
Resistance to Obesity in SOD1 Deficient Mice with a High-Fat/High-Sucrose Diet

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal, Genotyping, and Diets
2.2. Tissue Preparation and Histology
2.3. Measurement of Metabolites
2.4. Islet of Pancreas and Beta Cell
2.5. Oxygen Consumption and Food Intake
2.6. Measurement of Skeletal Muscle and Vascular Superoxide Production
2.7. ATP Level and ADP/ATP Ratio of Skeletal Muscle
2.8. Western Blotting of Skeletal Muscle and Epididymal Fat
2.9. Statistical Analysis
3. Results
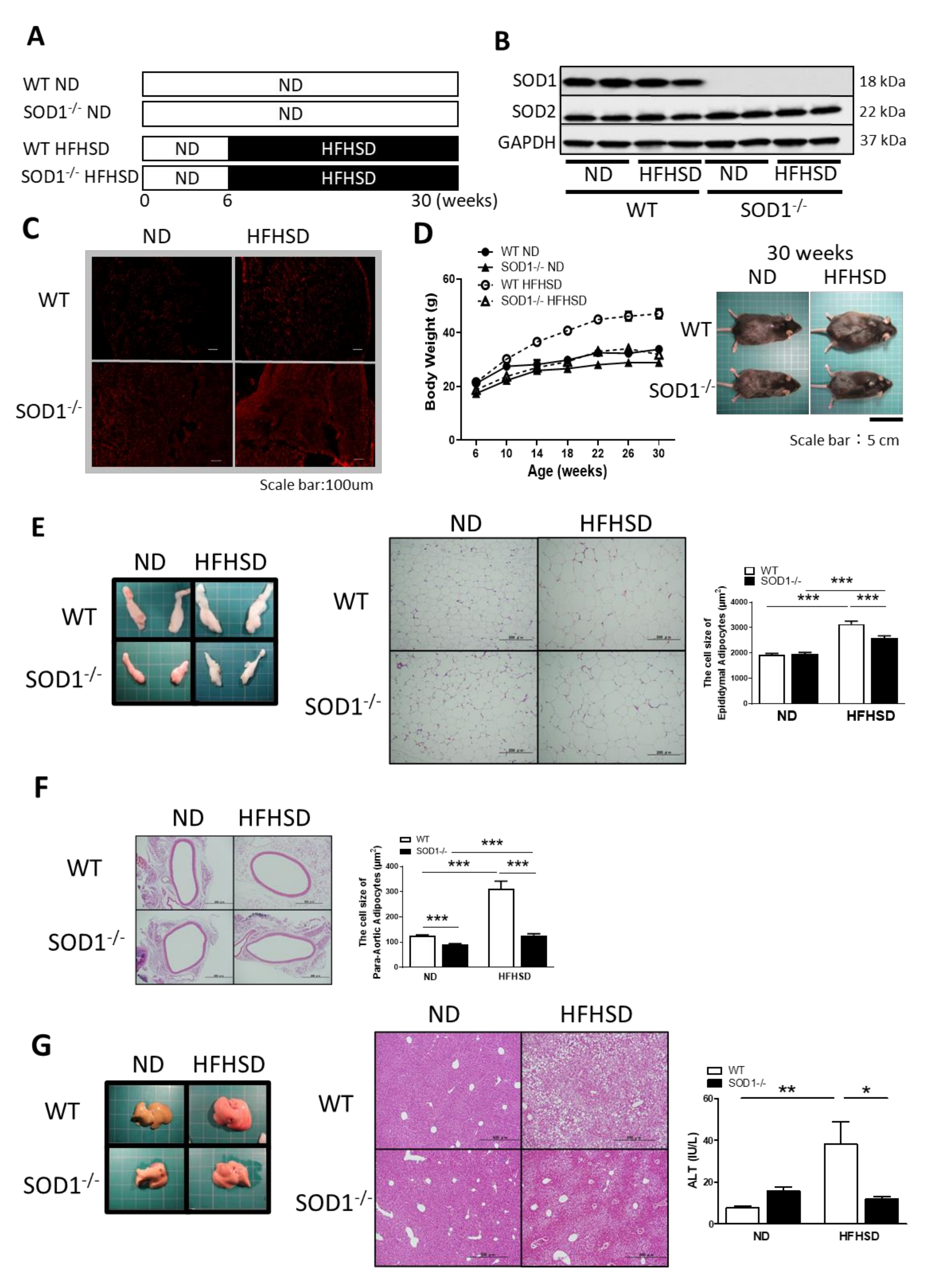
3.1. Study Design and Changes with ND or HFHSD
3.2. Blood Glucose and Insulin Levels
3.3. Weight of Pancreatic β-Cells
3.4. Oxygen Consumption and Intake of the Diet
3.5. ATP Levels and AMPK Expression in Skeletal Muscle
4. Discussion
4.1. Body Weight, Blood Glucose and Insulin Changes
4.2. Oxygen Consumption and AMPK Activation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reaven, G. The metabolic syndrome or the insulin resistance syndrome? Different names, different concepts, and different goals. Endocrinol. Metab. Clin. N. Am. 2004, 33, 283–303. [Google Scholar] [CrossRef] [PubMed]
- Eckel, R.; Grundy, S.; Zimmet, P. The metabolic syndrome. Lancet 2005, 365, 1415–1428. [Google Scholar] [CrossRef]
- Ando, K.; Fujita, T. Metabolic syndrome and oxidative stress. Free Radic. Biol. Med. 2009, 47, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Mahjoub, S.; Masrour-Roudsari, J. Role of oxidative stress in pathogenesis of metabolic syndrome. Caspian J. Intern. Med. 2012, 3, 386–396. [Google Scholar]
- Vincent, H.K.; Taylor, A.G. Biomarkers and potential mechanisms of obesity-induced oxidant stress in humans. Int. J. Obes. 2006, 30, 400–418. [Google Scholar] [CrossRef] [Green Version]
- Cross, C.E.; Halliwell, B.; Borish, E.T.; Pryor, W.A.; Ames, B.N.; Saul, R.L.; Harman, D. Oxygen Radicals and Human Disease. Ann. Intern. Med. 1987, 107, 526–545. [Google Scholar] [CrossRef]
- Finkel, T. Oxygen radicals and signaling. Curr. Opin. Cell Biol. 1998, 10, 248–253. [Google Scholar] [CrossRef]
- Hardie, D.G. AMP-activated protein kinase: An energy sensor that regulates all aspects of cell function. Genes Dev. 2011, 25, 1895–1908. [Google Scholar] [CrossRef] [Green Version]
- Ruderman, N.B.; Carling, D.; Prentki, M.; Cacicedo, J.M. AMPK, insulin resistance, and the metabolic syndrome. J. Clin. Investig. 2013, 123, 2764–2772. [Google Scholar] [CrossRef]
- Brunmair, B.; Staniek, K.; Gras, F.; Clara, R.; Roden, M.; Gnaiger, E.; Rnsinn, C.F. Thiazolidinediones, Like Metformin, Inhibit Respiratory Complex, I. Diabetes 2004, 53, 1052–1059. [Google Scholar] [CrossRef] [Green Version]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [Green Version]
- Shiraishi, Y.; Ishigami, N.; Kujiraoka, T.; Sato, A.; Fujita, M.; Ido, Y.; Adachi, T. Deletion of Superoxide Dismutase 1 Blunted Inflammatory Aortic Remodeling in Hypertensive Mice under Angiotensin II Infusion. Antioxidants 2021, 10, 471. [Google Scholar] [CrossRef]
- Hinchy, E.C.; Gruszczyk, A.V.; Willows, R.; Navaratnam, N.; Hall, A.R.; Bates, G.; Bright, T.P.; Krieg, T.; Carling, D.; Murphy, M.P. Mitochondria-derived ROS activate AMP-activated protein kinase (AMPK) indirectly. J. Biol. Chem. 2018, 293, 17208–17217. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, A.J.; Ellacott, K.L.; King, V.L.; Hasty, A.H. Mouse models of the metabolic syndrome. Dis. Model. Mech. 2010, 3, 156–166. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.K.; Chin, K.Y.; Suhaimi, F.H.; Fairus, A.; Ima-Nirwana, S. Animal models of metabolic syndrome: A review. Nutr. Metab. 2016, 13, 65. [Google Scholar] [CrossRef] [Green Version]
- Ishigami, N.; Isoda, K.; Adachi, T.; Niida, T.; Kujiraoka, T.; Hakuno, D.; Kondo, H.; Kusuhara, M.; Ohsuzu, F. Deficiency of CuZn superoxide dismutase promotes inflammation and alters medial structure following vascular injury. J. Atheroscler. Thromb. 2011, 18, 1009–1017. [Google Scholar] [CrossRef] [Green Version]
- Saisho, Y.; Manesso, E.; Butler, A.E.; Galasso, R.; Kavanagh, K.; Flynn, M.; Zhang, L.; Clark, P.; Gurlo, T.; Toffolo, G.M.; et al. Ongoing b-Cell Turnover in Adult NonhumanPrimates Is Not Adaptively Increased inStreptozotocin-Induced Diabetes. Diabetes 2011, 60, 848–856. [Google Scholar] [CrossRef] [Green Version]
- Butler, A.E.; Campbell-Thompson, M.; Gurlo, T.; Dawson, D.W.; Atkinson, M.; Butler, P.C. Marked Expansion of Exocrine and Endocrine Pancreas With Incretin Therapy in Humans With Increased Exocrine Pancreas Dysplasia and the Potential for Glucagon-Producing Neuroendocrine Tumors. Diabetes 2013, 62, 2595–2604. [Google Scholar] [CrossRef] [Green Version]
- Kujiraoka, T.; Satoh, Y.; Ayaori, M.; Shiraishi, Y.; Arai-Nakaya, Y.; Hakuno, D.; Yada, H.; Kuwada, N.; Endo, S.; Isoda, K.; et al. Hepatic extracellular signal-regulated kinase 2 suppresses endoplasmic reticulum stress and protects from oxidative stress and endothelial dysfunction. J. Am. Heart Assoc. 2013, 2, e000361. [Google Scholar] [CrossRef] [Green Version]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Huang, S.; Czech, M.P. The GLUT4 glucose transporter. Cell Metab. 2007, 5, 237–252. [Google Scholar] [CrossRef] [Green Version]
- Hall, J.C.; Fisher, K.C.; Stern, J.R. Stimulation of oxygen consumption by insulin in intact isolated frog muscle. Am. J. Physiol. 1954, 179, 29–35. [Google Scholar] [CrossRef]
- Brown, J.D.; Naples, S.P.; Booth, F.W. Effects of voluntary running on oxygen consumption, RQ, and energy expenditure during primary prevention of diet-induced obesity in C57BL/6N mice. J. Appl. Physiol. 2012, 113, 473–478. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Fink, B.D.; Herlein, J.A.; Oltman, C.L.; Lamping, K.G.; Sivitz, W.I. Dietary fat, fatty acid saturation and mitochondrial bioenergetics. J. Bioenerg. Biomembr. 2014, 46, 33–44. [Google Scholar] [CrossRef] [Green Version]
- Marvyn, P.M.; Bradley, R.M.; Mardian, E.B.; Marks, K.A.; Duncan, R.E. Data on oxygen consumption rate, respiratory exchange ratio, and movement in C57BL/6J female mice on the third day of consuming a high-fat diet. Data Brief 2016, 7, 472–475. [Google Scholar] [CrossRef]
- Auberval, N.; Dal, S.; Bietiger, W.; Pinget, M.; Jeandidier, N.; Maillard-Pedracini, E.; Schini-Kerth, V.; Sigrist, S. Metabolic and oxidative stress markers in Wistar rats after 2 months on a high-fat diet. Diabetol. Metab. Syndr. 2014, 6, 130. [Google Scholar] [CrossRef] [Green Version]
- Frikke-Schmidt, H.; Pedersen, T.; Fledelius, C.; Olsen, G.S.; Hellerstein, M. Adipose weight gain during chronic insulin treatment of mice results from changes in lipid storage without affecting de novo synthesis of palmitate. PLoS ONE 2013, 8, e76060. [Google Scholar] [CrossRef]
- Russell-Jones, D.; Khan, R. Insulin-associated weight gain in diabetes--causes, effects and coping strategies. Diabetes Obes. Metab. 2007, 9, 799–812. [Google Scholar] [CrossRef]
- Sagi, H.; Shibuya, S.; Kato, T.; Nakanishi, Y.; Tsuboi, A.; Moriya, S.; Ohno, H.; Miyamoto, H.; Kodama, H.; Shimizu, T. SOD1 deficiency alters gastrointestinal microbiota and metabolites in mice. Exp. Gerontol. 2020, 130, 110795. [Google Scholar] [CrossRef]
- Deepa, S.S.; Van Remmen, H.; Brooks, S.V.; Faulkner, J.A.; Larkin, L.; McArdle, A.; Jackson, M.J.; Vasilaki, A.; Richardson, A. Accelerated sarcopenia in Cu/Zn superoxide dismutase knockout mice. Free Radic. Biol. Med. 2019, 132, 19–23. [Google Scholar] [CrossRef]
- Morgan, B.J.; Chai, S.Y.; Albiston, A.L. GLUT4 Associated Proteins as Therapeutic Targets for Diabetes. Recent Pat. Endocr. Metab. Immune Drug Discov. 2011, 5, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Takaguri, A.; Inoue, S.; Kubo, T.; Satoh, K. AMPK activation by prolonged stimulation with interleukin-1beta contributes to the promotion of GLUT4 translocation in skeletal muscle cells. Cell Biol. Int. 2016, 40, 1204–1211. [Google Scholar] [CrossRef] [PubMed]
- Krauss, S.; Zhang, C.Y.; Scorrano, L.; Dalgaard, L.T.; St-Pierre, J.; Grey, S.T.; Lowell, B.B. Superoxide-mediated activation of uncoupling protein 2 causes pancreatic beta cell dysfunction. J. Clin. Investig. 2003, 112, 1831–1842. [Google Scholar] [CrossRef]
- Sakai, K.; Matsumoto, K.; Nishikawa, T.; Suefuji, M.; Nakamaru, K.; Hirashima, Y.; Kawashima, J.; Shirotani, T.; Ichinose, K.; Brownlee, M.; et al. Mitochondrial reactive oxygen species reduce insulin secretion by pancreatic b-cells. Biochem. Biophys. Res. Commun. 2003, 300, 216–222. [Google Scholar] [CrossRef]
- Bloch-Damti, A.; Bashan, N. Proposed mechanisms for the induction of insulin resistance by oxidative stress. Antioxid. Redox Signal. 2005, 7, 1553–1567. [Google Scholar] [CrossRef]
- Teratani, T.; Tomita, K.; Toma-Fukai, S.; Nakamura, Y.; Itoh, T.; Shimizu, H.; Shiraishi, Y.; Sugihara, N.; Higashiyama, M.; Shimizu, T.; et al. Redox-dependent PPARγ/Tnpo1 complex formation enhances PPARγ nuclear localization and signaling. Free Radic. Biol. Med. 2020, 156, 45–56. [Google Scholar] [CrossRef]
- Zmijewski, J.W.; Banerjee, S.; Bae, H.; Friggeri, A.; Lazarowski, E.R.; Abraham, E. Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J. Biol. Chem. 2010, 285, 33154–33164. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.; Oka, S.; Liu, T.; Zhai, P.; Ago, T.; Sciarretta, S.; Li, H.; Sadoshima, J. A redox-dependent mechanism for regulation of AMPK activation by Thioredoxin1 during energy starvation. Cell Metab. 2014, 19, 232–245. [Google Scholar] [CrossRef] [Green Version]
- Turrens, J.F.; Boveris, A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem. J. 1980, 191, 421–427. [Google Scholar]
- Cadenas, E.; Boveris, A.; Ragan, C.I.; Stoppani, A.O. Production of superoxide radicals and hydrogen peroxide by NADH-ubiquinone reductase and ubiquinol-cytochrome c reductase from beef-heart mitochondria. Arch. Biochem. Biophys. 1977, 180, 248–257. [Google Scholar] [CrossRef]
- Muller, F.L.; Liu, Y.; Van Remmen, H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 2004, 279, 49064–49073. [Google Scholar] [CrossRef] [Green Version]
- Echtay, K.S.; Roussel, D.; St-Pierre, J.; Jekabsons, M.B.; Cadenas, S.; Stuart, J.A.; Harper, J.A.; Roebuck, S.J.; Morrison, A.; Pickering, S.; et al. Superoxide activates mitochondrial uncoupling proteins. Nature 2002, 415, 96–99. [Google Scholar] [CrossRef]
- Zhang, C.Y.; Baffy, G.; Perret, P.; Krauss, S.; Peroni, O.; Grujic, D.; Hagen, T.; Vidal-Puig, A.J.; Boss, O.; Kim, Y.B.; et al. Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell 2001, 105, 745–755. [Google Scholar] [CrossRef] [Green Version]
- Kaludercic, N.; Giorgio, V. The Dual Function of Reactive Oxygen/Nitrogen Species in Bioenergetics and Cell Death: The Role of ATP Synthase. Oxidative Med. Cell. Longev. 2016, 2016, 3869610. [Google Scholar] [CrossRef] [Green Version]
- Winder, W.W.; Hardie, D.G. Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. Am. J. Physiol. 1996, 2, E299–E304. [Google Scholar] [CrossRef] [Green Version]
- Barnes, K.; Ingram, J.C.; Porras, O.H.; Barros, L.F.; Hudson, E.R.; Fryer, L.G.; Baldwin, S.A. Activation of GLUT1 by metabolic and osmotic stress: Potential involvement of AMP-activated protein kinase (AMPK). J. Cell Sci. 2002, 115, 2433–2442. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Normal Diet | High-Fat/High-Sucrose Diet (F2HFHSD) | |||
|---|---|---|---|---|
| (CE-7) | ||||
| g/100 g | kcal, % | g/100 g | kcal, % | |
| Total calories | 343 kcal | 481 kcal | ||
| Protein | 17.7 g | 20.6 | 20.7 g | 17.2 |
| Fat | 3.8 g | 10 | 29.1 g | 54.5 |
| Carbohydrate | 59.4 g | 69.4 | 34.0 g | 28.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sato, A.; Shiraishi, Y.; Kimura, T.; Osaki, A.; Kagami, K.; Ido, Y.; Adachi, T. Resistance to Obesity in SOD1 Deficient Mice with a High-Fat/High-Sucrose Diet. Antioxidants 2022, 11, 1403. https://doi.org/10.3390/antiox11071403
Sato A, Shiraishi Y, Kimura T, Osaki A, Kagami K, Ido Y, Adachi T. Resistance to Obesity in SOD1 Deficient Mice with a High-Fat/High-Sucrose Diet. Antioxidants. 2022; 11(7):1403. https://doi.org/10.3390/antiox11071403
Chicago/Turabian StyleSato, Atsushi, Yasunaga Shiraishi, Toyokazu Kimura, Ayumu Osaki, Kazuki Kagami, Yasuo Ido, and Takeshi Adachi. 2022. "Resistance to Obesity in SOD1 Deficient Mice with a High-Fat/High-Sucrose Diet" Antioxidants 11, no. 7: 1403. https://doi.org/10.3390/antiox11071403