate (mg/dL) | −0.25 | 0.10 | −0.3 | 0.07 | 0.29 | 0.06 | −0.18 | 0.22 | 0.10 | 0.64 |
| Serum XO activity (mU/mL) | 0.002 | 0.99 | 0.04 | 0.80 | 0.14 | 0.39 | 0.12 | 0.41 | 0.22 | 0.30 |
| Endothelial XO expression * | −0.27 | 0.41 | −0.61 | 0.15 | −0.17 | 0.70 | −0.21 | 0.54 | −0.1 | 0.90 |
| Measures of Kidney Disease | ||||||||||
| CKD-EPI eGFR | ACR | |||||||||
| r | p Value | r | p Value | |||||||
| Serum urate (mg/dL) | −0.58 | <0.0001 | 0.12 | 0.45 | ||||||
| Serum XO activity (mU/mL) | −0.01 | 0.95 | 0.004 | 0.98 | ||||||
| Endothelial XO expression * | 0.26 | 0.45 | 0.04 | 0.90 | ||||||
| Variable | Placebo | Allopurinol | p Value |
|---|---|---|---|
| Serum urate (mg/dL) | 0.26 (−0.38, 0.89) | −3.60 (−4.24, −2.96) | <0.0001 |
| Serum xanthine * | 1.02 (0.77, 1.33) | 7.54 (5.74, 9.90) | <0.0001 |
| Serum XO activity (mU/mL) | −0.04 (−0.39, 0.09) | −0.14 (−0.34, 0.064) | 0.70 |
| Placebo | Allopurinol | ||||
|---|---|---|---|---|---|
| Median (IQR) | p Value * | p Value * | In-Between Groups $ | ||
| 1-Octadecanol | 0.99 (0.93, 1.01) | 0.15 | 1.02 (0.98, 1.08) | 0.27 | 0.05 |
| 2-Hydroxybutyrate | 1.05 (0.87, 1.28) | 0.54 | 1.08 (0.82, 1.34) | 0.43 | 0.85 |
| 2-Hydroxyglutarate | 1.09 (0.87, 1.24) | 0.30 | 0.97 (0.77, 1.11) | 0.39 | 0.17 |
| 2-Oxoadipate | 1.05 (0.84, 1.25) | 0.33 | 1.02 (0.70, 1.21) | 0.86 | 0.29 |
| 6-Phosphogluconate | 0.92 (0.84, 1.12) | 0.67 | 0.89 (0.82, 0.97) | 0.04 | 0.43 |
| Adonitol | 1.05 (0.93, 1.16) | 0.24 | 0.89 (0.88, 1.02) | 0.30 | 0.08 |
| Alanine | 1.04 (0.93, 1.29) | 0.43 | 1.00 (0.82, 1.17) | 0.86 | 0.85 |
| α-Keto β-Methylvalerate | 0.98 (0.85, 1.16) | 0.90 | 0.97 (0.84, 1.21) | 0.81 | 0.85 |
| α-Ketoglutarate | 1.03 (0.97, 1.18) | 0.36 | 1.03 (0.93, 1.16) | 0.39 | 0.82 |
| α-Ketoisocaproate | 0.97 (0.88, 1.09) | 0.86 | 1.01 (0.91, 1.07) | 0.71 | 0.89 |
| α-Ketoisovalerate | 0.99 (0.83, 1.11) | 0.71 | 0.95 (0.82, 1.25) | 0.90 | 0.96 |
| Aminoadipate | 1.11 (0.93, 1.29) | 0.36 | 1.29 (1.11, 1.57) | 0.09 | 0.12 |
| Arachidic acid | 1.02 (0.82, 1.09) | 1.00 | 0.92 (0.85, 1.16) | 0.86 | 0.93 |
| Arachidonate | 0.98 (0.88, 1.04) | 0.50 | 0.90 (0.73, 1.06) | 0.08 | 0.43 |
| Asparagine | 1.08 (0.86, 1.11) | 0.90 | 1.03 (0.86, 1.25) | 0.58 | 1.00 |
| Aspartate | 0.99 (0.71, 1.28) | 0.76 | 1.09 (0.77, 1.53) | 0.46 | 0.58 |
| Behenic acid | 1.03 (0.79, 1.11) | 0.95 | 0.85 (0.71, 1.04) | 0.01 | 0.10 |
| β-Alanine | 1.02 (0.96, 1.06) | 0.58 | 1.01 (0.96, 1.08) | 0.67 | 1.00 |
| β-Hydroxy β-Methylbutyric.acid | 1.08 (0.89, 1.17) | 0.36 | 1.10 (0.94, 1.29) | 0.14 | 0.68 |
| β-Hydroxybutyrate-3 | 1.10 (0.73, 1.47) | 0.33 | 1.07 (0.66, 2.15) | 0.46 | 0.93 |
| Cholesterol | 1.04 (0.95, 1.09) | 0.43 | 0.94 (0.88, 1.01) | 0.12 | 0.05 |
| Citraconate | 0.97 (0.84, 1.11) | 0.46 | 0.98 (0.93, 1.10) | 0.86 | 0.61 |
| Citrate # | 1.05 (0.93, 1.26) | 0.33 | 0.86 (0.74, 1.00) | 0.07 | 0.04 |
| Citrulline | 0.92 (0.81, 1.12) | 0.50 | 0.88 (0.81, 1.03) | 0.63 | 0.82 |
| Cysteine | 0.95 (0.86, 1.24) | 0.81 | 1.04 (0.90, 1.12) | 0.76 | 0.65 |
| Cytidine | 1.07 (0.94, 1.24) | 0.19 | 1.06 (0.62, 1.20) | 0.95 | 0.46 |
| Cytosine | 1.06 (0.94, 1.15) | 0.36 | 0.98 (0.72, 1.28) | 0.95 | 0.61 |
| Dihydroxyphenylalanine # | 0.99 (0.92, 1.18) | 0.81 | 0.71 (0.58, 0.88) | 0.01 | <0.0001 |
| Fructose | 0.91 (0.47, 1.40) | 0.50 | 0.99 (0.40, 2.92) | 0.50 | 0.68 |
| Fumarate | 1.06 (0.77, 1.21) | 0.95 | 1.02 (0.86, 1.17) | 0.58 | 0.65 |
| γ-aminobutyric acid | 1.01 (0.73, 1.20) | 0.95 | 1.10 (0.58, 1.88) | 0.46 | 0.82 |
| Glucose | 0.95 (0.82, 1.17) | 0.86 | 1.01 (0.78, 1.38) | 0.71 | 0.85 |
| Glucose-6-phosphate | 0.77 (0.42, 1.28) | 0.43 | 1.49 (0.60, 2.58) | 0.08 | 0.10 |
| Glutamate | 1.00 (0.92, 1.18) | 0.76 | 1.17 (0.74, 1.64) | 0.30 | 0.52 |
| Glutamine | 1.08 (0.93, 1.17) | 0.43 | 1.01 (0.87, 1.17) | 0.81 | 0.71 |
| Glycerate # | 1.12 (0.86, 1.17) | 0.43 | 0.92 (0.76, 0.98) | 0.05 | 0.04 |
| Glycerol # | 1.16 (1.00, 1.49) | 0.01 | 0.93 (0.74, 1.34) | 0.63 | 0.03 |
| Glycerol Monolaurate | 0.96 (0.88, 1.04) | 0.63 | 0.93 (0.79, 1.01) | 0.17 | 0.49 |
| Glycine | 0.99 (0.87, 1.18) | 0.95 | 1.04 (0.78, 1.11) | 0.81 | 0.82 |
| Guanosine | 1.10 (0.51, 1.26) | 0.95 | 1.47 (0.91, 2.35) | 0.09 | 0.27 |
| Heneicosylic acid | 0.99 (0.93, 1.05) | 0.76 | 1.01 (0.94, 1.05) | 0.81 | 0.93 |
| Heptadecanoic acid | 1.00 (0.90, 1.22) | 0.76 | 1.03 (0.85, 1.15) | 0.95 | 0.78 |
| Histidine | 0.97 (0.81, 1.17) | 0.95 | 0.94 (0.85, 1.12) | 0.76 | 0.96 |
| Homocysteine | 1.05 (0.86, 1.34) | 0.33 | 1.00 (0.87, 1.55) | 0.58 | 0.89 |
| Homoserine | 1.03 (0.97, 1.18) | 0.15 | 0.98 (0.95, 1.13) | 0.86 | 0.27 |
| Hypotaurine | 1.02 (0.73, 1.41) | 0.71 | 0.93 (0.85, 1.40) | 0.71 | 0.82 |
| Hypoxanthine | 1.00 (0.66, 1.15) | 0.76 | 1.11 (0.83, 1.37) | 0.24 | 0.31 |
| Inotisol | 1.00 (0.89, 1.25) | 0.67 | 0.88 (0.78, 1.16) | 0.71 | 0.46 |
| Isoleucine | 1.01 (0.78, 1.33) | 0.71 | 1.00 (0.89, 1.47) | 0.58 | 0.52 |
| Itaconic acid # | 0.85 (0.68, 1.14) | 0.39 | 1.43 (0.82, 1.67) | 0.07 | 0.04 |
| Lactate | 1.06 (0.86, 1.16) | 0.50 | 0.92 (0.83, 1.10) | 0.30 | 0.25 |
| Lauric acid | 1.08 (0.87, 1.18) | 0.54 | 1.01 (0.62, 1.44) | 0.90 | 0.68 |
| Leucine | 1.02 (0.87, 1.23) | 0.67 | 0.98 (0.87, 1.26) | 0.67 | 1.00 |
| Linoleate | 1.06 (1.02, 1.32) | 0.09 | 0.92 (0.84, 1.10) | 0.43 | 0.13 |
| Linolenic acid # | 1.16 (0.82, 1.72) | 0.15 | 0.87 (0.64, 1.05) | 0.06 | 0.02 |
| Lysine | 1.05 (0.98, 1.12) | 0.12 | 0.95 (0.85, 1.24) | 0.71 | 0.52 |
| Malate | 1.00 (0.81, 1.28) | 0.50 | 0.97 (0.88, 1.09) | 0.58 | 0.68 |
| Malonate | 0.97 (0.91, 1.16) | 0.63 | 1.01 (0.97, 1.07) | 0.76 | 0.49 |
| Mannose | 1.10 (0.89, 1.33) | 0.24 | 1.06 (0.99, 1.24) | 0.24 | 0.82 |
| Methionine | 1.02 (0.91, 1.20) | 0.54 | 0.96 (0.79, 1.21) | 0.90 | 0.55 |
| Myristic.acid | 1.03 (0.83, 1.61) | 0.43 | 1.05 (0.75, 1.34) | 0.81 | 0.65 |
| N-acetyl aspartate # | 1.03 (0.97, 1.10) | 0.36 | 0.93 (0.90, 1.01) | 0.04 | 0.04 |
| N-acetyl glutamate | 1.00 (0.93, 1.28) | 0.76 | 0.91 (0.79, 1.03) | 0.39 | 0.18 |
| N-acetyl serine | 0.95 (0.78, 1.14) | 0.81 | 0.87 (0.78, 0.99) | 0.30 | 0.52 |
| N-acetyl tyrosine # | 0.97 (0.85, 1.18) | 0.86 | 0.41 (0.29, 0.45) | <0.0001 | <0.0001 |
| Oleic acid | 1.08 (0.91, 1.61) | 0.19 | 0.90 (0.76, 1.37) | 0.81 | 0.27 |
| O-Phosphoethanolamine | 0.99 (0.90, 1.15) | 0.76 | 1.01 (0.86, 1.21) | 0.63 | 0.71 |
| Ornithine | 1.08 (0.89, 1.27) | 0.39 | 1.07 (0.80, 1.19) | 0.81 | 0.89 |
| Orotate # | 0.90 (0.87, 1.14) | 0.81 | 11.82 (8.97, 18.04) | <0.0001 | <0.0001 |
| Palmitate | 1.05 (0.91, 1.31) | 0.33 | 0.97 (0.85, 1.19) | 0.86 | 0.38 |
| Phenylalanine | 1.02 (0.86, 1.20) | 0.76 | 0.95 (0.89, 1.20) | 1.00 | 0.96 |
| Phosphoenolpyruvate | 0.99 (0.92, 1.08) | 0.95 | 0.94 (0.87, 0.98) | 0.01 | 0.12 |
| Proline | 0.95 (0.79, 1.08) | 0.71 | 1.00 (0.69, 1.30) | 0.90 | 0.85 |
| Pyruvate | 1.10 (0.96, 1.46) | 0.14 | 1.01 (0.77, 1.28) | 0.67 | 0.38 |
| Ribose | 1.01 (0.83, 1.45) | 0.63 | 0.91 (0.66, 1.35) | 0.95 | 0.58 |
| Ribose-5-phosphate # | 0.98 (0.79, 1.28) | 0.81 | 0.58 (0.48, 0.61) | <0.0001 | <0.0001 |
| Sedoheptulose | 1.06 (0.76, 1.23) | 0.76 | 1.00 (0.91, 1.33) | 0.50 | 0.85 |
| Serine | 1.08 (0.83, 1.23) | 0.71 | 0.94 (0.80, 1.19) | 0.81 | 0.78 |
| Serotonin | 0.98 (0.85, 1.27) | 0.67 | 1.06 (0.90, 1.14) | 0.58 | 0.65 |
| Stearate | 0.97 (0.88, 1.13) | 0.95 | 1.02 (0.86, 1.14) | 0.90 | 1.00 |
| Succinate | 1.08 (0.93, 1.22) | 0.43 | 0.97 (0.86, 1.06) | 0.30 | 0.18 |
| Threonine | 0.97 (0.80, 1.19) | 1.00 | 0.80 (0.71, 1.45) | 0.90 | 0.36 |
| Thymine | 1.08 (1.05, 1.12) | 0.33 | 1.01 (0.84, 1.06) | 0.46 | 0.10 |
| Tryptophan | 1.03 (0.92, 1.08) | 0.50 | 0.97 (0.86, 1.08) | 0.67 | 0.75 |
| Tyrosine | 1.01 (0.98, 1.11) | 0.67 | 0.93 (0.82, 1.17) | 1.00 | 0.89 |
| Uracil | 1.02 (0.90, 1.15) | 0.36 | 0.98 (0.92, 1.11) | 0.71 | 0.75 |
| Urea | 1.07 (0.86, 1.21) | 0.58 | 0.99 (0.83, 1.14) | 0.76 | 0.46 |
| Uridine | 1.06 (0.92, 1.08) | 0.58 | 1.05 (0.90, 1.27) | 0.30 | 0.61 |
| Valine | 1.03 (0.89, 1.30) | 0.50 | 0.93 (0.88, 1.16) | 1.00 | 0.71 |
| Xanthine # | 1.04 (0.70, 1.26) | 1.00 | 6.14 (3.61, 8.44) | <0.0001 | <0.0001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, M.; Hines, N.; Scerbo, D.; Buchanan, J.; Wu, C.; Ten Eyck, P.; Zepeda-Orozco, D.; Taylor, E.B.; Jalal, D.I. Allopurinol Lowers Serum Urate but Does Not Reduce Oxidative Stress in CKD. Antioxidants 2022, 11, 1297. https://doi.org/10.3390/antiox11071297
Sun M, Hines N, Scerbo D, Buchanan J, Wu C, Ten Eyck P, Zepeda-Orozco D, Taylor EB, Jalal DI. Allopurinol Lowers Serum Urate but Does Not Reduce Oxidative Stress in CKD. Antioxidants. 2022; 11(7):1297. https://doi.org/10.3390/antiox11071297
Chicago/Turabian StyleSun, Mingyao, Nicole Hines, Diego Scerbo, Jane Buchanan, Chaorong Wu, Patrick Ten Eyck, Diana Zepeda-Orozco, Eric B. Taylor, and Diana I. Jalal. 2022. "Allopurinol Lowers Serum Urate but Does Not Reduce Oxidative Stress in CKD" Antioxidants 11, no. 7: 1297. https://doi.org/10.3390/antiox11071297
APA StyleSun, M., Hines, N., Scerbo, D., Buchanan, J., Wu, C., Ten Eyck, P., Zepeda-Orozco, D., Taylor, E. B., & Jalal, D. I. (2022). Allopurinol Lowers Serum Urate but Does Not Reduce Oxidative Stress in CKD. Antioxidants, 11(7), 1297. https://doi.org/10.3390/antiox11071297