
NRF2 Mediates Cellular Resistance to Transformation, Radiation, and Inflammation in Mice

 ,
, 
Abstract
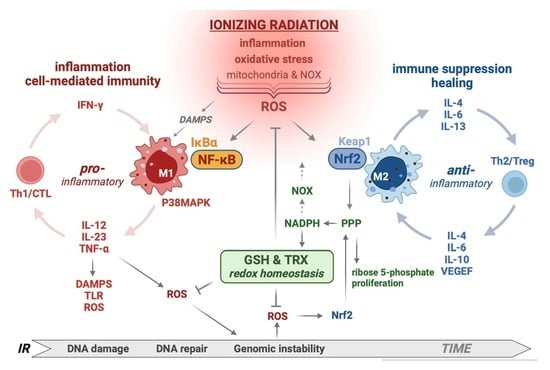
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture, In Vitro and In Vivo Irradiation
2.2. Foci Formation Assay
2.3. NF-κB Reporter Gene Assay
2.4. γ-H2AX Assay
2.5. Immune Profiling
2.6. B16-OVA Tumor Model and Enzyme-Linked Immunospot (ELISPOT) Assay
3. Results
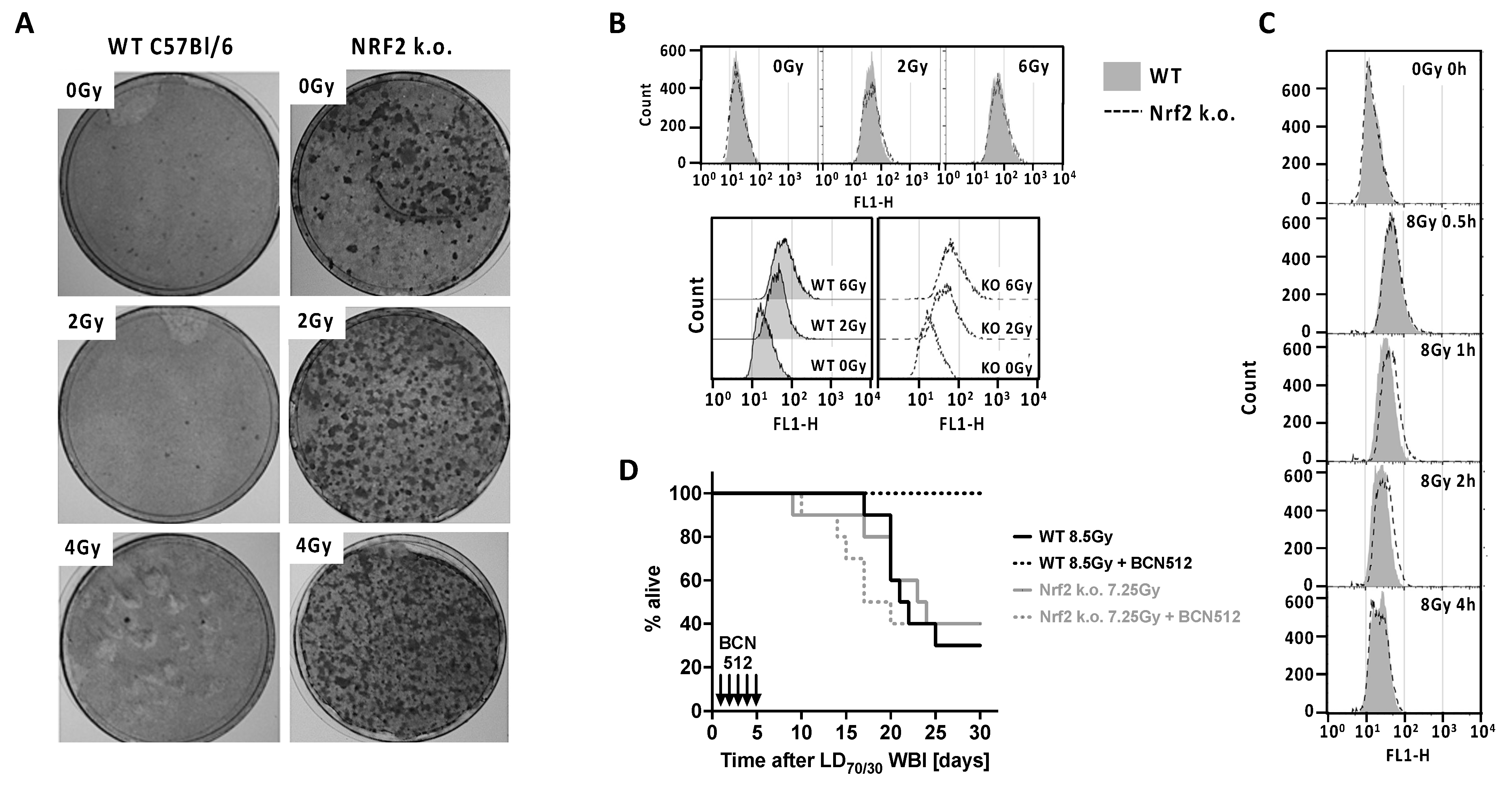
3.1. Nrf2 and IR Exposure
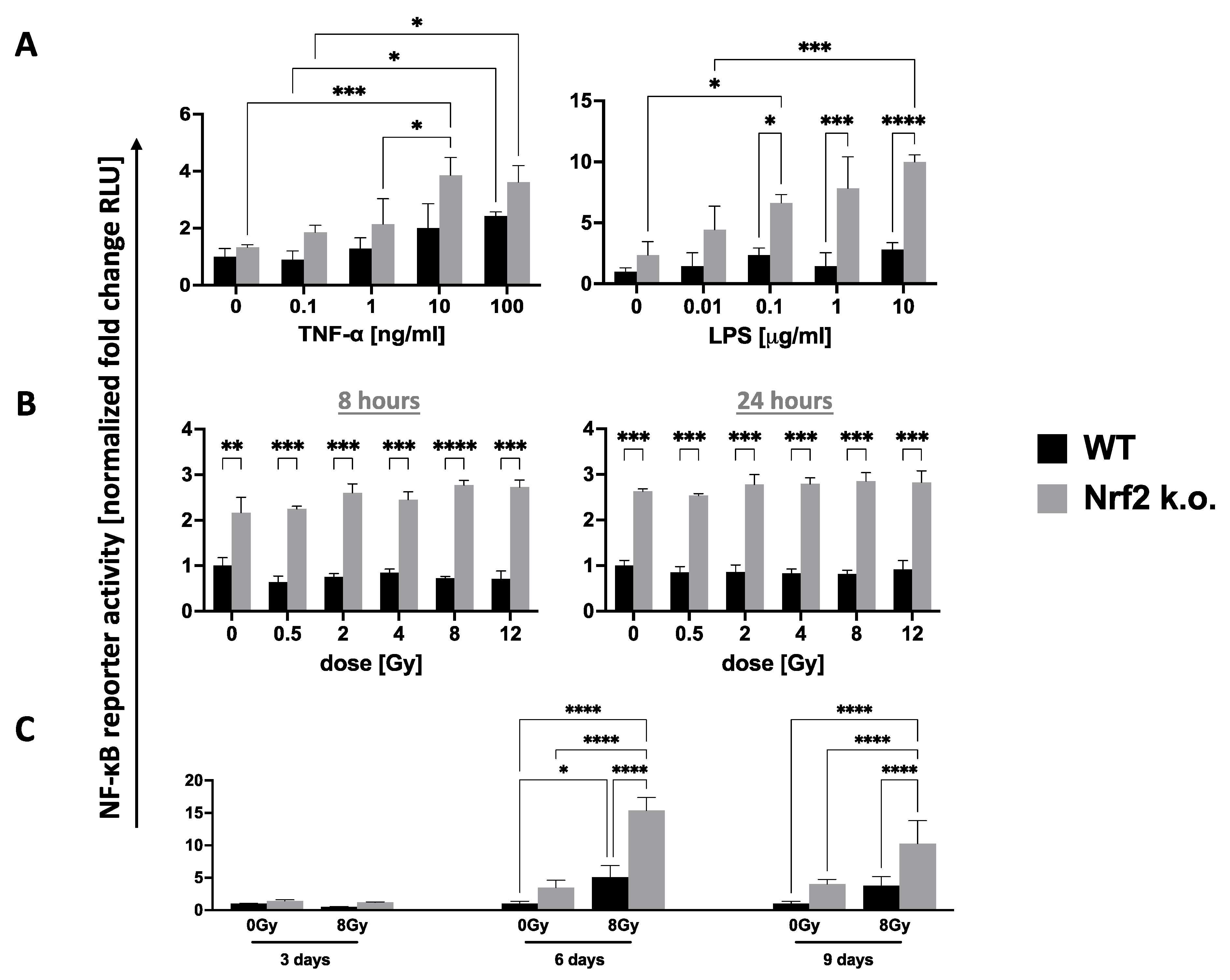
3.2. Nrf2 Control of Inflammation and Immunity
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef] [PubMed]
- Schaue, D.; Micewicz, E.D.; Ratikan, J.A.; Xie, M.W.; Cheng, G.; McBride, W.H. Radiation and inflammation. Semin. Radiat. Oncol. 2015, 25, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Cameron, B.D.; Sekhar, K.R.; Ofori, M.; Freeman, M.L. The Role of Nrf2 in the Response to Normal Tissue Radiation Injury. Radiat. Res. 2018, 190, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Acharya, A.; Das, I.; Chandhok, D.; Saha, T. Redox regulation in cancer: A double-edged sword with therapeutic potential. Oxid. Med. Cell Longev. 2010, 3, 23–34. [Google Scholar] [CrossRef]
- Juge, N.; Mithen, R.F.; Traka, M. Molecular basis for chemoprevention by sulforaphane: A comprehensive review. Cell Mol. Life Sci. 2007, 64, 1105–1127. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Raghunath, A.; Sundarraj, K.; Nagarajan, R.; Arfuso, F.; Bian, J.; Kumar, A.P.; Sethi, G.; Perumal, E. Antioxidant response elements: Discovery, classes, regulation and potential applications. Redox Biol. 2018, 17, 297–314. [Google Scholar] [CrossRef]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Gordan, J.D.; Jin, J.; Harper, J.W.; Diehl, J.A. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: Oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell Biol. 2004, 24, 8477–8486. [Google Scholar] [CrossRef] [PubMed]
- Kopacz, A.; Kloska, D.; Forman, H.J.; Jozkowicz, A.; Grochot-Przeczek, A. Beyond repression of Nrf2: An update on Keap1. Free Radic. Biol. Med. 2020, 157, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Yamamoto, M. Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv. Enzyme Regul. 2006, 46, 113–140. [Google Scholar] [CrossRef]
- DeBlasi, J.M.; DeNicola, G.M. Dissecting the Crosstalk between NRF2 Signaling and Metabolic Processes in Cancer. Cancers 2020, 12, 3023. [Google Scholar] [CrossRef]
- Harder, B.; Jiang, T.; Wu, T.; Tao, S.; Rojo de la Vega, M.; Tian, W.; Chapman, E.; Zhang, D.D. Molecular mechanisms of Nrf2 regulation and how these influence chemical modulation for disease intervention. Biochem. Soc. Trans. 2015, 43, 680–686. [Google Scholar] [CrossRef]
- Liu, K.; Singer, E.; Cohn, W.; Micewicz, E.D.; McBride, W.H.; Whitelegge, J.P.; Loo, J.A. Time-Dependent Measurement of Nrf2-Regulated Antioxidant Response to Ionizing Radiation Toward Identifying Potential Protein Biomarkers for Acute Radiation Injury. Proteom. Clin. Appl. 2019, 13, e1900035. [Google Scholar] [CrossRef]
- Wakabayashi, N.; Itoh, K.; Wakabayashi, J.; Motohashi, H.; Noda, S.; Takahashi, S.; Imakado, S.; Kotsuji, T.; Otsuka, F.; Roop, D.R.; et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat. Genet. 2003, 35, 238–245. [Google Scholar] [CrossRef]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Nilsa, R.D.; Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef]
- Gutierrez-Cuevas, J.; Galicia-Moreno, M.; Monroy-Ramirez, H.C.; Sandoval-Rodriguez, A.; Garcia-Banuelos, J.; Santos, A.; Armendariz-Borunda, J. The Role of NRF2 in Obesity-Associated Cardiovascular Risk Factors. Antioxidants 2022, 11, 235. [Google Scholar] [CrossRef]
- Cykowiak, M.; Krajka-Kuzniak, V. Role of Nrf2 in Pancreatic Cancer. Antioxidants 2021, 11, 98. [Google Scholar] [CrossRef]
- Hamada, S.; Matsumoto, R.; Masamune, A. HIF-1 and NRF2; Key Molecules for Malignant Phenotypes of Pancreatic Cancer. Cancers 2022, 14, 411. [Google Scholar] [CrossRef]
- Jiang, T.; Chen, N.; Zhao, F.; Wang, X.J.; Kong, B.; Zheng, W.; Zhang, D.D. High levels of Nrf2 determine chemoresistance in type II endometrial cancer. Cancer Res. 2010, 70, 5486–5496. [Google Scholar] [CrossRef]
- Tanaka, Y.; Hamada, S.; Matsumoto, R.; Taguchi, K.; Yamamoto, M.; Masamune, A. Nrf2 expression in pancreatic stellate cells promotes progression of cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 321, G378–G388. [Google Scholar] [CrossRef]
- Tao, S.; Wang, S.; Moghaddam, S.J.; Ooi, A.; Chapman, E.; Wong, P.K.; Zhang, D.D. Oncogenic KRAS confers chemoresistance by upregulating NRF2. Cancer Res. 2014, 74, 7430–7441. [Google Scholar] [CrossRef]
- Matsuoka, Y.; Yoshida, R.; Kawahara, K.; Sakata, J.; Arita, H.; Nkashima, H.; Takahashi, N.; Hirayama, M.; Nagata, M.; Hirosue, A.; et al. The antioxidative stress regulator Nrf2 potentiates radioresistance of oral squamous cell carcinoma accompanied with metabolic modulation. Lab. Investig. 2022, 102, 896–907. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef]
- Clinical Lung Cancer Genome Project; Network Genomic Medicine. A genomics-based classification of human lung tumors. Sci. Transl. Med. 2013, 5, 209ra153. [Google Scholar]
- Malhotra, D.; Portales-Casamar, E.; Singh, A.; Srivastava, S.; Arenillas, D.; Happel, C.; Shyr, C.; Wakabayashi, N.; Kensler, T.W.; Wasserman, W.W.; et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010, 38, 5718–5734. [Google Scholar] [CrossRef]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef]
- McBride, W.H.; Dougherty, G.J. Radiotherapy for genes that cause cancer. Nat. Med. 1995, 1, 1215–1217. [Google Scholar] [CrossRef]
- Boutten, A.; Goven, D.; Boczkowski, J.; Bonay, M. Oxidative stress targets in pulmonary emphysema: Focus on the Nrf2 pathway. Expert Opin. Ther. Targets 2010, 14, 329–346. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol. Sci. 2011, 123, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Gray, G.R.; Stamatoyannopoulos, G.; Naiman, S.C.; Kliman, M.R.; Klebanoff, S.J.; Austin, T.; Yoshida, A.; Robinson, G.C. Neutrophil dysfunction, chronic granulomatous disease, and non-spherocytic haemolytic anaemia caused by complete deficiency of glucose-6-phosphate dehydrogenase. Lancet 1973, 2, 530–534. [Google Scholar] [CrossRef]
- Azevedo, E.P.; Rochael, N.C.; Guimaraes-Costa, A.B.; de Souza-Vieira, T.S.; Ganilho, J.; Saraiva, E.M.; Palhano, F.L.; Foguel, D. A Metabolic Shift toward Pentose Phosphate Pathway Is Necessary for Amyloid Fibril- and Phorbol 12-Myristate 13-Acetate-induced Neutrophil Extracellular Trap (NET) Formation. J. Biol. Chem. 2015, 290, 22174–22183. [Google Scholar] [CrossRef] [PubMed]
- Kovac, S.; Angelova, P.R.; Holmstrom, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta 2015, 1850, 794–801. [Google Scholar] [CrossRef] [PubMed]
- Sakai, Y.; Yamamori, T.; Yoshikawa, Y.; Bo, T.; Suzuki, M.; Yamamoto, K.; Ago, T.; Inanami, O. NADPH oxidase 4 mediates ROS production in radiation-induced senescent cells and promotes migration of inflammatory cells. Free Radic. Res. 2018, 52, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Ameziane El Hassani, R.; Buffet, C.; Leboulleux, S.; Dupuy, C. Oxidative stress in thyroid carcinomas: Biological and clinical significance. Endocr. Relat. Cancer 2019, 26, R131–R143. [Google Scholar] [CrossRef]
- Najafi, M.; Shirazi, A.; Motevaseli, E.; Geraily, G.; Amini, P.; Shabeeb, D.; Eleojo Musa, A. Evaluating the Expression of NOX2 and NOX4 Signaling Pathways in Rats’ Lung Tissues Following Local Chest Irradiation; Modulatory Effect of Melatonin. Int. J. Mol. Cell Med. 2018, 7, 220–225. [Google Scholar] [PubMed]
- Owusu, S.B.; Hudik, E.; Ferard, C.; Dupre-Crochet, S.; Addison, E.; Preko, K.; Bizouarn, T.; Houee-Levin, C.; Baciou, L. Radiation-induced reactive oxygen species partially assemble neutrophil NADPH oxidase. Free Radic. Biol. Med. 2021, 164, 76–84. [Google Scholar] [CrossRef]
- McDonald, J.T.; Kim, K.; Norris, A.J.; Vlashi, E.; Phillips, T.M.; Lagadec, C.; Della Donna, L.; Ratikan, J.; Szelag, H.; Hlatky, L.; et al. Ionizing radiation activates the Nrf2 antioxidant response. Cancer Res. 2010, 70, 8886–8895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuttle, S.; Stamato, T.; Perez, M.L.; Biaglow, J. Glucose-6-phosphate dehydrogenase and the oxidative pentose phosphate cycle protect cells against apoptosis induced by low doses of ionizing radiation. Radiat. Res. 2000, 153, 781–787. [Google Scholar] [CrossRef]
- Tuttle, S.W.; Varnes, M.E.; Mitchell, J.B.; Biaglow, J.E. Sensitivity to chemical oxidants and radiation in CHO cell lines deficient in oxidative pentose cycle activity. Int. J. Radiat. Oncol. Biol. Phys. 1992, 22, 671–675. [Google Scholar] [CrossRef]
- Kim, J.H.; Thimmulappa, R.K.; Kumar, V.; Cui, W.; Kumar, S.; Kombairaju, P.; Zhang, H.; Margolick, J.; Matsui, W.; Macvittie, T.; et al. NRF2-mediated Notch pathway activation enhances hematopoietic reconstitution following myelosuppressive radiation. J. Clin. Investig. 2014, 124, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Takahashi, K.; Monzen, S.; Yamamoto, H.; Maruyama, A.; Itoh, K.; Kashiwakura, I. Relationship between radiosensitivity and Nrf2 target gene expression in human hematopoietic stem cells. Radiat. Res. 2010, 174, 177–184. [Google Scholar] [CrossRef]
- Micewicz, E.D.; Damoiseaux, R.D.; Deng, G.; Gomez, A.; Iwamoto, K.S.; Jung, M.E.; Nguyen, C.; Norris, A.J.; Ratikan, J.A.; Ruchala, P.; et al. Classes of Drugs that Mitigate Radiation Syndromes. Front. Pharmacol. 2021, 12, 666776. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Hayes, J.D.; Wolf, C.R. Generation of a stable antioxidant response element-driven reporter gene cell line and its use to show redox-dependent activation of nrf2 by cancer chemotherapeutic agents. Cancer Res. 2006, 66, 10983–10994. [Google Scholar] [CrossRef]
- Chen, X.L.; Dodd, G.; Thomas, S.; Zhang, X.; Wasserman, M.A.; Rovin, B.H.; Kunsch, C. Activation of Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1862–H1870. [Google Scholar] [CrossRef]
- Kong, X.; Thimmulappa, R.; Kombairaju, P.; Biswal, S. NADPH oxidase-dependent reactive oxygen species mediate amplified TLR4 signaling and sepsis-induced mortality in Nrf2-deficient mice. J. Immunol. 2010, 185, 569–577. [Google Scholar] [CrossRef]
- Thimmulappa, R.K.; Scollick, C.; Traore, K.; Yates, M.; Trush, M.A.; Liby, K.T.; Sporn, M.B.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2-dependent protection from LPS induced inflammatory response and mortality by CDDO-Imidazolide. Biochem. Biophys. Res. Commun. 2006, 351, 883–889. [Google Scholar] [CrossRef]
- Ma, Q.; Battelli, L.; Hubbs, A.F. Multiorgan autoimmune inflammation, enhanced lymphoproliferation, and impaired homeostasis of reactive oxygen species in mice lacking the antioxidant-activated transcription factor Nrf2. Am. J. Pathol. 2006, 168, 1960–1974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terzaghi, M.; Little, J.B. Repair of potentially lethal radiation damage in mammalian cells is associated with enhancement of malignant transformation. Nature 1975, 253, 548–549. [Google Scholar] [CrossRef] [PubMed]
- Borek, C.; Sachs, L. Cell susceptibility to transformation by x-irradiation and fixation of the transformed state. Proc. Natl. Acad. Sci. USA 1967, 57, 1522–1527. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Wakabayashi, N.; Misra, V.; Biswal, S.; Lee, G.H.; Agoston, E.S.; Yamamoto, M.; Kensler, T.W. NRF2 modulates aryl hydrocarbon receptor signaling: Influence on adipogenesis. Mol. Cell Biol. 2007, 27, 7188–7197. [Google Scholar] [CrossRef] [PubMed]
- Johansson, P.; Fasth, A.; Ek, T.; Hammarsten, O. Validation of a flow cytometry-based detection of gamma-H2AX, to measure DNA damage for clinical applications. Cytom. B Clin. Cytom. 2017, 92, 534–540. [Google Scholar] [CrossRef]
- Elgart, S.R.; Bostani, M.; Mok, K.C.; Adibi, A.; Ruehm, S.; Enzmann, D.; McNitt-Gray, M.; Iwamoto, K.S. Investigation of DNA Damage Dose-Response Kinetics after Ionizing Radiation Schemes Similar to CT Protocols. Radiat. Res. 2015, 183, 701–707. [Google Scholar] [CrossRef]
- Schaue, D.; Ratikan, J.A.; Iwamoto, K.S.; McBride, W.H. Maximizing tumor immunity with fractionated radiation. Int. J. Radiat. Oncol. Biol. Phys. 2012, 83, 1306–1310. [Google Scholar] [CrossRef]
- Jeong, Y.; Hoang, N.T.; Lovejoy, A.; Stehr, H.; Newman, A.M.; Gentles, A.J.; Kong, W.; Truong, D.; Martin, S.; Chaudhuri, A.; et al. Role of KEAP1/NRF2 and TP53 Mutations in Lung Squamous Cell Carcinoma Development and Radiation Resistance. Cancer Discov. 2017, 7, 86–101. [Google Scholar] [CrossRef]
- Kim, S.B.; Pandita, R.K.; Eskiocak, U.; Ly, P.; Kaisani, A.; Kumar, R.; Cornelius, C.; Wright, W.E.; Pandita, T.K.; Shay, J.W. Targeting of Nrf2 induces DNA damage signaling and protects colonic epithelial cells from ionizing radiation. Proc. Natl. Acad. Sci. USA 2012, 109, E2949–E2955. [Google Scholar] [CrossRef]
- Reddy, N.M.; Kleeberger, S.R.; Cho, H.Y.; Yamamoto, M.; Kensler, T.W.; Biswal, S.; Reddy, S.P. Deficiency in Nrf2-GSH signaling impairs type II cell growth and enhances sensitivity to oxidants. Am. J. Respir. Cell Mol. Biol. 2007, 37, 3–8. [Google Scholar] [CrossRef]
- Shrishrimal, S.; Chatterjee, A.; Kosmacek, E.A.; Davis, P.J.; McDonald, J.T.; Oberley-Deegan, R.E. Manganese porphyrin, MnTE-2-PyP, treatment protects the prostate from radiation-induced fibrosis (RIF) by activating the NRF2 signaling pathway and enhancing SOD2 and sirtuin activity. Free Radic. Biol. Med. 2020, 152, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Zhao, Q.; Zhang, Y.; Shi, W.; Wang, H.; Zheng, Z.; Meng, L.; Xin, Y.; Jiang, X. Sulforaphane-Mediated Nrf2 Activation Prevents Radiation-Induced Skin Injury through Inhibiting the Oxidative-Stress-Activated DNA Damage and NLRP3 Inflammasome. Antioxidants 2021, 10, 1850. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.F.; Yan, P.W.; Wang, L.J.; Liu, Y.T.; Wen, J.; Zhang, Q.; Fan, Y.X.; Luo, Y.H. Protective properties of Huperzine A through activation Nrf2/ARE-mediated transcriptional response in X-rays radiation-induced NIH3T3 cells. J. Cell Biochem. 2018, 119, 8359–8367. [Google Scholar] [CrossRef]
- Zou, G.L.; Zhang, X.R.; Ma, Y.L.; Lu, Q.; Zhao, R.; Zhu, Y.Z.; Wang, Y.Y. The role of Nrf2/PIWIL2/purine metabolism axis in controlling radiation-induced lung fibrosis. Am. J. Cancer Res. 2020, 10, 2752–2767. [Google Scholar] [PubMed]
- McBride, W.H.; Chiang, C.S.; Olson, J.L.; Wang, C.C.; Hong, J.H.; Pajonk, F.; Dougherty, G.J.; Iwamoto, K.S.; Pervan, M.; Liao, Y.P. A sense of danger from radiation. Radiat. Res. 2004, 162, 1–19. [Google Scholar] [CrossRef]
- Chiang, C.S.; McBride, W.H.; Withers, H.R. Radiation-induced astrocytic and microglial responses in mouse brain. Radiother. Oncol. 1993, 29, 60–68. [Google Scholar] [CrossRef]
- Hong, J.H.; Chiang, C.S.; Campbell, I.L.; Sun, J.R.; Withers, H.R.; McBride, W.H. Induction of acute phase gene expression by brain irradiation. Int. J. Radiat. Oncol. Biol. Phys. 1995, 33, 619–626. [Google Scholar] [CrossRef]
- Sherman, M.L.; Datta, R.; Hallahan, D.E.; Weichselbaum, R.R.; Kufe, D.W. Regulation of tumor necrosis factor gene expression by ionizing radiation in human myeloid leukemia cells and peripheral blood monocytes. J. Clin. Investig. 1991, 87, 1794–1797. [Google Scholar] [CrossRef]
- Morito, N.; Yoh, K.; Itoh, K.; Hirayama, A.; Koyama, A.; Yamamoto, M.; Takahashi, S. Nrf2 regulates the sensitivity of death receptor signals by affecting intracellular glutathione levels. Oncogene 2003, 22, 9275–9281. [Google Scholar] [CrossRef]
- Yoh, K.; Itoh, K.; Enomoto, A.; Hirayama, A.; Yamaguchi, N.; Kobayashi, M.; Morito, N.; Koyama, A.; Yamamoto, M.; Takahashi, S. Nrf2-deficient female mice develop lupus-like autoimmune nephritis. Kidney Int. 2001, 60, 1343–1353. [Google Scholar] [CrossRef]
- Zaki-Dizaji, M.; Akrami, S.M.; Azizi, G.; Abolhassani, H.; Aghamohammadi, A. Inflammation, a significant player of Ataxia-Telangiectasia pathogenesis? Inflamm. Res. 2018, 67, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, C.; Grieco, D.; Costanzo, V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J. 2011, 30, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.F. Ataxia-telangiectasia: From a rare disorder to a paradigm for cell signalling and cancer. Nat. Rev. Mol. Cell Biol. 2008, 9, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Baltimore, D. Dual roles of ATM in the cellular response to radiation and in cell growth control. Genes Dev. 1996, 10, 2401–2410. [Google Scholar] [CrossRef]
- Hartlova, A.; Erttmann, S.F.; Raffi, F.A.; Schmalz, A.M.; Resch, U.; Anugula, S.; Lienenklaus, S.; Nilsson, L.M.; Kroger, A.; Nilsson, J.A.; et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 2015, 42, 332–343. [Google Scholar] [CrossRef]
- Sun, X.; Wang, Y.; Ji, K.; Liu, Y.; Kong, Y.; Nie, S.; Li, N.; Hao, J.; Xie, Y.; Xu, C.; et al. NRF2 preserves genomic integrity by facilitating ATR activation and G2 cell cycle arr.rest. Nucleic Acids Res. 2020, 48, 9109–9123. [Google Scholar] [CrossRef]
- Khalil, H.; Deeni, Y. NRF2 inhibition causes repression of ATM and ATR expression leading to aberrant DNA Damage Response. BioDiscovery 2015, 15, e8964. [Google Scholar] [CrossRef]
- Li, B.; Wang, X.; Rasheed, N.; Hu, Y.; Boast, S.; Ishii, T.; Nakayama, K.; Nakayama, K.I.; Goff, S.P. Distinct roles of c-Abl and Atm in oxidative stress response are mediated by protein kinase C delta. Genes Dev. 2004, 18, 1824–1837. [Google Scholar] [CrossRef]
- Su, F.; Smilenov, L.B.; Ludwig, T.; Zhou, L.; Zhu, J.; Zhou, G.; Hall, E.J. Hemizygosity for Atm and Brca1 influence the balance between cell transformation and apoptosis. Radiat. Oncol. 2010, 5, 15. [Google Scholar] [CrossRef]
- Jayakumar, S.; Pal, D.; Sandur, S.K. Nrf2 facilitates repair of radiation induced DNA damage through homologous recombination repair pathway in a ROS independent manner in cancer cells. Mutat. Res. 2015, 779, 33–45. [Google Scholar] [CrossRef]
- Limoli, C.L.; Giedzinski, E. Induction of chromosomal instability by chronic oxidative stress. Neoplasia 2003, 5, 339–346. [Google Scholar] [CrossRef] [Green Version]
- Marder, B.A.; Morgan, W.F. Delayed chromosomal instability induced by DNA damage. Mol. Cell Biol. 1993, 13, 6667–6677. [Google Scholar] [PubMed]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Martin-Moldes, Z.; Ye, J.; Lastres-Becker, I. Transcription factors NRF2 and NF-kappaB are coordinated effectors of the Rho family, GTP-binding protein RAC1 during inflammation. J. Biol. Chem. 2014, 289, 15244–15258. [Google Scholar] [CrossRef]
- Chen, H.; Fang, Y.; Li, W.; Orlando, R.C.; Shaheen, N.; Chen, X.L. NFkB and Nrf2 in esophageal epithelial barrier function. Tissue Barriers 2013, 1, e27463. [Google Scholar] [CrossRef]
- Rubio, V.; Garcia-Perez, A.I.; Herraez, A.; Diez, J.C. Different roles of Nrf2 and NFKB in the antioxidant imbalance produced by esculetin or quercetin on NB4 leukemia cells. Chem. Biol. Interact. 2018, 294, 158–166. [Google Scholar] [CrossRef]
- Song, Z.; Zhong, X.; Li, M.; Gao, P.; Ning, Z.; Sun, Z.; Song, X. 1-MNA Ameliorates High Fat Diet-Induced Heart Injury by Upregulating Nrf2 Expression and Inhibiting NF-kappaB in vivo and in vitro. Front. Cardiovasc. Med. 2021, 8, 721814. [Google Scholar] [CrossRef]
- Girotti, M.J.; Moon, B.; Patterson, G.A.; Hong, K.; Todd, T.R. Effect on cardiopulmonary changes of gram-negative endotoxemia in sheep after type-specific, cross-reactive, and nonspecific immune stimulation. Circ. Shock 1986, 18, 171–178. [Google Scholar]
- Chiang, C.S.; Hong, J.H.; Stalder, A.; Sun, J.R.; Withers, H.R.; McBride, W.H. Delayed molecular responses to brain irradiation. Int. J. Radiat. Biol. 1997, 72, 45–53. [Google Scholar] [CrossRef]
- Hong, J.H.; Chiang, C.S.; Tsao, C.Y.; Lin, P.Y.; McBride, W.H.; Wu, C.J. Rapid induction of cytokine gene expression in the lung after single and fractionated doses of radiation. Int. J. Radiat. Biol. 1999, 75, 1421–1427. [Google Scholar]
- Purbey, P.K.; Scumpia, P.O.; Kim, P.J.; Tong, A.-J.; Iwamoto, K.S.; McBride, W.H.; Smale, S.T. Defined Sensing Mechanisms and Signaling Pathways Contribute to the Global Inflammatory Gene Expression Output Elicited by Ionizing Radiation. Immunity 2017, 47, 421–434.e3. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues-Moreira, S.; Moreno, S.G.; Ghinatti, G.; Lewandowski, D.; Hoffschir, F.; Ferri, F.; Gallouet, A.S.; Gay, D.; Motohashi, H.; Yamamoto, M.; et al. Low-Dose Irradiation Promotes Persistent Oxidative Stress and Decreases Self-Renewal in Hematopoietic Stem Cells. Cell Rep. 2017, 20, 3199–3211. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Murakami, S.; Suzuki, T.; Harigae, H.; Romeo, P.H.; Yamamoto, M.; Motohashi, H. NRF2 Activation Impairs Quiescence and Bone Marrow Reconstitution Capacity of Hematopoietic Stem Cells. Mol. Cell Biol. 2017, 37, e00086-17. [Google Scholar] [CrossRef]
- Ito, K.; Hirao, A.; Arai, F.; Takubo, K.; Matsuoka, S.; Miyamoto, K.; Ohmura, M.; Naka, K.; Hosokawa, K.; Ikeda, Y.; et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med. 2006, 12, 446–451. [Google Scholar] [CrossRef]
- Varnes, M.E.; Biaglow, J.E.; Roizin-Towle, L.; Hall, E.J. Depletion of intracellular GSH and NPSH by buthionine sulfoximine and diethyl maleate: Factors that influence enhancement of aerobic radiation response. Int. J. Radiat. Oncol. Biol. Phys. 1984, 10, 1229–1233. [Google Scholar] [CrossRef]
- Micewicz, E.D.; Kim, K.; Iwamoto, K.S.; Ratikan, J.A.; Cheng, G.; Boxx, G.M.; Damoiseaux, R.D.; Whitelegge, J.P.; Ruchala, P.; Nguyen, C.; et al. 4-(Nitrophenylsulfonyl) piperazines mitigate radiation damage to multiple tissues. PLoS ONE 2017, 12, e0181577. [Google Scholar] [CrossRef]
- Feng, R.; Morine, Y.; Ikemoto, T.; Imura, S.; Iwahashi, S.; Saito, Y.; Shimada, M. Nrf2 activation drive macrophages polarization and cancer cell epithelial-mesenchymal transition during interaction. Cell Commun. Signal. 2018, 16, 54. [Google Scholar] [CrossRef] [PubMed]
- Beury, D.W.; Carter, K.A.; Nelson, C.; Sinha, P.; Hanson, E.; Nyandjo, M.; Fitzgerald, P.J.; Majeed, A.; Wali, N.; Ostrand-Rosenberg, S. Myeloid-Derived Suppressor Cell Survival and Function Are Regulated by the Transcription Factor Nrf2. J. Immunol. 2016, 196, 3470–3478. [Google Scholar] [CrossRef]
- Micewicz, E.D.; Iwamoto, K.S.; Ratikan, J.A.; Nguyen, C.; Xie, M.W.; Cheng, G.; Boxx, G.M.; Deriu, E.; Damoiseaux, R.D.; Whitelegge, J.P.; et al. The Aftermath of Surviving Acute Radiation Hematopoietic Syndrome and its Mitigation. Radiat. Res. 2019, 191, 323–334. [Google Scholar] [CrossRef]
- Rockwell, C.E.; Zhang, M.; Fields, P.E.; Klaassen, C.D. Th2 skewing by activation of Nrf2 in CD4(+) T cells. J. Immunol. 2012, 188, 1630–1637. [Google Scholar] [CrossRef] [PubMed]
- Murata, Y.; Shimamura, T.; Hamuro, J. The polarization of T(h)1/T(h)2 balance is dependent on the intracellular thiol redox status of macrophages due to the distinctive cytokine production. Int. Immunol. 2002, 14, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Nesseler, J.P.; Lee, M.H.; Nguyen, C.; Kalbasi, A.; Sayre, J.W.; Romero, T.; Nickers, P.; McBride, W.H.; Schaue, D. Tumor Size Matters-Understanding Concomitant Tumor Immunity in the Context of Hypofractionated Radiotherapy with Immunotherapy. Cancers 2020, 12, 714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaue, D. A Century of Radiation Therapy and Adaptive Immunity. Front. Immunol. 2017, 8, 431. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schaue, D.; Micewicz, E.D.; Ratikan, J.A.; Iwamoto, K.S.; Vlashi, E.; McDonald, J.T.; McBride, W.H. NRF2 Mediates Cellular Resistance to Transformation, Radiation, and Inflammation in Mice. Antioxidants 2022, 11, 1649. https://doi.org/10.3390/antiox11091649
Schaue D, Micewicz ED, Ratikan JA, Iwamoto KS, Vlashi E, McDonald JT, McBride WH. NRF2 Mediates Cellular Resistance to Transformation, Radiation, and Inflammation in Mice. Antioxidants. 2022; 11(9):1649. https://doi.org/10.3390/antiox11091649
Chicago/Turabian StyleSchaue, Dörthe, Ewa D. Micewicz, Josephine A. Ratikan, Keisuke S. Iwamoto, Erina Vlashi, J. Tyson McDonald, and William H. McBride. 2022. "NRF2 Mediates Cellular Resistance to Transformation, Radiation, and Inflammation in Mice" Antioxidants 11, no. 9: 1649. https://doi.org/10.3390/antiox11091649
APA StyleSchaue, D., Micewicz, E. D., Ratikan, J. A., Iwamoto, K. S., Vlashi, E., McDonald, J. T., & McBride, W. H. (2022). NRF2 Mediates Cellular Resistance to Transformation, Radiation, and Inflammation in Mice. Antioxidants, 11(9), 1649. https://doi.org/10.3390/antiox11091649