
Computation-Assisted Identification of Bioactive Compounds in Botanical Extracts: A Case Study of Anti-Inflammatory Natural Products from Hops

 ,
,  , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Plant Material
2.3. Extraction and Extract Fractionation
2.4. Nitric Oxide (NO) Assay
2.5. Mass Spectroscopic Analysis of the Crude Extract and Extract Fractions
2.6. Elastic Net Analysis
2.7. Random Forests Analysis
2.8. Spectral Network Analysis
2.9. Bioassay Validation of Xanthohumol
2.10. Quantitative Analysis of Inhibitory Fractions
2.11. Analysis of Late Fractions
3. Results
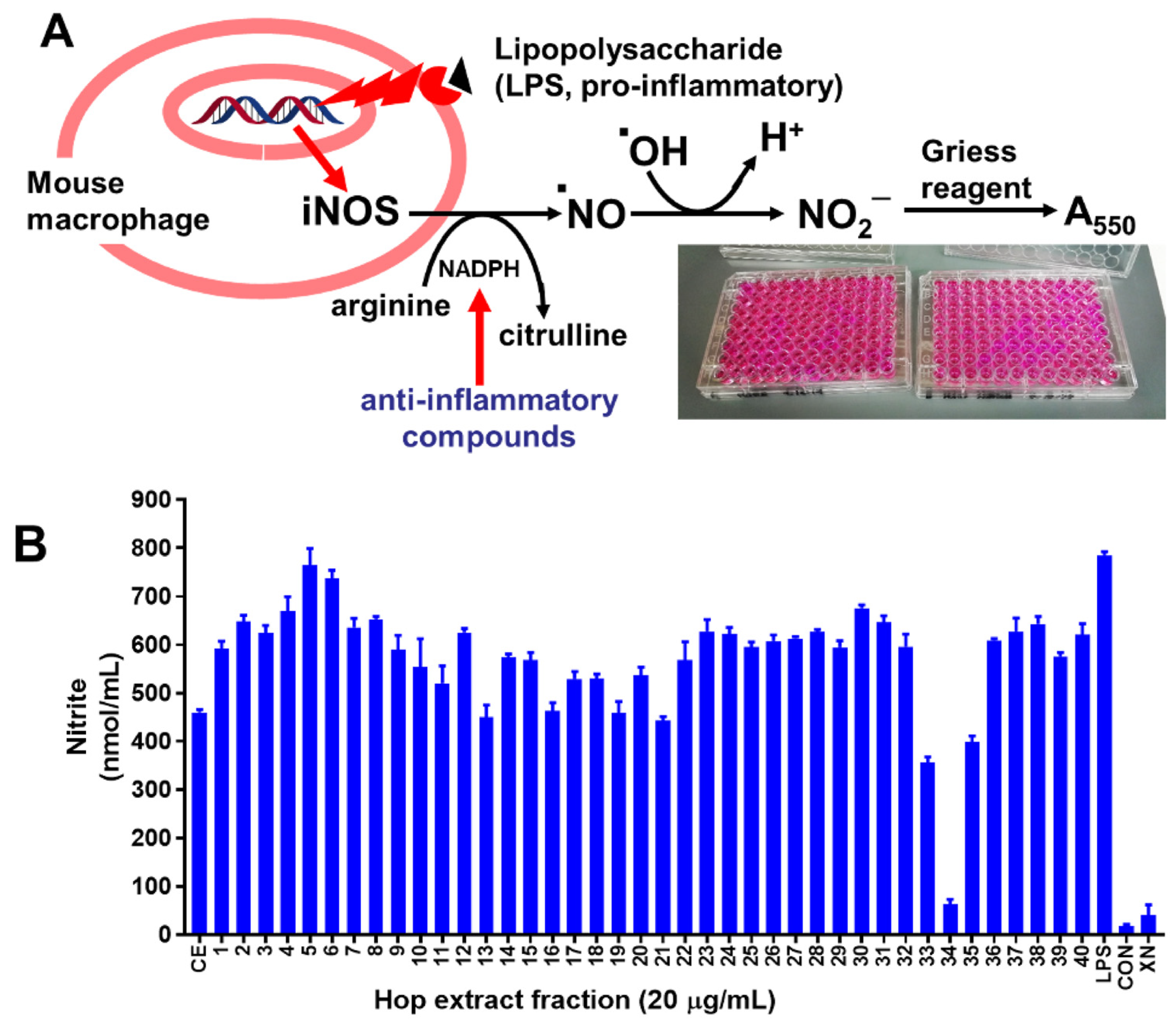
3.1. Bioactivity Testing of Fractions and Model Input
3.1.1. Bioactivity Testing of Fractions
3.1.2. Input Data to the Model
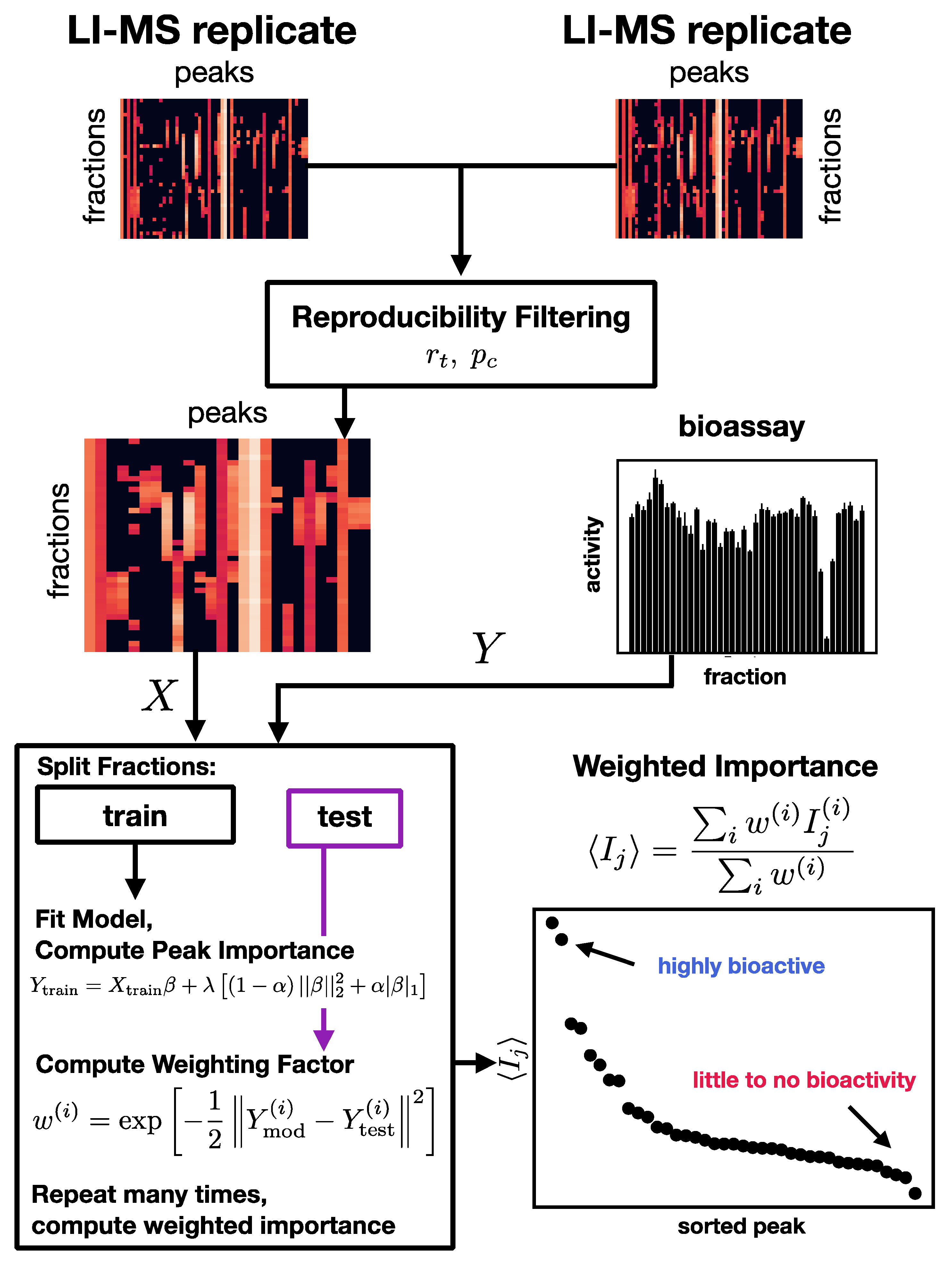
3.2. Computational Discovery Pipeline
3.2.1. Challenges
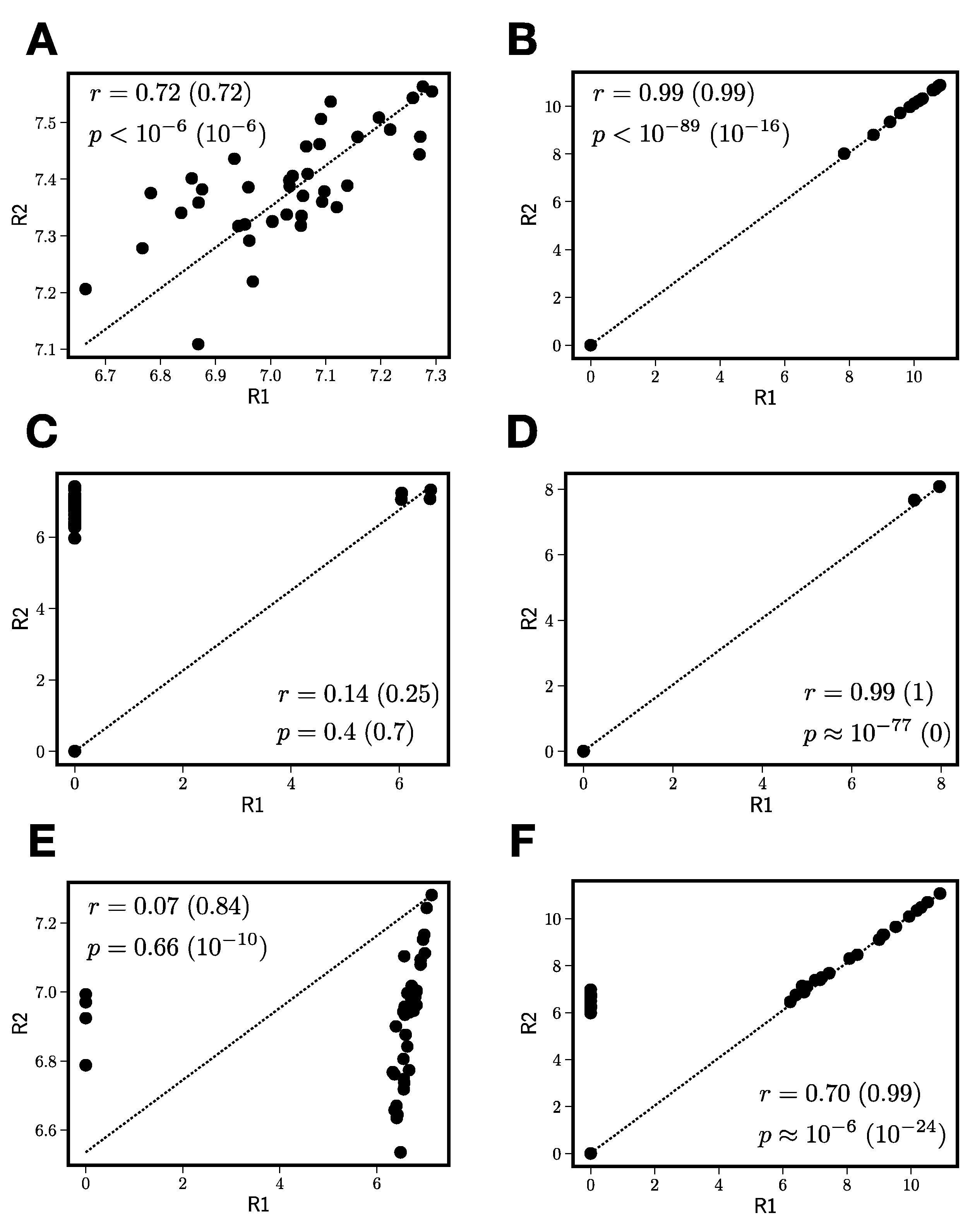
- In order to keep the full-scan QTOF-MS data in the linear range (so peak intensity is linearly proportional to concentration), the samples submitted to QTOF-MS analysis must be relatively dilute. This is essential for the modeling but can result in poor replicate-to-replicate reproducibility for some peaks that can lead to suboptimal model results.
- The standard method for parameter estimation in machine learning is cross-validation [30]. The data is divided into training and testing data, and the model parameters are then determined by fitting to only the training data. Then, the model goodness-of-fit is assessed relative to the test data, which the model has never seen. While this is generally a sound strategy, it relies on the assumption that the testing and training data come from the same underlying distribution—with thousands of samples or more that are split into training and test sets, one can be relatively certain this is the case. However, with a small number of samples—especially the lupulin extract in which bioactivity is not broadly shared across all fractions (see Figure 1B)—this can be easily violated and lead to unstable model estimates, even if cross-validation is used.
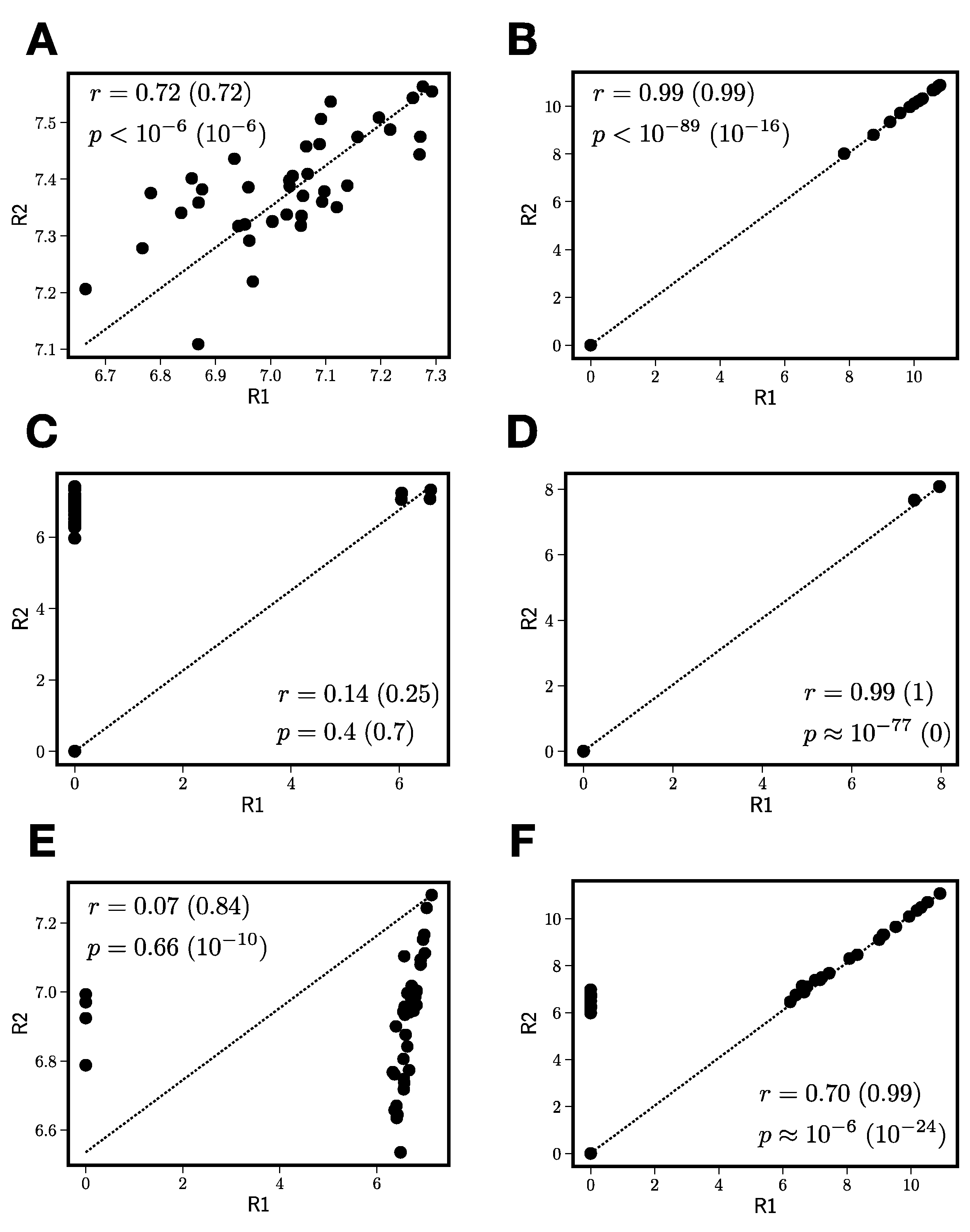
3.2.2. Reproducibility Filtering
- Compute the number of jointly nonzero elements nG.
- If nG is less than 3, exclude this peak from further processing.
- For peaks that pass the nonzero element criterion, compute zero-censored correlations r and associated -values between and .
- Correct zero-censored -values for multiple testing using the Benjamini–Hochberg false discovery rate [32].
- If < 0.05 and > , construct the replicate average peak by averaging only over the jointly nonzero values in the two replicates.
3.2.3. Wide Problems
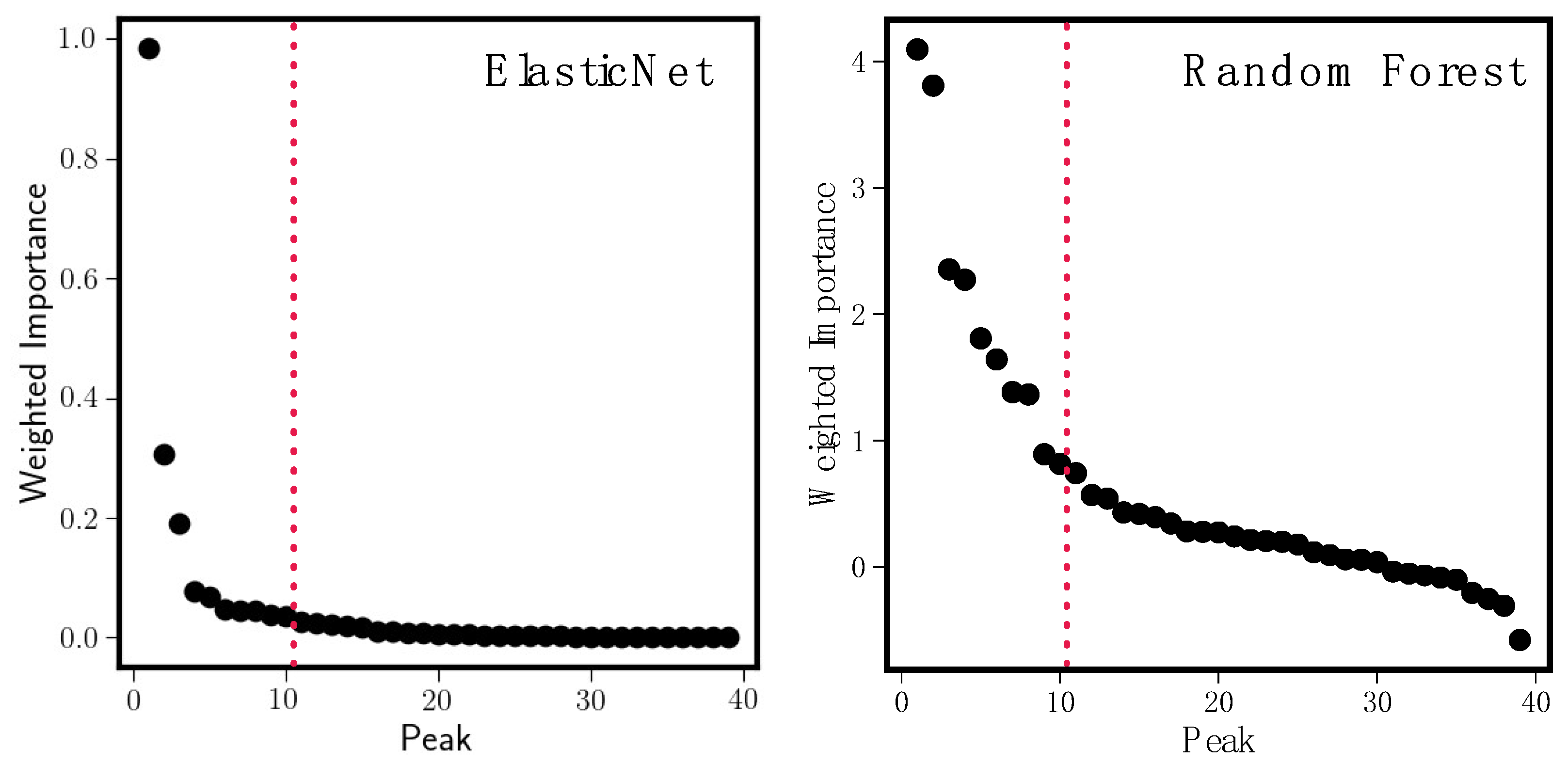
3.2.4. Variable Importance
3.2.5. Model Ensembles
3.3. Model Results and Validation
3.3.1. Model Results
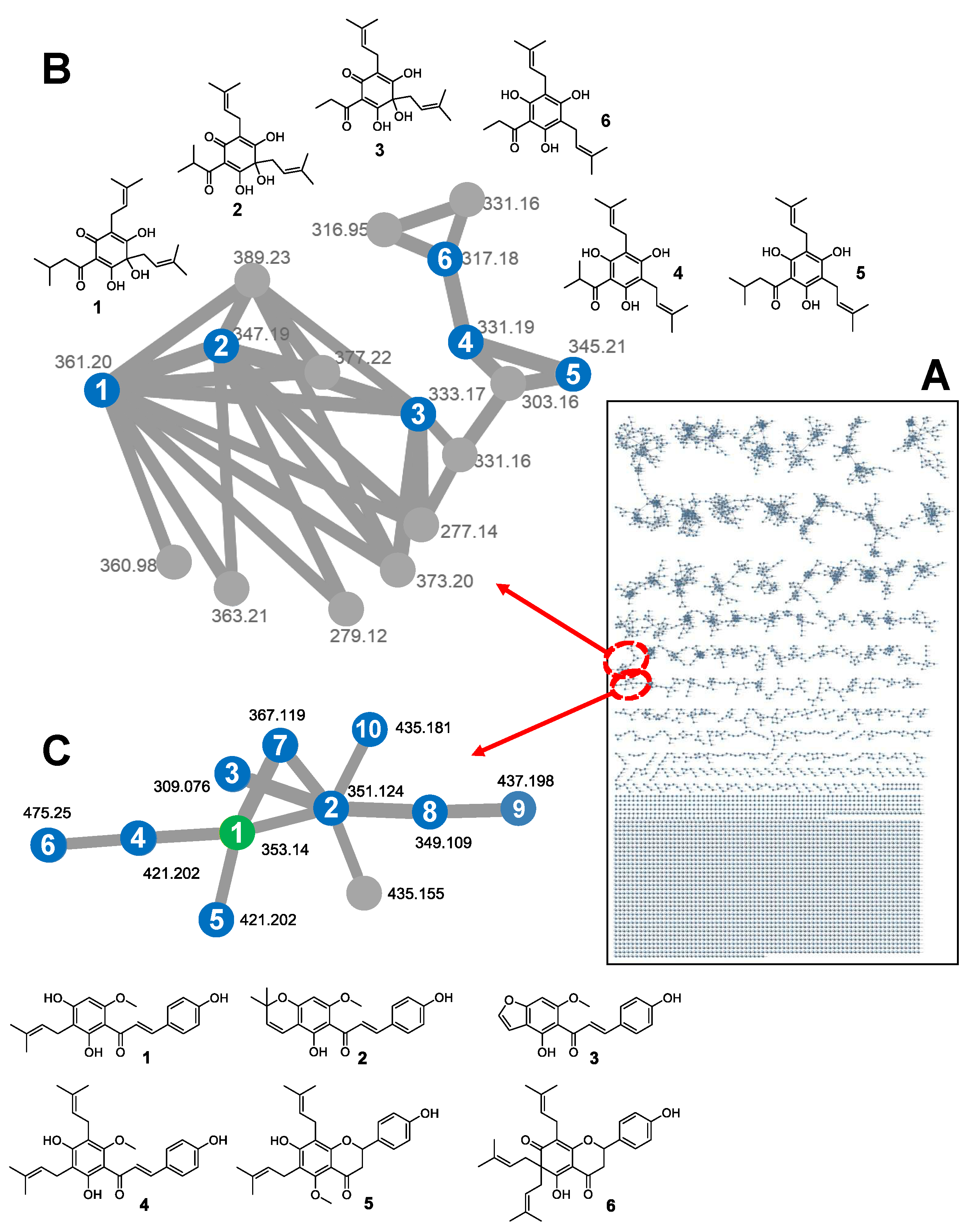
3.3.2. Global Natural Products Social (GNPS) Molecular Network
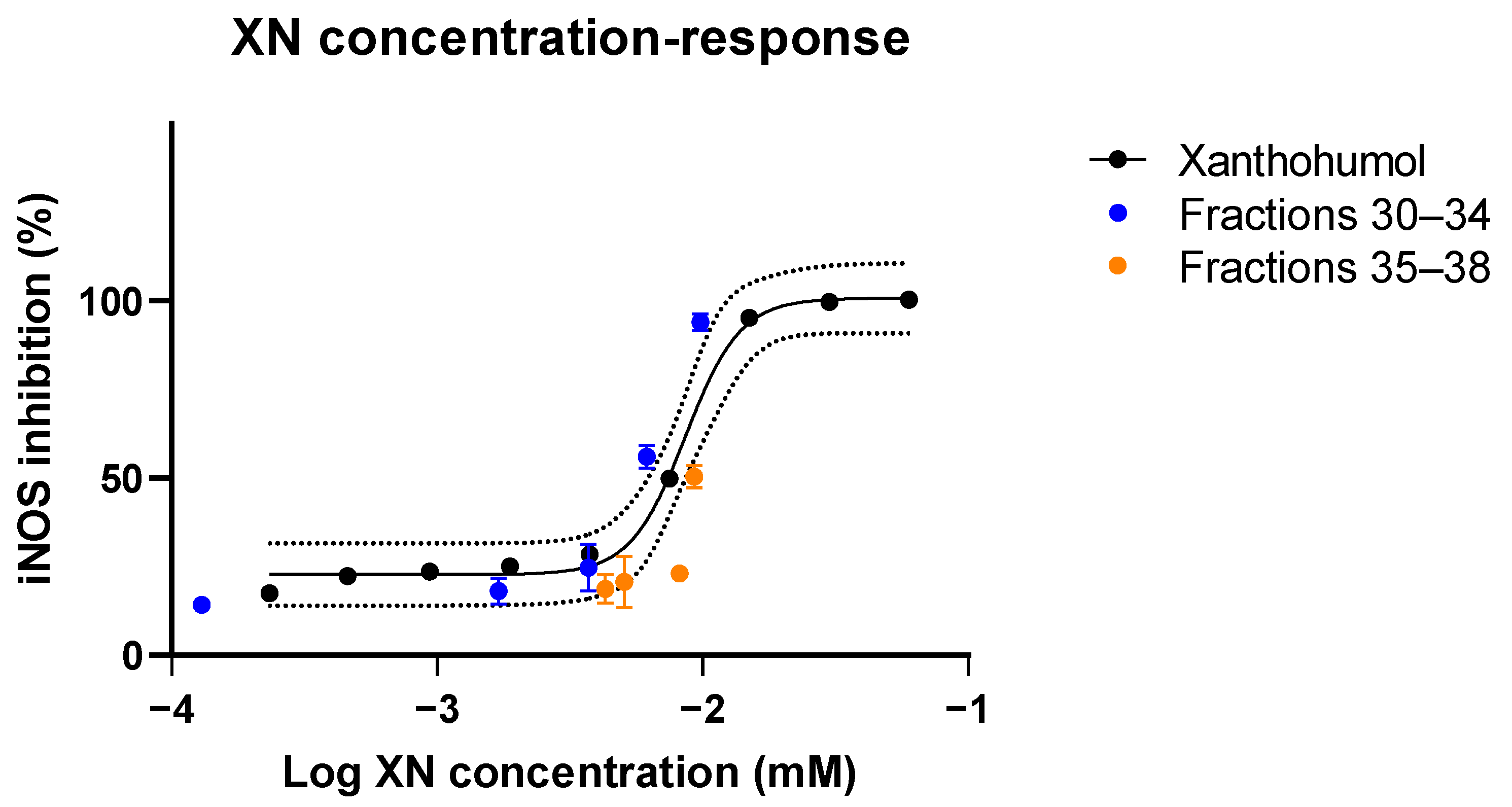
3.3.3. Validation Study
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stevens, J.F.; Revel, J. Xanthohumol, what a delightful problem child! In American Chemical Society Book: Chemistry and Biological Activities of Phenolic Compounds from Fruits and Vegetables. ACS Symposium Series, “Advances in Plant Phenolics: From Chemistry to Human Health”; ACS: Washington, DC, USA, 2018; Chapter 15; Volume 1286, pp. 283–304. [Google Scholar]
- Stevens, J.F. Xanthohumol and Structurally Related Prenylflavonoids for Cancer Chemoprevention and Control. In Natural Products for Cancer Chemoprevention; Springer: Cham, Switzerland, 2020; pp. 319–350. [Google Scholar]
- Stevens, J.F.; Taylor, A.W.; Deinzer, M.L. Quantitative analysis of xanthohumol and related prenylflavonoids in hops and beer by liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 1999, 832, 97–107. [Google Scholar] [CrossRef]
- Peluso, M.R.; Miranda, C.L.; Hobbs, D.J.; Proteau, R.R.; Stevens, J.F. Xanthohumol and related prenylated flavonoids inhibit inflammatory cytokine production in LPS-activated THP-1 monocytes: Structure-activity relationships and in silico binding to myeloid differentiation protein-2 (MD-2). Planta Med. 2010, 76, 1536–1543. [Google Scholar] [CrossRef]
- Miranda, C.L.; Elias, V.D.; Hay, J.J.; Choi, J.; Reed, R.L.; Stevens, J.F. Xanthohumol improves dysfunctional glucose and lipid metabolism in diet-induced obese C57BL/6J mice. Arch. Biochem. Biophys. 2016, 599, 22–30. [Google Scholar] [CrossRef] [Green Version]
- Miranda, C.L.; Johnson, L.A.; de Montgolfier, O.; Elias, V.D.; Ullrich, L.S.; Hay, J.J.; Paraiso, I.L.; Choi, J.; Reed, R.L.; Revel, J.S.; et al. Non-estrogenic Xanthohumol Derivatives Mitigate Insulin Resistance and Cognitive Impairment in High-Fat Diet-induced Obese Mice. Sci. Rep. 2018, 8, 613. [Google Scholar] [CrossRef] [Green Version]
- Wold, S.; Sjöström, M.; Eriksson, L. PLS-regression: A basic tool of chemometrics. Chemom. Intell. Lab. Syst. 2001, 58, 109–130. [Google Scholar] [CrossRef]
- Wold, H. Partial least squares. In Encyclopedia of Statistical Sciences; John Wiley & Sons: New York, NY, USA, 1985; pp. 581–591. [Google Scholar]
- Caesar, L.K.; Nogo, S.; Naphen, C.N.; Cech, N.B. Simplify: A Mass Spectrometry Metabolomics Approach to Identify Additives and Synergists from Complex Mixtures. Anal. Chem. 2019, 91, 11297–11305. [Google Scholar] [CrossRef]
- Kellogg, J.J.; Todd, D.A.; Egan, J.M.; Raja, H.A.; Oberlies, N.H.; Kvalheim, O.M.; Cech, N.B. Biochemometrics for Natural Products Research: Comparison of Data Analysis Approaches and Application to Identification of Bioactive Compounds. J. Nat. Prod. 2016, 79, 376–386. [Google Scholar] [CrossRef] [Green Version]
- Zou, H.; Hastie, T. Regularization and Variable Selection via the Elastic Net. J. Royal. Stat. Soc. B 2005, 67, 301–320. [Google Scholar] [CrossRef] [Green Version]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Colgate, E.C.; Miranda, C.L.; Stevens, J.F.; Bray, T.M.; Ho, E. Xanthohumol, a prenylflavonoid derived from hops induces apoptosis and inhibits NF-kappaB activation in prostate epithelial cells. Cancer Lett. 2007, 246, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Henderson, M.C.; Miranda, C.L.; Stevens, J.F.; Deinzer, M.L.; Buhler, D.R. In vitro inhibition of human P450 enzymes by prenylated flavonoids from hops, Humulus lupulus. Xenobiotica 2000, 30, 235–251. [Google Scholar] [CrossRef] [PubMed]
- Miranda, C.L.; Aponso, G.L.; Stevens, J.F.; Deinzer, M.L.; Buhler, D.R. Prenylated chalcones and flavanones as inducers of quinone reductase in mouse Hepa 1c1c7 cells. Cancer Lett. 2000, 149, 21–29. [Google Scholar] [CrossRef]
- Miranda, C.L.; Stevens, J.F.; Helmrich, A.; Henderson, M.C.; Rodriguez, R.J.; Yang, Y.H.; Deinzer, M.L.; Barnes, D.W.; Buhler, D.R. Antiproliferative and cytotoxic effects of prenylated flavonoids from hops (Humulus lupulus) in human cancer cell lines. Food Chem. Toxicol. 1999, 37, 271–285. [Google Scholar] [CrossRef]
- Kirkwood, J.S.; Legette, L.L.; Miranda, C.L.; Jiang, Y.; Stevens, J.F. A metabolomics-driven elucidation of the anti-obesity mechanisms of xanthohumol. J. Biol. Chem. 2013, 288, 19000–19013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legette, L.; Ma, L.; Reed, R.L.; Miranda, C.L.; Christensen, J.M.; Rodriguez-Proteau, R.; Stevens, J.F. Pharmacokinetics of xanthohumol and metabolites in rats after oral and intravenous administration. Mol. Nutr. Food Res. 2012, 56, 466–474. [Google Scholar] [CrossRef] [Green Version]
- Legette, L.L.; Luna, A.Y.; Reed, R.L.; Miranda, C.L.; Bobe, G.; Christensen, J.M.; Rodriguez-Proteau, R.; Stevens, J.F. Xanthohumol lowers body weight and fasting plasma glucose in obese male Zucker fa/fa rats. Phytochemistry 2013, 91, 236–241. [Google Scholar] [CrossRef]
- Legette, L.; Karnpracha, C.; Reed, R.L.; Choi, J.; Bobe, G.; Christensen, J.M.; Rodriguez-Proteau, R.; Purnell, J.Q.; Stevens, J.F. Human pharmacokinetics of xanthohumol, an antihyperglycemic flavonoid from hops. Mol. Nutr. Food Res. 2014, 58, 248–255. [Google Scholar] [CrossRef]
- Ellinwood, D.C.; El-Mansy, M.F.; Plagmann, L.S.; Stevens, J.F.; Maier, C.S.; Gombart, A.F.; Blakemore, P.R. Total synthesis of [(13) C]2-, [(13) C]3-, and [(13) C]5-isotopomers of xanthohumol, the principal prenylflavonoid from hops. J. Label. Compd. Radiopharm. 2017, 60, 639–648. [Google Scholar] [CrossRef]
- Kuhn, M. Building predictive models in R using the caret package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Liaw, A.; Wiener, M. Classification and regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- Olivon, F.; Grelier, G.; Roussi, F.; Litaudon, M.; Touboul, D. MZmine 2 Data-Preprocessing To Enhance Molecular Networking Reliability. Anal. Chem. 2017, 89, 7836–7840. [Google Scholar] [CrossRef] [PubMed]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Oresic, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanches, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [Green Version]
- Hevel, J.M.; Marietta, M.A. [25] Nitric-oxide synthase assays. Methods Enzymol. 1994, 233, 250–258. [Google Scholar] [PubMed]
- Donoho, D.L. High-dimensional data analysis: The curses and blessings of dimensionality. AMS Math. Chall. Lect. 2000, 1, 32. [Google Scholar]
- Hastie, T.; Tibshirani, R.; Friedman, J. The Elements of Statistical Learning: Data Mining, Inference, and Prediction, 2nd ed.; Springer: New York, NY, USA, 2016. [Google Scholar]
- Stone, M. Cross-validatory choice and assessment of statistical predictions. J. R. Stat. Soc. Series B Stat. Methodol 1974, 36, 111–133. [Google Scholar] [CrossRef]
- Fisher, R.A. Statistical Methods for Research Workers, 12th ed.; Oliver and Boyd: Edinburgh, UK; Scotland, UK, 1954. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A new and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat. Methodol. 1995, 57, 289–300. [Google Scholar]
- Hoerl, A.E.; Kennard, R.W. Ridge regression: Biased estimation for nonorthogonal problems. Technometrics 1970, 12, 55–67. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Series B Stat. Methodol. 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Olden, J.D.; Joy, M.K.; Death, R.G. An accurate comparison of methods for quantifying variable importance in artificial neural networks using simulated data. Ecol. Model. 2004, 178, 389–397. [Google Scholar] [CrossRef]
- Garson, D.G. Interpreting neural network connection weights. AI Expert 1991, 6, 47–51. [Google Scholar]
- Mehmood, T.; Sæbø, S.; Liland, K.H. Comparison of variable selection methods in partial least squares regression. J. Chemom. 2020, 34, e3226. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z. Ensemble Methods: Foundations and Algorithms. Chapman & Hall/CRC Machine Learning: Boca Raton, FL, USA, 2012. [Google Scholar]
- Stevens, J.F.; Ivancic, M.; Hsu, V.L.; Deinzer, M.L. Prenylflavonoids from Humulus lupulus. Phytochemistry 1997, 44, 1575–1585. [Google Scholar] [CrossRef]
- Stevens, J.F.; Taylor, A.W.; Nickerson, G.B.; Ivancic, M.; Henning, J.; Haunold, A.; Deinzer, M.L. Prenylflavonoid variation in Humulus lupulus: Distribution and taxonomic significance of xanthogalenol and 4′-O-methylxanthohumol. Phytochemistry 2000, 53, 759–775. [Google Scholar] [PubMed]
- Andrés-Iglesias, C.; Blanco, C.A.; Blanco, J.; Montero, O. Mass spectrometry-based metabolomics approach to determine differential metabolites between regular and non-alcohol beers. Food Chem. 2014, 157, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.Y.; Kao, T.-H. Evaluation of prenylflavonoids and hop bitter acids in surplus yeast. J. Food Sci. Technol. 2019, 56, 1939–1953. [Google Scholar] [CrossRef]
- Taniguchi, Y.; Matsukura, Y.; Taniguchi, H.; Koizumi, H.; Katayama, M. Development of preparative and analytical methods of the hop bitter acid oxide fraction and chemical properties of its components. Biosci. Biotechnol. Biochem. 2015, 79, 1684–1694. [Google Scholar] [CrossRef] [Green Version]
- Dresel, M.C.; Vogt, C.; Dunkel, A.; Hofmann, T. The bitter chemodiversity of hops (Humulus lupus L.). J. Agric. Food Chem. 2016, 64, 7789–7799. [Google Scholar] [CrossRef]
- Gagne, S.J.; Stout, J.M.; Liu, E.; Page, J.E. Identification of olivetolic acid cyclase from Cannabis sativa reveals a unique catalytic route to plant polyketides. Proc. Natl. Acad. Sci. USA 2012, 109, 12811–12816. [Google Scholar] [CrossRef] [Green Version]
- Gnanaprakasam, J.N.R.; Estrada-Muñiz, E.; Vega, L. The anacardic 6-pentadecyl salicylic acid induces macrophage activation via the phosphorylation of of ERK1/2, JNK, p38 kinases and NF-κB. Int. Immunopharmacol. 2015, 29, 808–817. [Google Scholar] [CrossRef]
- Eriksson, L.; Johansson, E.; Kettaneh-Wold, N.; Trygg, J.; Wikstrom, C.; Wold, S. Multi-and Megavariate Data Analysis Part I: Basic Principles and Applications; Umetrics: Umeå, Sweden, 2006. [Google Scholar]
- Rajalahti, T.; Arneberg, R.; Berven, F.S.; Myhr, K.-M.; Ulvik, R.J.; Kvalheim, O.M. Biomarker discovery in mass spectral profiles by means of selectivity ratio plot. Chemom. Intell. Lab. Syst. 2009, 95, 35–48. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
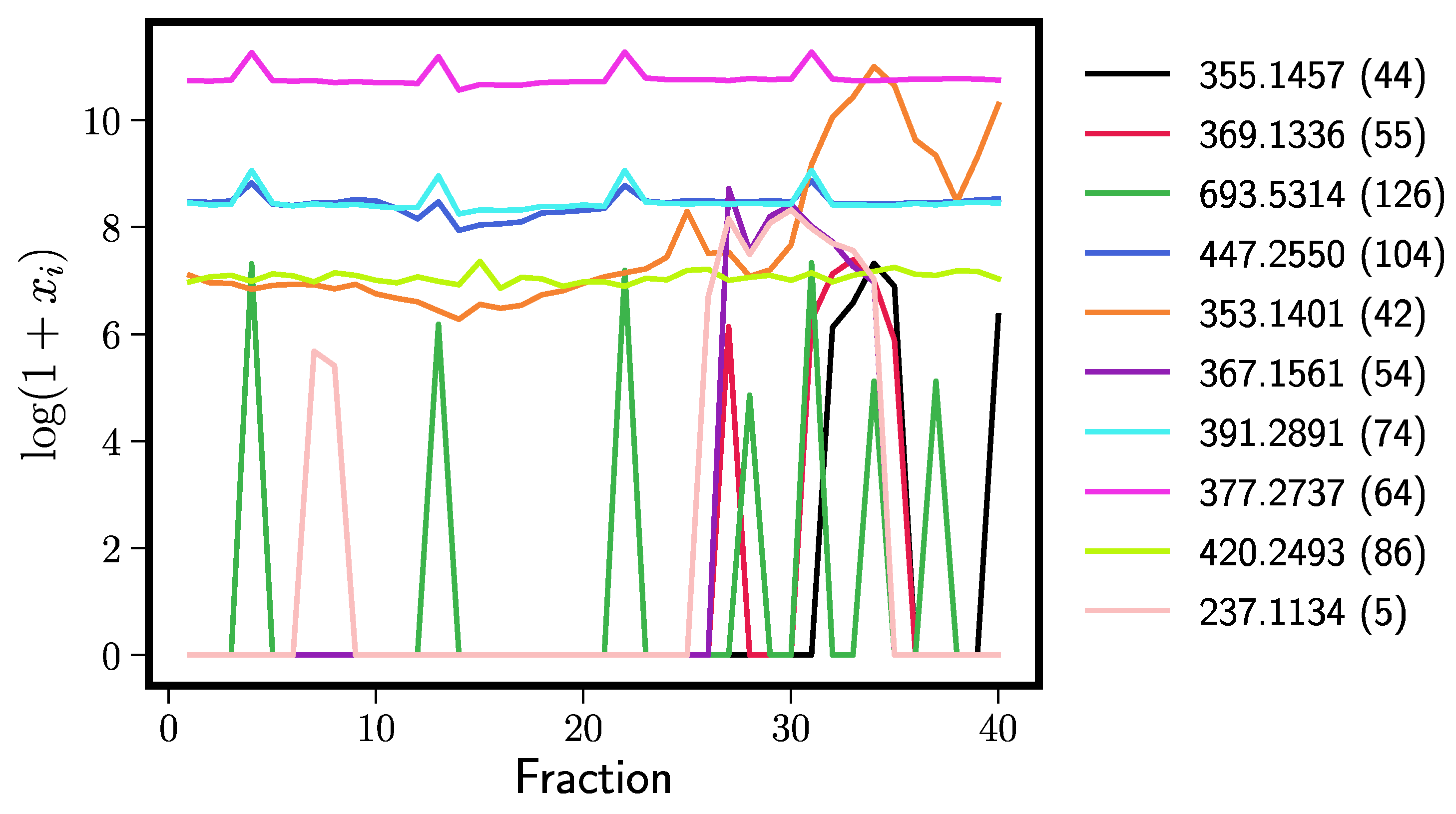
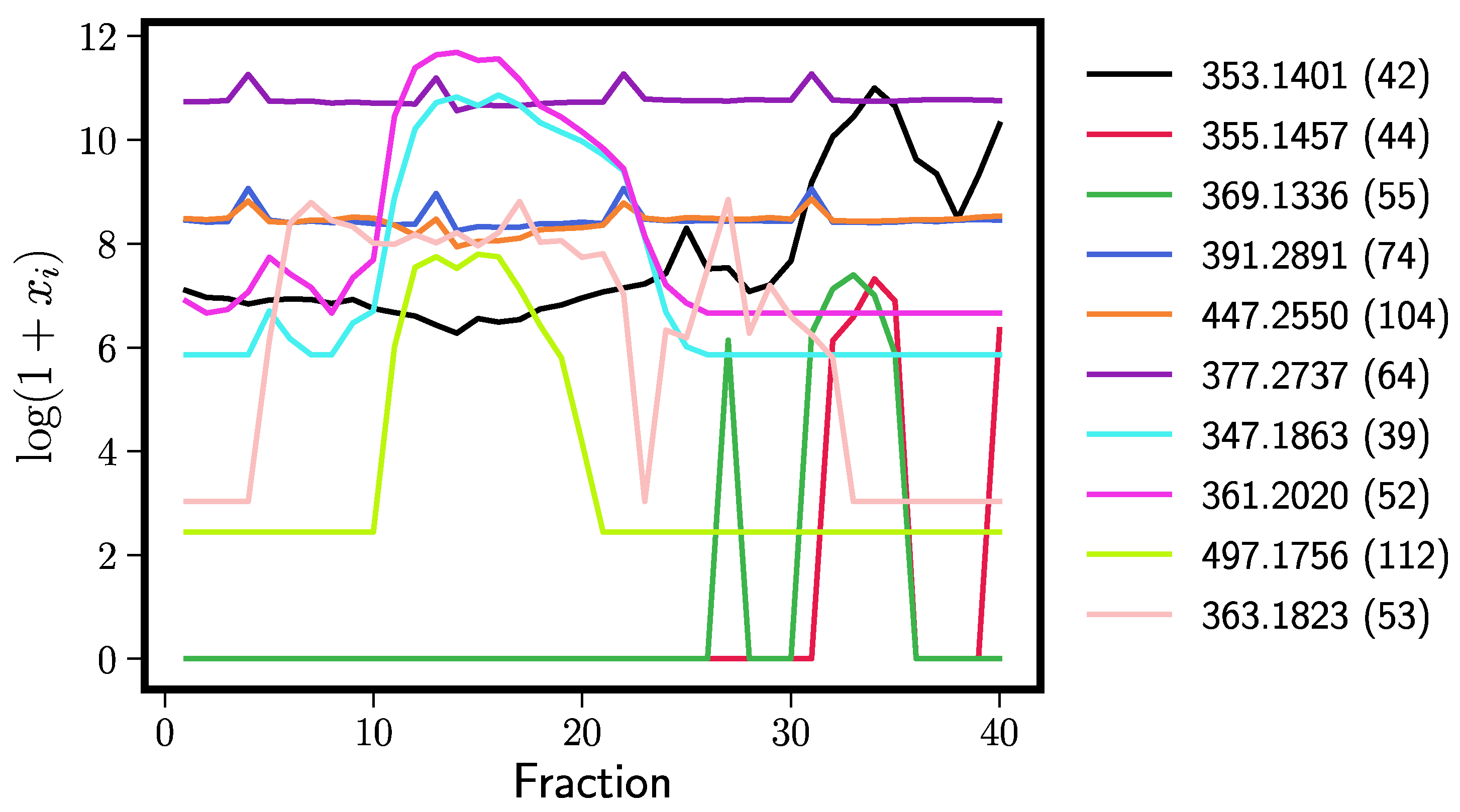
| Rank | Rt = 0.71, 250 Elastic Nets | Rt = 0.71, 250 Random Forests | ||
|---|---|---|---|---|
| m/z (Feature Tag) | Feature Assignment | m/z (Feature Tag) | ||
| 1 | 355.1457 (44) | dihydroxanthohumol | 353.1401 (42) | xanthohumol |
| 2 | 369.1336 (55) | xanthohumol B | 355.1457 (44) | dihydroxanthohumol |
| 3 | 693.5314 (126) | noise | 369.1336 (55) | xanthohumol B |
| 4 | 447.2550 (104) | noise | 391.2891 (74) | noise |
| 5 | 353.1401 (42) | xanthohumol | 447.2550 (104) | noise |
| 6 | 367.1561 (54) | 4′-O-methyl xanthohumol | 377.2737 (64) | noise |
| 7 | 391.2891 (74) | noise | 347.1863 (39) | cohumulone |
| 8 | 377.2737 (64) | noise | 361.2020 (52) | (ad)humulone |
| 9 | 420.2493 (86) | noise | 497.1756 (112) | (ad)humulone adduct/derivative |
| 10 | 237.1134 (5) | unknown | 363.1823 (53) | cohumulinone |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brown, K.S.; Jamieson, P.; Wu, W.; Vaswani, A.; Alcazar Magana, A.; Choi, J.; Mattio, L.M.; Cheong, P.H.-Y.; Nelson, D.; Reardon, P.N.; et al. Computation-Assisted Identification of Bioactive Compounds in Botanical Extracts: A Case Study of Anti-Inflammatory Natural Products from Hops. Antioxidants 2022, 11, 1400. https://doi.org/10.3390/antiox11071400
Brown KS, Jamieson P, Wu W, Vaswani A, Alcazar Magana A, Choi J, Mattio LM, Cheong PH-Y, Nelson D, Reardon PN, et al. Computation-Assisted Identification of Bioactive Compounds in Botanical Extracts: A Case Study of Anti-Inflammatory Natural Products from Hops. Antioxidants. 2022; 11(7):1400. https://doi.org/10.3390/antiox11071400
Chicago/Turabian StyleBrown, Kevin S., Paige Jamieson, Wenbin Wu, Ashish Vaswani, Armando Alcazar Magana, Jaewoo Choi, Luce M. Mattio, Paul Ha-Yeon Cheong, Dylan Nelson, Patrick N. Reardon, and et al. 2022. "Computation-Assisted Identification of Bioactive Compounds in Botanical Extracts: A Case Study of Anti-Inflammatory Natural Products from Hops" Antioxidants 11, no. 7: 1400. https://doi.org/10.3390/antiox11071400
APA StyleBrown, K. S., Jamieson, P., Wu, W., Vaswani, A., Alcazar Magana, A., Choi, J., Mattio, L. M., Cheong, P. H.-Y., Nelson, D., Reardon, P. N., Miranda, C. L., Maier, C. S., & Stevens, J. F. (2022). Computation-Assisted Identification of Bioactive Compounds in Botanical Extracts: A Case Study of Anti-Inflammatory Natural Products from Hops. Antioxidants, 11(7), 1400. https://doi.org/10.3390/antiox11071400