

The Role of Nrf2 in Pulmonary Fibrosis: Molecular Mechanisms and Treatment Approaches


Abstract
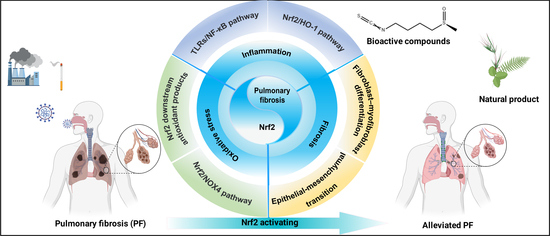
:
1. Introduction
2. Nrf2
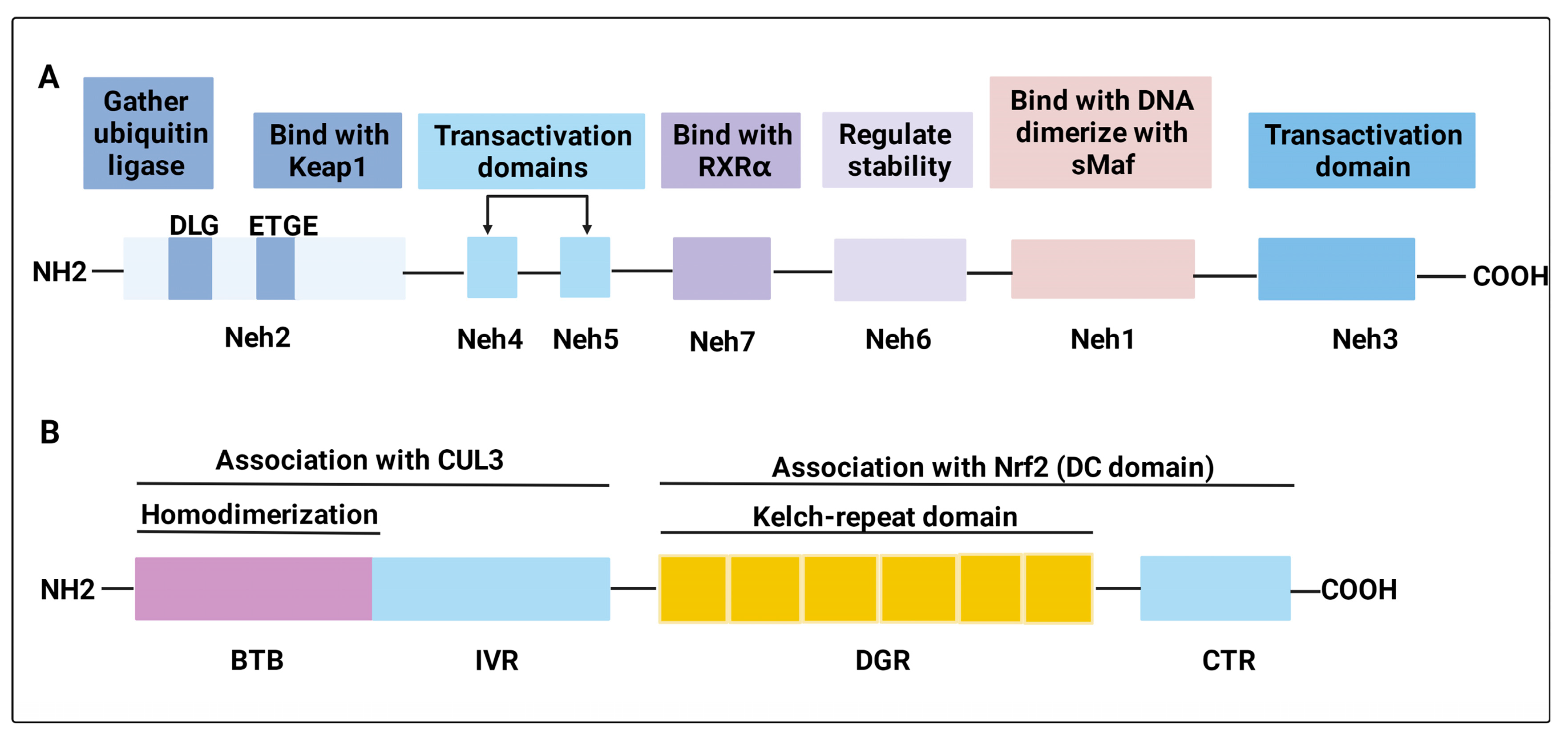
2.1. Structure of Nrf2 and Keap1
2.2. Nrf2 Activation
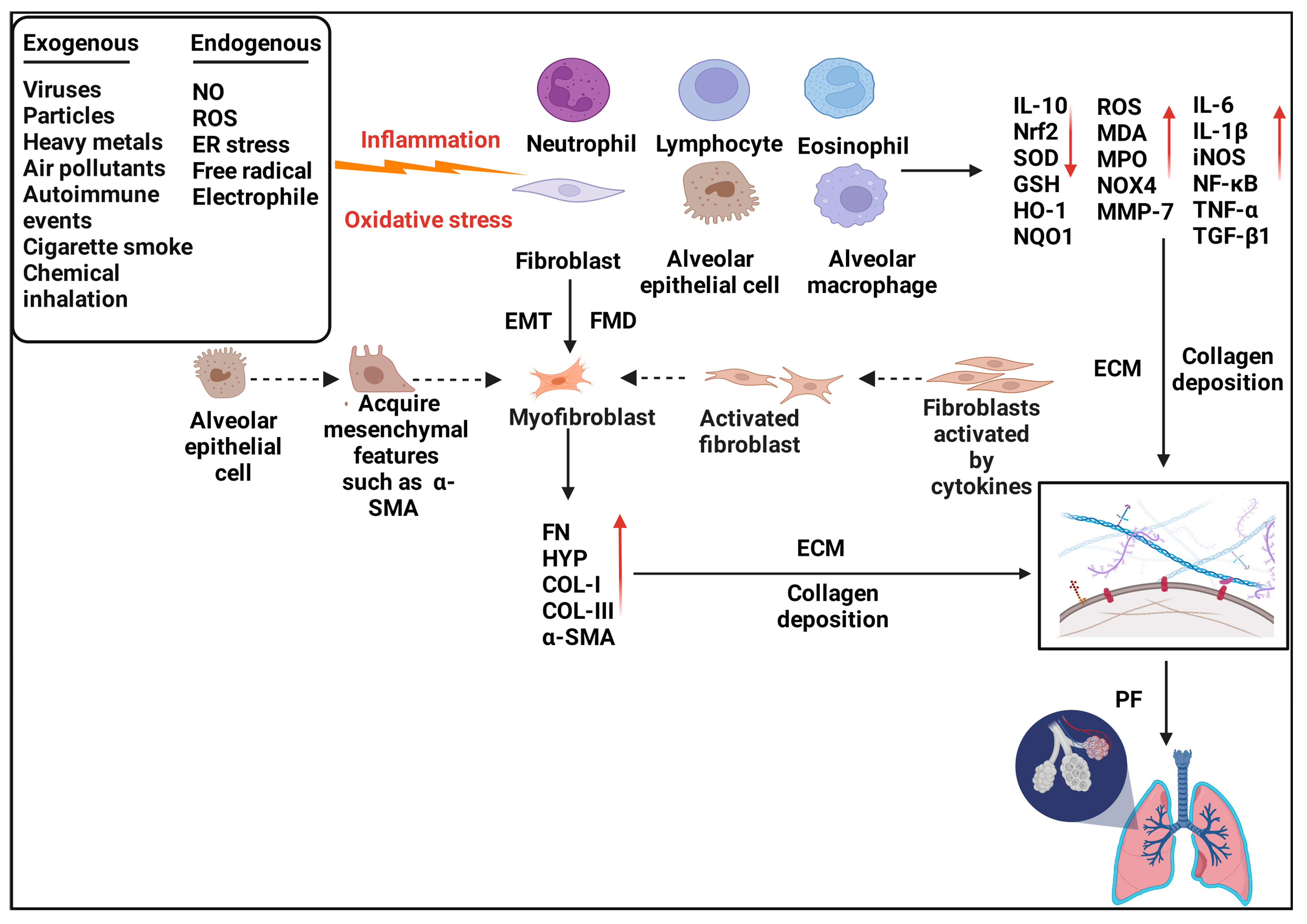
3. Nrf2 and Inflammation in Pulmonary Fibrosis
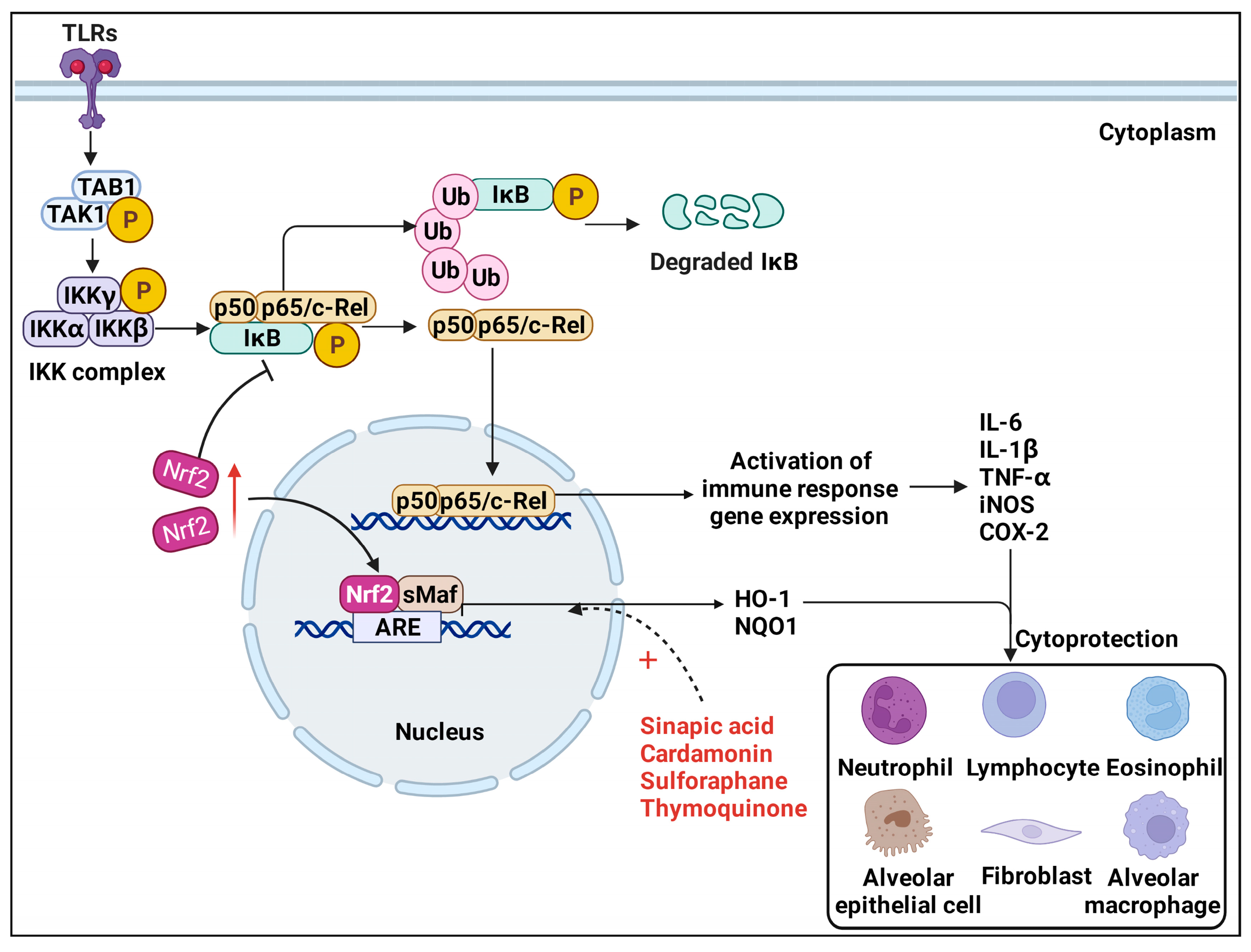
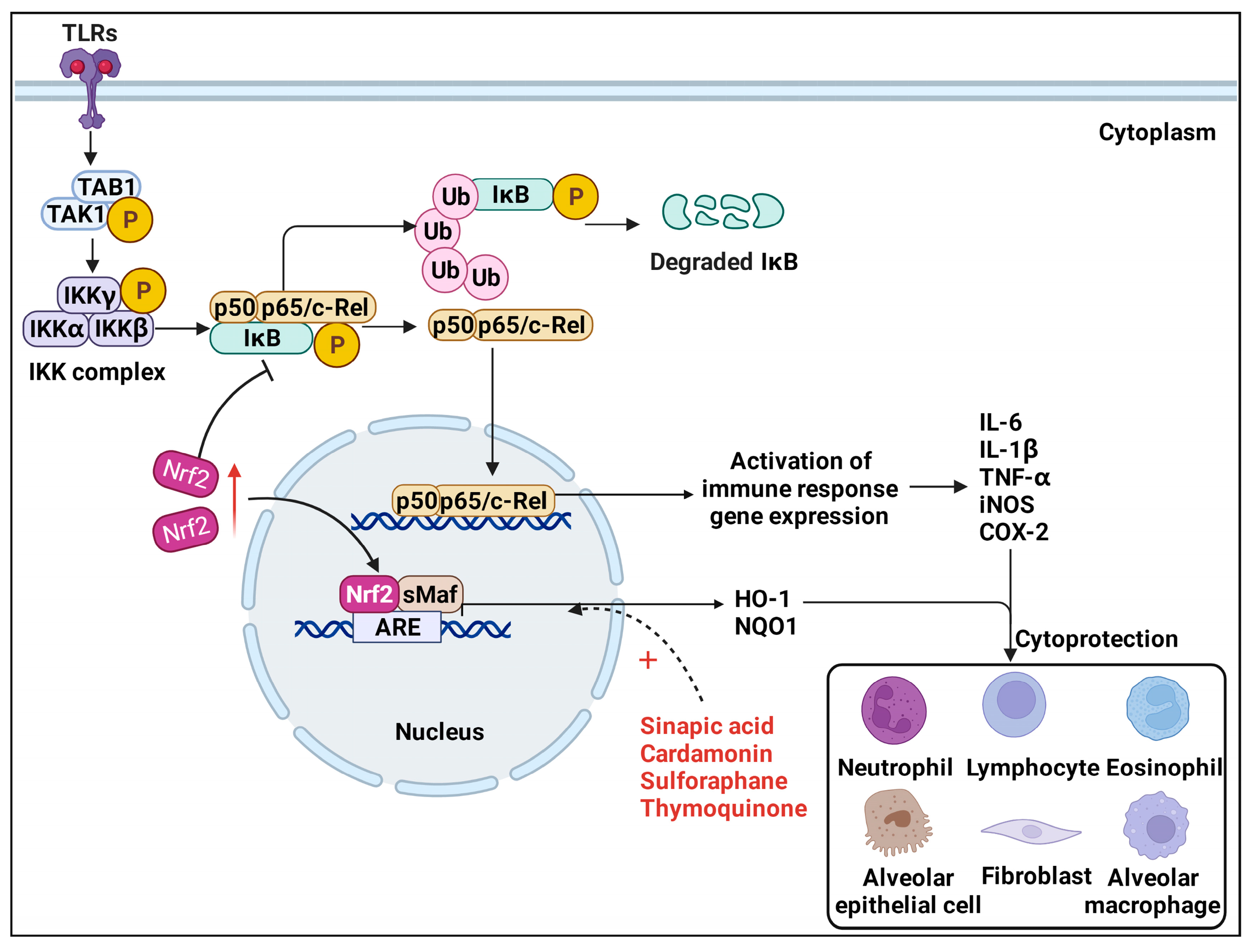
3.1. TLRs/NF-κB Pathway
3.2. Nrf2/HO−1 Pathway
4. Nrf2 and Oxidative Stress in Pulmonary Fibrosis
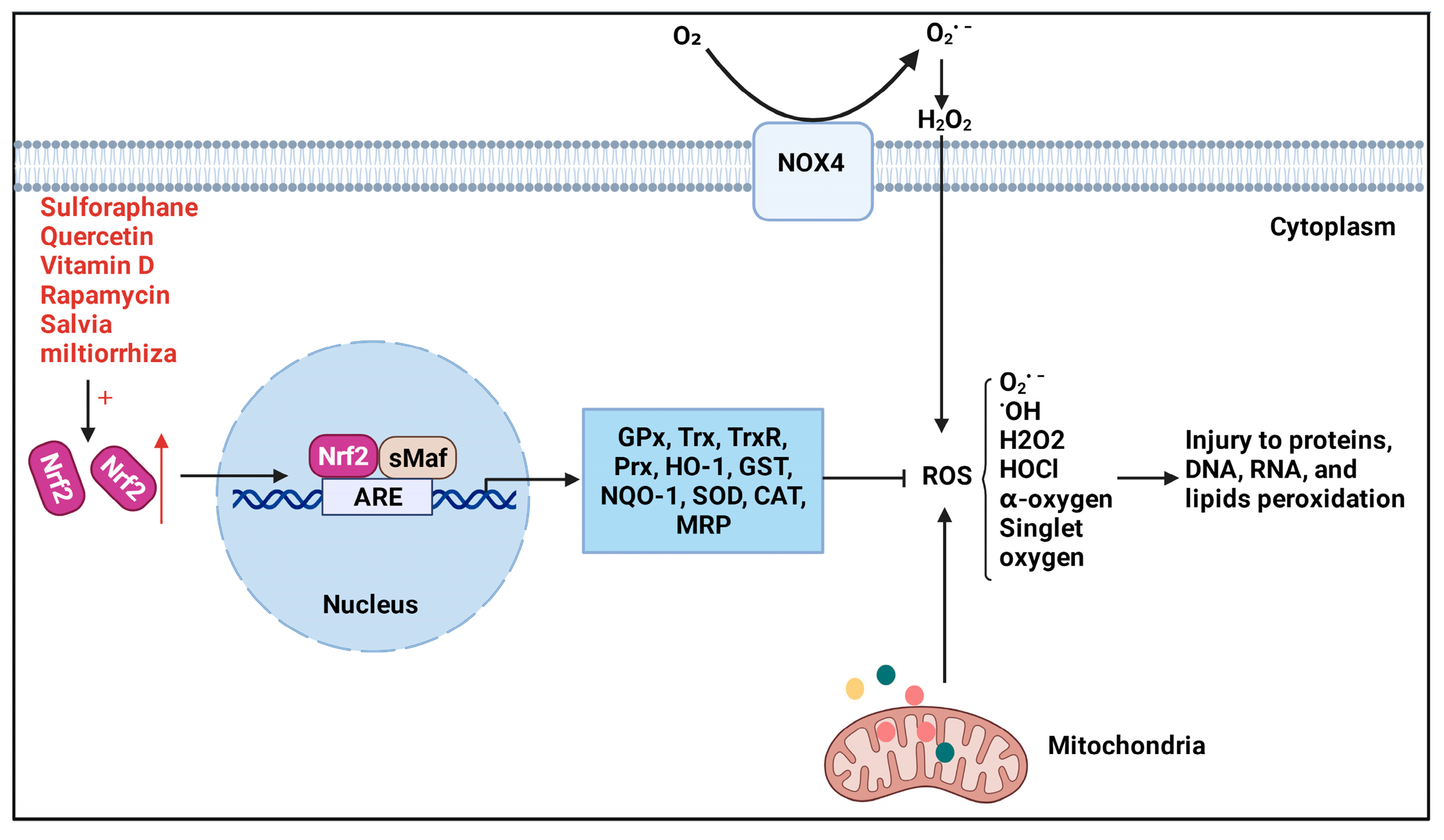
4.1. Nrf2 Downstream Antioxidant Products
4.2. Nrf2/NOX4 Pathway
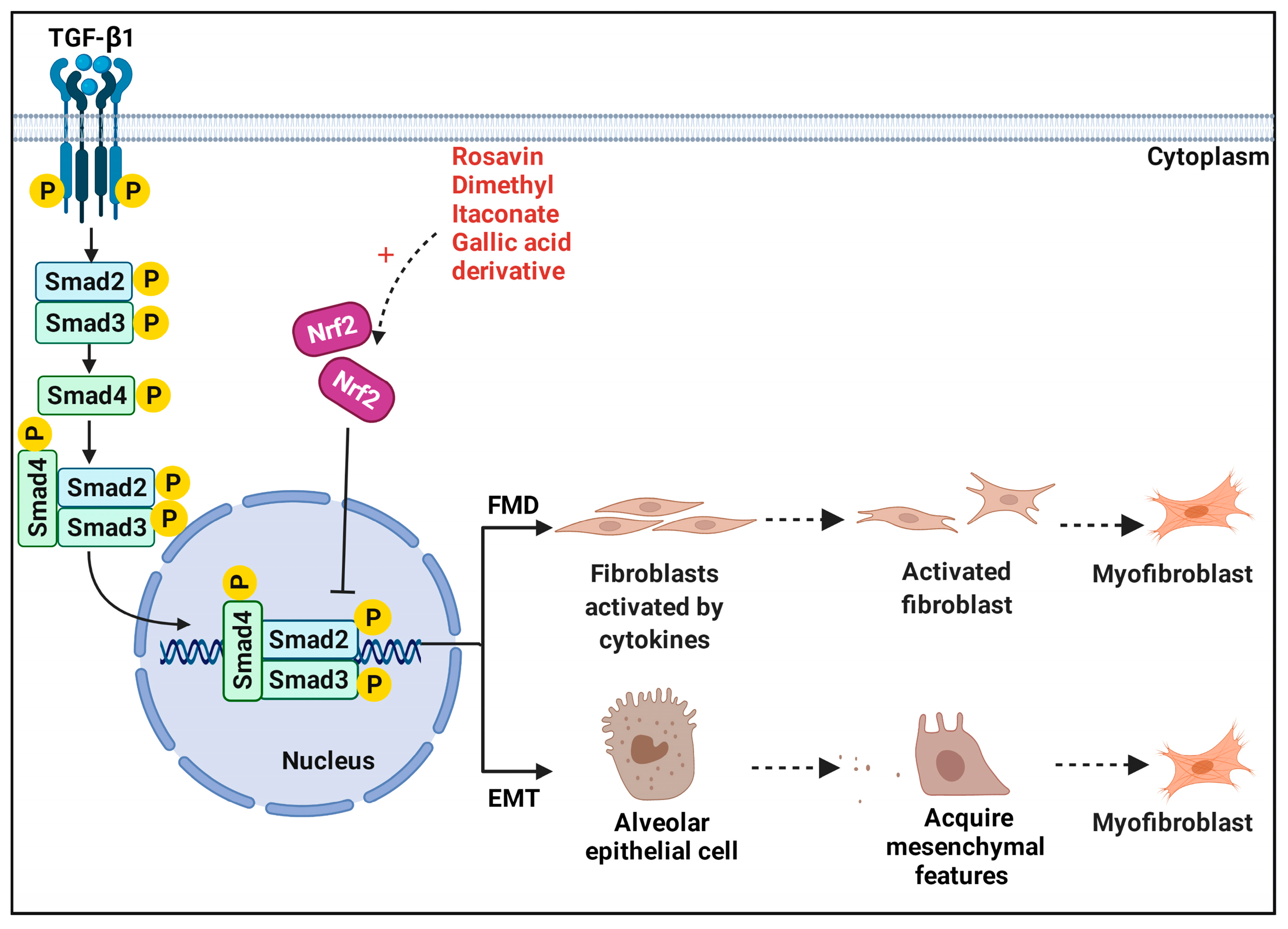
5. Nrf2 and Fibroblasts in Pulmonary Fibrosis—TGF-β1/Smad Pathway
5.1. Fibroblast–Myofibroblast Differentiation
5.2. Epithelial–Mesenchymal Transition
6. Potential Therapies and Nrf2 Activators

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Model | Target | Function and Detection Index | Refs |
|---|---|---|---|---|
| Rapamycin1 | PQ-treated male rats and LFs | Nrf2 activating | Suppressed PQ-induced oxidant stress, cell death and apoptosis, fibrosis-related factors, reversed PQ-induced FMD and PF induced by PQ. | [61] |
| Tanshinone IIA2 | Silica-treated silicosis rat and NIH-3T3 cells | TGFβ1/Smad Nrf2/ NOX4 Nrf2/GSH | Reduced the levels of collagen deposition, TGF-β1, α-SMA, Col-I, Col-III, NOX4, ROS; increased the levels of Nrf2, HO−1, NQO1, Gclc, Gclm, and GSH; regulated myofibroblast activation, protected Nrf2 from protein ubiquitination, promoted Keap1 degradation. | [113,114,115] |
| Pterostilbene3 | Lps-treated female BALB/C mice | Nrf2 activating | Decreased lung injury and fibrosis scores; reduced levels of Col-I, TGF-β1, HYP, IL-1β, IL-6, TNF-α; increased the levels of Nrf2, HO−1, NQO1, GSH, SOD. | [64] |
| Sinapic acid4 | BLM-treated SD rats | Nrf2/HO−1 NF-κB | Increased the levels of Nrf2, inflammatory cell population, GPx, CAT, Bcl-2; reduced the levels of MDA, TNF-α, IL-1β, MPO, MMP-7, HYP, TGF-β1, NF-κB; restore the antioxidant system, inflammatory or fibrotic alterations. | [96] |
| Thymoquinone5 | BLM-treated Wistar rats | Nrf2/HO−1 | Decreased levels of HYP, LDH, total and differential leukocytes, MDA, TNF-α, IL-1β, MPO, MMP-7, caspase-3, Bax, NF-κB; upregulate Nrf2, HO−1, Bcl2; ameliorated severe hemorrhage, thickening of alveolar septa, emphysema, infiltration of leukocytes in walls alveoli and fibroplasia, inflammation, and PF. | [99] |
| Dihydroartemisinin6 | BLM-treated SD rats and AECs | Nrf2/HO−1 | Reduced the levels of α-SMA, MDA; increased the levels of E-cadherin, Nrf2, HO−1, SOD, and GSH; mitigated alveolitis severity, relieved fibrosis scores, inhibited the increase in the myofibroblasts–like processes of the AECs. | [100] |
| Atractylenolide III7 | BLM-treated SD rats | Nrf2/NQO1/HO−1 | Reduced the expression of Caspase-3 and Caspase-9, IL-6, iNOS, TNF-α, MDA, LDH; upregulated the levels of Nrf2, NQO1, HO−1, SOD, GSH, IL-10; improved lung function alleviated PF and oxidative stress. | [101] |
| Rosavin8 | BLM-treated Kunming mice | Nrf2/NF-κB TGF-β1 | Inhibited inflammatory cells, MDA, HYP, NF-κB-p65, α-SMA TGF-β1 levels; improved Nrf2, SOD, GSH-Px levels; ameliorated PF, alveolar inflammatory cell contents. | [121] |
| Vitamin D39 | Particles-treated Nrf2+/+ and Nrf2−/− C57BL/6 mice, HFLIII cells | Nrf2 activating | Reduced the levels of α-SMA, FN, E-cadherin; increased the levels of N-cadherin, Nrf2, VDR; limited fibroblast cells’ migration, FDM, ECM. | [122] |
| S-Allylmercaptocysteine10 | BLM-treated C57/BL6 mice | Nrf2/NOX4 TGF-β1/Smad | Increased antioxidants such as HO−1, GSH, and SOD; decreased HYP, SMA; ameliorated the pathological structure, and decrease inflammatory cell infiltration and pro-inflammatory cytokines in BALF. | [131] |
| Gallic acid derivative (GAD)11 | BLM-treated C57/BL6 mice | Nrf2/NOX4 TGF-β1/Smad | Reduced the levels of α-SMA, HYP, collagen type I/III, IL-6, TGF-β1, NOX4; increase the levels of SOD and GSH; increased body weight, survival rate, and alleviated alveolar structure, alveolar inflammation, and the degree of PF. | [132] |
| Salvia miltiorrhiza12 | BLM-treated C57/BL6 mice and NIH-3T3 cells | Nrf2/GSH Nrf2/Keap1 Nrf2/Nox4 | Reduced the levels of TGF-β1, α-SMA, ECM, COL-1, NOX4, ROS, PKC-δ, Smad3; increase the levels of Nrf2, NQO1, HO−1; protected Nrf2 from protein ubiquitination, PF; regulated myofibroblasts activation, Increased the sensitivity of fibroblasts to the loss of GSH. | [133] |
| Dimethyl itaconate13 | TGF-β1-induced FMD in vitro and BLM-treated mouse | Nrf2 activating | Nrf2 decreased TXNIP expression and alleviated FMD in PF; Nrf2 inhibited TGF-β1-induced FMD and the increase of ROS. | [141] |
| Sulforaphane14 | BLM-treated C57/BL6 mice | Nrf2 activating | Reduced the levels of caspase-3, IL-1β, TNF-α, TGF-β, HYP, 3-NT, and 4-HNE; increased the levels of Nrf2, HO−1, NQO1, SOD1, and CAT; alleviated BLM-induced alveolar epithelial cell apoptosis, alveolitis, collagen accumulation, lung oxidative stress, and lung fibrosis. | [168] |
| Bletilla striata15 | SiO2-treated C57BL/6 mice and A549 cells line | Nrf2/HO−1/γ-GCSc | Reduced the levels of MDA, ROS; increased the levels of γ-GCSc, Nrf2, SOD, HO−1; protective effect of lung injury, lung cell viability, apoptosis, and ROS accumulation. | [10] |
| Sarcodon aspratus16 | BLM-treated Kunming mice and A549 cells | MAPK/Nrf2/HO−1 TGF-β1/MAPK TLR4/NF-κB | Reduced the levels of ROS, MDA, TNF-α, IL-1β, IL-6, CTGF, MMP-2, HYP, α-SMA, ECM, TLR4, MyD88, NF-κB-p65; increased the levels of GSH-Px, SOD, Nrf2, HO−1, CAT, Smad7; inhibited H2O2-induced cell apoptosis, oxidative stress, fibrosis, phosphorylation of JNK, ERK and P38, weight loss. | [169] |
| Arenaria kansuensis17 | PQ-treated C57BL mice | TGF-β1/Smad NF-κB-p65 Nrf2/NOX4 | Downregulated α-SMA, TGF-β1, TNF-α, IL-6, IL-1β1, HYP, ROS, collagen deposition, NOX4; upregulate Nrf2, SOD, and GSH; improved mice survival rate, body weight, lung pathological lesion, and the lung index. | [170] |
| Quercetin18 | BLM-treated BEAS-2B cells | Nrf2 activating | Reduced the expression levels of ROS, TNF-α, and IL-8; increased Nrf2-ARE binding, HO−1, and γ-GCS; restored the disturbed redox balance and reduce inflammation. | [171] |
| Chelerythrine19 | BLM-treated C57/BL6 mice | Nrf2/ARE | Reduced the expression levels of fibronectin, α-SMA, TGF-β, 4-HNE, and HYP; upregulated the levels of SOD, GSH, Nrf2, HO−1, and NQO1; alleviates collagen deposition, oxidative stress, and PF. | [172] |
| Bergenin20 | BLM-treated C57/BL6 mice and NIH3T3 cells | p62/Nrf2 | Decreased content of α-SMA, COL-1, HYP, ROS, MDA; increased the levels and activity of Nrf2, GSH, SOD, HO−1, NQO1; inhibited the TGF-β1 induced FDM, oxidative stress, and PF. | [173] |
| Jinshui Huanxian formula21 | BLM-treated SD rats, MRC-5 cells and NIH-3T3 cells | Nrf2/NOX4 TGF-β1 | Reduced the levels of TGF-β1, collagen deposition, HYP, α-SMA, COL-I, COL-III, MDA, MPO, NOX4, FN1; increased the levels of Nrf2, GSH, SOD, CAT, NQO1, HO−1; suppressed the increases of lung coefficient, TGF-β1-induced FDM, ROS production | [174] |
| Dimethyl fumarate22 | BLM-treated C57/BL6 mice; RAW264.7 and NIH-3T3 cells coculture | Nrf2 activating | Attenuated macrophage activity and fibrosis in mice; promoted Nrf2 and HO−1 expression and suppress TGF-β and ROS production; reduced fibroblast-to-myofibroblast transition and collagen production by NIH-3T3 cells. | [175] |
| Chloroquine23 | PQ-treated male C57BL/6 mice | Nrf2/NQO1/HO−1 TGF-β | Reduced the levels of TNF-α, IL-1β, IL-6, NO, iNOS, MDA, α-SMA, TGF-β; increased the levels of SOD, NQO1, Nrf2, HO−1; attenuated lung injury, oxidative stress, decreases protein, inflammatory cells. | [176] |
| Esomeprazole24 | BLM- or TGF-β-treated PHLE cells and fibroblasts | MAPK/Nrf2/HO−1 DDAH/iNOS | Reduced the levels of DDAH, iNOS, IL-1β, IL-6, TNF-α, COL-I, COL-III, COL-V; increased the levels of HO−1, NQO1, Nrf2; downregulates pro-inflammatory and profibrotic molecules, collagen expression; activates MAPK via phosphorylation. | [156] |
7. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef]
- Martinez, F.; Collard, H.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.; Taniguchi, H.; Wells, A. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Primers 2017, 3, 17074. [Google Scholar] [CrossRef] [PubMed]
- Wolters, P.J.; Blackwell, T.S.; Eickelberg, O.; Loyd, J.E.; Kaminski, N.; Jenkins, G.; Maher, T.M.; Molina-Molina, M.; Noble, P.W.; Raghu, G.; et al. Time for a change: Is idiopathic pulmonary fibrosis still idiopathic and only fibrotic? Lancet Respir. Med. 2018, 6, 154–160. [Google Scholar] [CrossRef]
- Wynn, T.A. Integrating mechanisms of pulmonary fibrosis. J. Exp. Med. 2011, 208, 1339–1350. [Google Scholar] [CrossRef]
- Wilson, M.S.; Wynn, T.A. Pulmonary fibrosis: Pathogenesis, etiology and regulation. Mucosal Immunol. 2009, 2, 103–121. [Google Scholar] [CrossRef]
- Rackow, A.R.; Nagel, D.J.; McCarthy, C.; Judge, J.; Lacy, S.; Freeberg, M.A.T.; Thatcher, T.H.; Kottmann, R.M.; Sime, P.J. The self-fulfilling prophecy of pulmonary fibrosis: A selective inspection of pathological signalling loops. Eur. Respir. J. 2020, 56, 2000075. [Google Scholar] [CrossRef]
- Moore, B.B.; Fry, C.; Zhou, Y.; Murray, S.; Han, M.K.; Martinez, F.J.; Flaherty, K.R.; The, C.I. Inflammatory leukocyte phenotypes correlate with disease progression in idiopathic pulmonary fibrosis. Front. Med. 2014, 1, 56. [Google Scholar] [CrossRef]
- Liu, P.; Luo, G.; Dodson, M.; Schmidlin, C.; Wei, Y.; Kerimoglu, B.; Ooi, A.; Chapman, E.; Garcia, J.; Zhang, D. The NRF2-LOC344887 signaling axis suppresses pulmonary fibrosis. Redox Biol. 2021, 38, 101766. [Google Scholar] [CrossRef]
- Deng, G.; Li, L.; Ouyang, Y.C. Elegans Modeling paraquat-induced lung fibrosis in reveals KRIT1 as a key regulator of collagen gene transcription. Aging 2021, 13, 4452–4467. [Google Scholar] [CrossRef]
- Chen, G.; Chang, W.; Li, X.; Han, L.; Zhou, D.; Feng, Y.; Li, B.; Zhu, F.; Li, N. n-BuOH extract of Bletilla striata exerts chemopreventive effects on lung against SiO nanoparticles through activation of Nrf2 pathway. Phytomedicine 2021, 82, 153445. [Google Scholar] [CrossRef]
- Yu, G.; Tzouvelekis, A.; Wang, R.; Herazo-Maya, J.D.; Ibarra, G.H.; Srivastava, A.; de Castro, J.P.W.; DeIuliis, G.; Ahangari, F.; Woolard, T.; et al. Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nat. Med. 2018, 24, 39–49. [Google Scholar] [CrossRef]
- D’Amico, R.; Monaco, F.; Fusco, R.; Siracusa, R.; Impellizzeri, D.; Peritore, A.; Crupi, R.; Gugliandolo, E.; Cuzzocrea, S.; Di Paola, R.; et al. Atrazine Inhalation Worsen Pulmonary Fibrosis Regulating the Nuclear Factor-Erythroid 2-Related Factor (Nrf2) Pathways Inducing Brain Comorbidities. Cell Physiol. Biochem. 2021, 55, 704–725. [Google Scholar] [CrossRef]
- Kreuter, M.; Picker, N.; Schwarzkopf, L.; Baumann, S.; Cerani, A.; Postema, R.; Maywald, U.; Dittmar, A.; Langley, J.; Patel, H. Epidemiology, healthcare utilization, and related costs among patients with IPF: Results from a German claims database analysis. Respir. Res. 2022, 23, 62. [Google Scholar] [CrossRef]
- Maher, T.; Bendstrup, E.; Dron, L.; Langley, J.; Smith, G.; Khalid, J.; Patel, H.; Kreuter, M. Global incidence and prevalence of idiopathic pulmonary fibrosis. Respir. Res. 2021, 22, 197. [Google Scholar] [CrossRef]
- Cox, I.; Otahal, P.; de Graaff, B.; Corte, T.; Moodley, Y.; Zappala, C.; Glaspole, I.; Hopkins, P.; Macansh, S.; Walters, E.; et al. Incidence, prevalence and mortality of idiopathic pulmonary fibrosis in Australia. Respirology 2021, 27, 209–216. [Google Scholar] [CrossRef]
- Novelli, L.; Ruggiero, R.; De Giacomi, F.; Biffi, A.; Faverio, P.; Bilucaglia, L.; Gamberini, S.; Messinesi, G.; Pesci, A. Corticosteroid and cyclophosphamide in acute exacerbation of idiopathic pulmonary fibrosis: A single center experience and literature review. Sarcoidosis Vasc. Diffus. Lung Dis. 2016, 33, 385–391. [Google Scholar]
- Luppi, F.; Cerri, S.; Beghè, B.; Fabbri, L.M.; Richeldi, L. Corticosteroid and immunomodulatory agents in idiopathic pulmonary fibrosis. Respir. Med. 2004, 98, 1035–1044. [Google Scholar] [CrossRef]
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef]
- King, T.E., Jr.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef]
- Woodcock, H.V.; Maher, T.M. The treatment of idiopathic pulmonary fibrosis. F1000Prime Rep. 2014, 6, 16. [Google Scholar] [CrossRef]
- George, P.; Patterson, C.; Reed, A.; Thillai, M. Lung transplantation for idiopathic pulmonary fibrosis. Lancet Respir. Med. 2019, 7, 271–282. [Google Scholar] [CrossRef]
- Christie, J.D.; Edwards, L.B.; Aurora, P.; Dobbels, F.; Kirk, R.; Rahmel, A.O.; Stehlik, J.; Taylor, D.O.; Kucheryavaya, A.Y.; Hertz, M.I. The Registry of the International Society for Heart and Lung Transplantation: Twenty-sixth Official Adult Lung and Heart-Lung Transplantation Report-2009. J. Heart Lung Transplant. 2009, 28, 1031–1049. [Google Scholar] [CrossRef]
- Sontake, V.; Gajjala, P.; Kasam, R.; Madala, S. New therapeutics based on emerging concepts in pulmonary fibrosis. Expert Opin. Ther. Targets. 2019, 23, 69–81. [Google Scholar] [CrossRef]
- Audousset, C.; McGovern, T.; Martin, J. Role of Nrf2 in Disease: Novel Molecular Mechanisms and Therapeutic Approaches—Pulmonary Disease/Asthma. Front. Physiol. 2021, 12, 727806. [Google Scholar] [CrossRef]
- Torrente, L.; DeNicola, G. Targeting NRF2 and Its Downstream Processes: Opportunities and Challenges. Annu. Rev. Pharmacol. Toxicol. 2022, 62, 279–300. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef]
- Chen, X.; Zhu, X.; Wei, A.; Chen, F.; Gao, Q.; Lu, K.; Jiang, Q.; Cao, W. Nrf2 epigenetic derepression induced by running exercise protects against osteoporosis. Bone Res. 2021, 9, 15. [Google Scholar] [CrossRef]
- Zhang, W.; Feng, C.; Jiang, H. Novel target for treating Alzheimer’s Diseases: Crosstalk between the Nrf2 pathway and autophagy. Ageing Res. Rev. 2021, 65, 101207. [Google Scholar] [CrossRef]
- Qu, J.; Zhang, Z.; Zhang, P.; Zheng, C.; Zhou, W.; Cui, W.; Xu, L.; Gao, J. Downregulation of HMGB1 is required for the protective role of Nrf2 in EMT-mediated PF. J. Cell Physiol. 2019, 234, 8862–8872. [Google Scholar] [CrossRef]
- Gu, X.; Liu, Y.; Wang, N.; Zhen, J.; Zhang, B.; Hou, S.; Cui, Z.; Wan, Q.; Feng, H. Transcription of MRPL12 regulated by Nrf2 contributes to the mitochondrial dysfunction in diabetic kidney disease. Free Radic. Biol. Med. 2021, 164, 329–340. [Google Scholar] [CrossRef]
- Sun, W.; Wang, Z.; Sun, M.; Huang, W.; Wang, Y.; Wang, Y. Aloin antagonizes stimulated ischemia/reperfusion-induced damage and inflammatory response in cardiomyocytes by activating the Nrf2/HO-1 defense pathway. Cell Tissue Res. 2021, 384, 735–744. [Google Scholar] [CrossRef]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J. Biol. Chem. 2004, 279, 31556–31567. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef]
- Tong, K.; Padmanabhan, B.; Kobayashi, A.; Shang, C.; Hirotsu, Y.; Yokoyama, S.; Yamamoto, M. Different electrostatic potentials define ETGE and DLG motifs as hinge and latch in oxidative stress response. Mol. Cell. Biol. 2007, 27, 7511–7521. [Google Scholar] [CrossRef]
- Plafker, K.S.; Nguyen, L.; Barneche, M.; Mirza, S.; Crawford, D.; Plafker, S.M. The ubiquitin-conjugating enzyme UbcM2 can regulate the stability and activity of the antioxidant transcription factor Nrf2. J. Biol. Chem. 2010, 285, 23064–23074. [Google Scholar] [CrossRef]
- Katoh, Y.; Itoh, K.; Yoshida, E.; Miyagishi, M.; Fukamizu, A.; Yamamoto, M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 2001, 6, 857–868. [Google Scholar]
- Nioi, P.; Nguyen, T.; Sherratt, P.J.; Pickett, C.B. The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol. Cell. Biol. 2005, 25, 10895–10906. [Google Scholar] [CrossRef]
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781. [Google Scholar] [CrossRef]
- Liu, Q.; Gao, Y.; Ci, X. Role of Nrf2 and Its Activators in Respiratory Diseases. Oxidative Med. Cell. Longev. 2019, 2019, 7090534. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, K.; Geng, M.; Gao, P.; Wu, X.; Hai, Y.; Li, Y.; Li, Y.; Luo, L.; Hayes, J.D.; et al. RXRalpha inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res. 2013, 73, 3097–3108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, J.D.; Chowdhry, S.; Dinkova-Kostova, A.T.; Sutherland, C. Dual regulation of transcription factor Nrf2 by Keap1 and by the combined actions of beta-TrCP and GSK-3. Biochem. Soc. Trans. 2015, 43, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Sotolongo, K.; Ghiso, J.; Rostagno, A. Nrf2 activation through the PI3K/GSK-3 axis protects neuronal cells from Abeta-mediated oxidative and metabolic damage. Alzheimers Res. Ther. 2020, 12, 13. [Google Scholar] [CrossRef]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef]
- Zipper, L.M.; Mulcahy, R.T. The Keap1 BTB/POZ dimerization function is required to sequester Nrf2 in cytoplasm. J. Biol. Chem. 2002, 277, 36544–36552. [Google Scholar] [CrossRef]
- Kobayashi, A.; Kang, M.I.; Watai, Y.; Tong, K.I.; Shibata, T.; Uchida, K.; Yamamoto, M. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol. Cell. Biol. 2006, 26, 221–229. [Google Scholar] [CrossRef]
- Furukawa, M.; Xiong, Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol. Cell. Biol. 2005, 25, 162–171. [Google Scholar] [CrossRef]
- Adams, J.; Kelso, R.; Cooley, L. The kelch repeat superfamily of proteins: Propellers of cell function. Trends Cell Biol. 2000, 10, 17–24. [Google Scholar] [CrossRef]
- Zhang, D.D.; Lo, S.C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 2004, 24, 10941–10953. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Kügler, S.; Lastres-Becker, I. Pharmacological targeting of GSK-3 and NRF2 provides neuroprotection in a preclinical model of tauopathy. Redox Biol. 2018, 14, 522–534. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A. Structural and functional characterization of Nrf2 degradation by glycogen synthase kinase 3/beta-TrCP. Free Radic. Biol. Med. 2015, 88, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell. Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Cao, Z.; Guo, Y.; Tong, C.; Yang, S.; Long, M.; Li, P.; He, J. Selenium Yeast Alleviates Ochratoxin A-Induced Apoptosis and Oxidative Stress via Modulation of the PI3K/AKT and Nrf2/Keap1 Signaling Pathways in the Kidneys of Chickens. Oxidative Med. Cell. Longev. 2020, 2020, 4048706. [Google Scholar] [CrossRef]
- Xiao, Q.; Piao, R.; Wang, H.; Li, C.; Song, L. Orientin-mediated Nrf2/HO-1 signal alleviates H2O2-induced oxidative damage via induction of JNK and PI3K/AKT activation. Int. J. Biol. Macromol. 2018, 118, 747–755. [Google Scholar] [CrossRef]
- Motohashi, H.; Yamamoto, M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef]
- Nguyen, T.; Sherratt, P.J.; Pickett, C.B. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 233–260. [Google Scholar] [CrossRef]
- Rushmore, T.H.; Morton, M.R.; Pickett, C.B. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J. Biol. Chem. 1991, 266, 11632–11639. [Google Scholar] [CrossRef]
- Tai, W.; Deng, S.; Wu, W.; Li, Z.; Lei, W.; Wang, Y.; Vongphouttha, C.; Zhang, T.; Dong, Z. Rapamycin attenuates the paraquat-induced pulmonary fibrosis through activating Nrf2 pathway. J. Cell. Physiol. 2020, 235, 1759–1768. [Google Scholar] [CrossRef] [PubMed]
- Oztay, F.; Tunali, S.; Kayalar, O.; Yanardag, R. The protective effect of vitamin U on valproic acid-induced lung toxicity in rats via amelioration of oxidative stress. J. Biochem. Mol. Toxicol. 2020, 34, e22602. [Google Scholar] [CrossRef] [PubMed]
- An, L.; Peng, L.; Sun, N.; Yang, Y.; Zhang, X.; Li, B.; Liu, B.; Li, P.; Chen, J. Tanshinone IIA Activates Nuclear Factor-Erythroid 2-Related Factor 2 to Restrain Pulmonary Fibrosis via Regulation of Redox Homeostasis and Glutaminolysis. Antioxid. Redox Signal. 2019, 30, 1831–1848. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Hua, C.; Yang, X.; Fan, X.; Song, H.; Peng, L.; Ci, X. Pterostilbene prevents LPS-induced early pulmonary fibrosis by suppressing oxidative stress, inflammation and apoptosis in vivo. Food Funct. 2020, 11, 4471–4484. [Google Scholar] [CrossRef]
- Kikuchi, N.; Ishii, Y.; Morishima, Y.; Yageta, Y.; Haraguchi, N.; Itoh, K.; Yamamoto, M.; Hizawa, N. Nrf2 protects against pulmonary fibrosis by regulating the lung oxidant level and Th1/Th2 balance. Respir. Res. 2010, 11, 31. [Google Scholar] [CrossRef]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef]
- Maher, J.; Dieter, M.; Aleksunes, L.; Slitt, A.; Guo, G.; Tanaka, Y.; Scheffer, G.; Chan, J.; Manautou, J.; Chen, Y.; et al. Oxidative and electrophilic stress induces multidrug resistance-associated protein transporters via the nuclear factor-E2-related factor-2 transcriptional pathway. Hepatology 2007, 46, 1597–1610. [Google Scholar] [CrossRef]
- Moldoveanu, B.; Otmishi, P.; Jani, P.; Walker, J.; Sarmiento, X.; Guardiola, J.; Saad, M.; Yu, J. Inflammatory mechanisms in the lung. J. Inflamm. Res. 2009, 2. [Google Scholar]
- Robb, C.T.; Regan, K.H.; Dorward, D.A.; Rossi, A.G. Key mechanisms governing resolution of lung inflammation. Semin. Immunopathol. 2016, 38, 425–448. [Google Scholar] [CrossRef]
- Harari, S.; Caminati, A. IPF: New insight on pathogenesis and treatment. Allergy 2010, 65, 537–553. [Google Scholar] [CrossRef]
- Chen, Y.; Cheng, Y.; Chen, J.; Tsai, C.; Wang, T.; Wu, C.; Chang, P.; Yeh, W. Cardamonin attenuates phorbol 12-myristate 13-acetate-induced pulmonary inflammation in alveolar macrophages. Food Chem. Toxicol. 2021, 159, 112761. [Google Scholar] [CrossRef] [PubMed]
- Sauler, M.; Bazan, I.S.; Lee, P.J. Cell Death in the Lung: The Apoptosis-Necroptosis Axis. Annu. Rev. Physiol. 2019, 81, 375–402. [Google Scholar] [CrossRef]
- Wu, L.; Luo, Z.; Zheng, J.; Yao, P.; Yuan, Z.; Lv, X.; Zhao, J.; Wang, M. IL-33 Can Promote the Process of Pulmonary Fibrosis by Inducing the Imbalance Between MMP-9 and TIMP-1. Inflammation 2018, 41, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef] [PubMed]
- Parimon, T.; Yao, C.; Stripp, B.R.; Noble, P.W.; Chen, P. Alveolar Epithelial Type II Cells as Drivers of Lung Fibrosis in Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2020, 21, 2269. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Yu, Y.; Huang, H.; Hu, Y.; Fu, S.; Wang, Z.; Shi, M.; Zhao, X.; Yuan, J.; Li, J.; et al. Progressive Pulmonary Fibrosis Is Caused by Elevated Mechanical Tension on Alveolar Stem Cells. Cell 2020, 180, 107–121.e17. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Christman, J.W. Editorial: Alveolar Macrophages in Lung Inflammation and Resolution. Front. Immunol. 2019, 10, 2275. [Google Scholar] [CrossRef]
- Wu, J.; Yan, Z.; Schwartz, D.E.; Yu, J.; Malik, A.B.; Hu, G. Activation of NLRP3 inflammasome in alveolar macrophages contributes to mechanical stretch-induced lung inflammation and injury. J. Immunol. 2013, 190, 3590–3599. [Google Scholar] [CrossRef]
- Misharin, A.V.; Morales-Nebreda, L.; Reyfman, P.A.; Cuda, C.M.; Walter, J.M.; McQuattie-Pimentel, A.C.; Chen, C.I.; Anekalla, K.R.; Joshi, N.; Williams, K.J.N.; et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med. 2017, 214, 2387–2404. [Google Scholar] [CrossRef]
- Joshi, N.; Watanabe, S.; Verma, R.; Jablonski, R.P.; Chen, C.I.; Cheresh, P.; Markov, N.S.; Reyfman, P.A.; McQuattie-Pimentel, A.C.; Sichizya, L.; et al. A spatially restricted fibrotic niche in pulmonary fibrosis is sustained by M-CSF/M-CSFR signalling in monocyte-derived alveolar macrophages. Eur. Respir. J. 2020, 55, 1900646. [Google Scholar] [CrossRef]
- Kobayashi, E.; Suzuki, T.; Yamamoto, M. Roles nrf2 plays in myeloid cells and related disorders. Oxidative Med. Cell. Longev. 2013, 2013, 529219. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Du, R.; Yin, S.; Liu, X.; Xu, G.; Cao, W. Nrf2 is essential for the anti-inflammatory effect of carbon monoxide in LPS-induced inflammation. Inflamm. Res. 2015, 64, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-Y.; Reddy, S.P.M.; Yamamoto, M.; Kleeberger, S.R. The transcription factor NRF2 protects against pulmonary fibrosis. FASEB J. 2004, 18, 1258–1260. [Google Scholar] [PubMed]
- Reddy, N.M.; Kleeberger, S.R.; Kensler, T.W.; Yamamoto, M.; Hassoun, P.M.; Reddy, S.P. Disruption of Nrf2 impairs the resolution of hyperoxia-induced acute lung injury and inflammation in mice. J. Immunol. 2009, 182, 7264–7271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traver, G.; Mont, S.; Gius, D.; Lawson, W.; Ding, G.; Sekhar, K.; Freeman, M. Loss of Nrf2 promotes alveolar type 2 cell loss in irradiated, fibrotic lung. Free Radic. Biol. Med. 2017, 112, 578–586. [Google Scholar] [CrossRef]
- Reddy, N.; Tamatam, C.; Aparna, A.; Reddy, S. Nrf2 Is Required for Optimal Alveolar-Macrophage-Mediated Apoptotic Neutrophil Clearance after Oxidant Injury. Antioxidants 2022, 11, 212. [Google Scholar] [CrossRef]
- Sehsah, R.; Wu, W.; Ichihara, S.; Hashimoto, N.; Hasegawa, Y.; Zong, C.; Itoh, K.; Yamamoto, M.; Elsayed, A.A.; El-Bestar, S.; et al. Role of Nrf2 in inflammatory response in lung of mice exposed to zinc oxide nanoparticles. Part Fibre Toxicol. 2019, 16, 47. [Google Scholar] [CrossRef]
- Dong, J.; Ma, Q. Suppression of basal and carbon nanotube-induced oxidative stress, inflammation and fibrosis in mouse lungs by Nrf2. Nanotoxicology 2016, 10, 699–709. [Google Scholar] [CrossRef]
- Zhou, E.; Li, Y.; Wei, Z.; Fu, Y.; Lei, H.; Zhang, N.; Yang, Z.; Xie, G. Schisantherin A protects lipopolysaccharide-induced acute respiratory distress syndrome in mice through inhibiting NF-kappaB and MAPKs signaling pathways. Int. Immunopharmacol. 2014, 22, 133–140. [Google Scholar] [CrossRef]
- Pedruzzi, L.M.; Stockler-Pinto, M.B.; Leite, M., Jr.; Mafra, D. Nrf2-keap1 system versus NF-kappaB: The good and the evil in chronic kidney disease? Biochimie 2012, 94, 2461–2466. [Google Scholar] [CrossRef]
- Liu, S.; Chen, Z.J. Expanding role of ubiquitination in NF-kappaB signaling. Cell Res. 2011, 21, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Wang, C.; Spencer, E.; Yang, L.; Braun, A.; You, J.; Slaughter, C.; Pickart, C.; Chen, Z.J. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 2000, 103, 351–361. [Google Scholar] [PubMed]
- Wang, C.; Deng, L.; Hong, M.; Akkaraju, G.R.; Inoue, J.; Chen, Z.J. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 2001, 412, 346–351. [Google Scholar] [PubMed]
- Chen, Z.J.; Parent, L.; Maniatis, T. Site-specific phosphorylation of IkappaBalpha by a novel ubiquitination-dependent protein kinase activity. Cell 1996, 84, 853–862. [Google Scholar]
- Kang, H.H.; Kim, I.K.; Yeo, C.D.; Kim, S.W.; Lee, H.Y.; Im, J.H.; Kwon, H.Y.; Lee, S.H. The Effects of Chronic Intermittent Hypoxia in Bleomycin-Induced Lung Injury on Pulmonary Fibrosis via Regulating the NF-kappaB/Nrf2 Signaling Pathway. Tuberc. Respir. Dis. 2020, 83, S63–S74. [Google Scholar] [CrossRef]
- Raish, M.; Ahmad, A.; Ahmad Ansari, M.; Ahad, A.; Al-Jenoobi, F.I.; Al-Mohizea, A.M.; Khan, A.; Ali, N. Sinapic acid ameliorates bleomycin-induced lung fibrosis in rats. Biomed. Pharmacother. 2018, 108, 224–231. [Google Scholar] [CrossRef]
- Ryter, S.W.; Alam, J.; Choi, A.M. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef]
- Alam, J.; Stewart, D.; Touchard, C.; Boinapally, S.; Choi, A.M.; Cook, J.L. Nrf2, a Cap’n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J. Biol. Chem. 1999, 274, 26071–26078. [Google Scholar]
- Ahmad, A.; Alkharfy, K.M.; Jan, B.L.; Ahad, A.; Ansari, M.A.; Al-Jenoobi, F.I.; Raish, M. Thymoquinone treatment modulates the Nrf2/HO-1 signaling pathway and abrogates the inflammatory response in an animal model of lung fibrosis. Exp. Lung Res. 2020, 46, 53–63. [Google Scholar] [CrossRef]
- Yang, D.; Qiu, J.; Zhou, H.; Yu, Y.; Zhou, D.; Xu, Y.; Zhu, M.; Ge, X.; Li, J.; Lv, C.; et al. Dihydroartemisinin alleviates oxidative stress in bleomycin-induced pulmonary fibrosis. Life Sci. 2018, 205, 176–183. [Google Scholar] [CrossRef]
- Huai, B.; Ding, J. Atractylenolide III attenuates bleomycin-induced experimental pulmonary fibrosis and oxidative stress in rat model via Nrf2/NQO1/HO-1 pathway activation. Immunopharmacol. Immunotoxicol. 2020, 42, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cai, Z.; Mehmood, S.; Wang, Y.; Pan, W.; Zhang, W.; Lu, Y.; Chen, Y. Polysaccharide FMP-1 from Morchella esculenta attenuates cellular oxidative damage in human alveolar epithelial A549 cells through PI3K/AKT/Nrf2/HO-1 pathway. Int. J. Biol. Macromol. 2018, 120, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Li, J.; Zhang, L.; Sun, Y.; Jiang, J.; Huang, Y.; Xu, H.; Jiang, H.; Hu, R. Nrf2 protects against acute lung injury and inflammation by modulating TLR4 and Akt signaling. Free Radic. Biol. Med. 2018, 121, 78–85. [Google Scholar] [CrossRef]
- Cheresh, P.; Kim, S.J.; Tulasiram, S.; Kamp, D.W. Oxidative stress and pulmonary fibrosis. Biochim. Biophys. Acta 2013, 1832, 1028–1040. [Google Scholar] [CrossRef]
- Kinnula, V.L.; Fattman, C.L.; Tan, R.J.; Oury, T.D. Oxidative Stress in Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2005, 172, 417–422. [Google Scholar] [CrossRef] [Green Version]
- Kinnula, V.L.; Crapo, J.D. Superoxide dismutases in the lung and human lung diseases. Am. J. Respir. Crit. Care Med. 2003, 167, 1600–1619. [Google Scholar] [CrossRef]
- Walters, D.M.; Cho, H.Y.; Kleeberger, S.R. Oxidative stress and antioxidants in the pathogenesis of pulmonary fibrosis: A potential role for Nrf2. Antioxid. Redox Signal. 2008, 10, 321–332. [Google Scholar] [CrossRef]
- Wang, T.; Dai, F.; Li, G.; Chen, X.; Li, Y.; Wang, S.; Ren, D.; Wang, X.; Lou, H.; Zhou, B.; et al. Trans-4,4’-dihydroxystilbene ameliorates cigarette smoke-induced progression of chronic obstructive pulmonary disease via inhibiting oxidative stress and inflammatory response. Free Radic. Biol. Med. 2020, 152, 525–539. [Google Scholar] [CrossRef]
- Wang, S.; Sarria, B.; Mateos, R.; Goya, L.; Bravo-Clemente, L. TNF-alpha-induced oxidative stress and endothelial dysfunction in EA.hy926 cells is prevented by mate and green coffee extracts, 5-caffeoylquinic acid and its microbial metabolite, dihydrocaffeic acid. Int. J. Food Sci. Nutr. 2019, 70, 267–284. [Google Scholar] [CrossRef]
- Leung, C.C.; Yu, I.T.S.; Chen, W. Silicosis. Lancet 2012, 379, 2008–2018. [Google Scholar] [CrossRef]
- Koga, Y.; Satoh, T.; Kaira, K.; Hachisu, Y.; Ishii, Y.; Yajima, T.; Hisada, T.; Yokoo, H.; Dobashi, K. Progression of Idiopathic Pulmonary Fibrosis Is Associated with Silica/Silicate Inhalation. Environ. Sci. Technol. Lett. 2021, 8, 903–910. [Google Scholar] [CrossRef]
- Zhu, Z.; Li, Q.; Xu, C.; Zhao, J.; Li, S.; Wang, Y.; Tian, L. Sodium tanshinone IIA sulfonate attenuates silica-induced pulmonary fibrosis in rats via activation of the Nrf2 and thioredoxin system. Environ. Toxicol. Pharmacol. 2020, 80, 103461. [Google Scholar] [CrossRef]
- Feng, F.; Cheng, P.; Xu, S.; Li, N.; Wang, H.; Zhang, Y.; Wang, W. Tanshinone IIA attenuates silica-induced pulmonary fibrosis via Nrf2-mediated inhibition of EMT and TGF-β1/Smad signaling. Chem. Biol. Interact. 2020, 319, 109024. [Google Scholar] [CrossRef] [PubMed]
- Feng, F.; Cheng, P.; Zhang, H.; Li, N.; Qi, Y.; Wang, H.; Wang, Y.; Wang, W. The Protective Role of Tanshinone IIA in Silicosis Rat Model via TGF-β1/Smad Signaling Suppression, NOX4 Inhibition and Nrf2/ARE Signaling Activation. Drug Des. Dev. Ther. 2019, 13, 4275–4290. [Google Scholar] [CrossRef]
- Paliogiannis, P.; Fois, A.G.; Collu, C.; Bandinu, A.; Zinellu, E.; Carru, C.; Pirina, P.; Mangoni, A.A.; Zinellu, A. Oxidative stress-linked biomarkers in idiopathic pulmonary fibrosis: A systematic review and meta-analysis. Biomark. Med. 2018, 12, 1175–1184. [Google Scholar] [CrossRef]
- Fois, A.; Sotgiu, E.; Scano, V.; Negri, S.; Mellino, S.; Zinellu, E.; Pirina, P.; Pintus, G.; Carru, C.; Mangoni, A.; et al. Effects of Pirfenidone and Nintedanib on Markers of Systemic Oxidative Stress and Inflammation in Patients with Idiopathic Pulmonary Fibrosis: A Preliminary Report. Antioxidants 2020, 9, 1064. [Google Scholar] [CrossRef]
- Kovac, S.; Angelova, P.R.; Holmstrom, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta 2015, 1850, 794–801. [Google Scholar] [CrossRef]
- Han, B.; Li, S.; Lv, Y.; Yang, D.; Li, J.; Yang, Q.; Wu, P.; Lv, Z.; Zhang, Z. Dietary melatonin attenuates chromium-induced lung injury via activating the Sirt1/Pgc-1α/Nrf2 pathway. Food Funct. 2019, 10, 5555–5565. [Google Scholar] [CrossRef]
- Zhao, H.; Eguchi, S.; Alam, A.; Ma, D. The role of nuclear factor-erythroid 2 related factor 2 (Nrf-2) in the protection against lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L155–L162. [Google Scholar] [CrossRef]
- Xin, X.; Yao, D.; Zhang, K.; Han, S.; Liu, D.; Wang, H.; Liu, X.; Li, G.; Huang, J.; Wang, J. Protective effects of Rosavin on bleomycin-induced pulmonary fibrosis via suppressing fibrotic and inflammatory signaling pathways in mice. Biomed. Pharmacother. 2019, 115, 108870. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Deng, W.; Yang, Y.; Wei, S.; Xue, L.; Tao, S. Pharmaceutic application of vitamin D3 on particle-induced fibrotic effects through induction of Nrf2 signals. Toxicol. Res. 2020, 9, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Hecker, L.; Logsdon, N.J.; Kurundkar, D.; Kurundkar, A.; Bernard, K.; Hock, T.; Meldrum, E.; Sanders, Y.Y.; Thannickal, V.J. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci. Transl. Med. 2014, 6, 231ra247. [Google Scholar] [CrossRef] [PubMed]
- Veith, C.; Boots, A.W.; Idris, M.; van Schooten, F.J.; van der Vliet, A. Redox Imbalance in Idiopathic Pulmonary Fibrosis: A Role for Oxidant Cross-Talk Between NADPH Oxidase Enzymes and Mitochondria. Antioxid. Redox Signal. 2019, 31, 1092–1115. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.C.; Jiang, F.; Peshavariya, H.M.; Dusting, G.J. Regulation of cell proliferation by NADPH oxidase-mediated signaling: Potential roles in tissue repair, regenerative medicine and tissue engineering. Pharmacol. Ther. 2009, 122, 97–108. [Google Scholar] [CrossRef]
- Sirokmany, G.; Donko, A.; Geiszt, M. Nox/Duox Family of NADPH Oxidases: Lessons from Knockout Mouse Models. Trends Pharmacol. Sci. 2016, 37, 318–327. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.M.; Xu, S.Y.; Feng, Y.Z.; Cheng, Y.R.; Xiong, J.B.; Zhou, Y.; Guan, C.X. The role of NOX4 in pulmonary diseases. J. Cell. Physiol. 2021, 236, 1628–1637. [Google Scholar] [CrossRef]
- Kawahara, T.; Quinn, M.T.; Lambeth, J.D. Molecular evolution of the reactive oxygen-generating NADPH oxidase (Nox/Duox) family of enzymes. BMC Evol. Biol. 2007, 7, 109. [Google Scholar] [CrossRef]
- Hecker, L.; Cheng, J.; Thannickal, V.J. Targeting NOX enzymes in pulmonary fibrosis. Cell. Mol. Life Sci. 2012, 69, 2365–2371. [Google Scholar] [CrossRef]
- Zeng, H.; Wang, Y.; Gu, Y.; Wang, J.; Zhang, H.; Gao, H.; Jin, Q.; Zhao, L. Polydatin attenuates reactive oxygen species-induced airway remodeling by promoting Nrf2-mediated antioxidant signaling in asthma mouse model. Life Sci. 2019, 218, 25–30. [Google Scholar] [CrossRef]
- Li, C.; Sun, X.; Li, A.; Mo, M.; Zhao, Z. S-Allylmercaptocysteine attenuates Bleomycin-induced pulmonary fibrosis in mice via suppressing TGF-β1/Smad and oxidative stress pathways. Int. Immunopharmacol. 2020, 79, 106110. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Cao, B.; Liu, B.; Li, W.; Chen, Y.; Chen, H.; Liu, Y.; Liu, T. A novel Gallic acid derivative attenuates BLM-induced pulmonary fibrosis in mice. Int. Immunopharmacol. 2018, 64, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; An, L.; Sun, N.; Ma, Y.; Zhang, X.; Liu, W.; Liu, B.; Li, P.; Chen, J. Salvia miltiorrhiza Restrains Reactive Oxygen Species-Associated Pulmonary Fibrosis via Targeting Nrf2-Nox4 Redox Balance. Am. J. Chin. Med. 2019, 47, 1113–1131. [Google Scholar] [CrossRef]
- Wu, Y.; Zheng, L.; Yang, H.; Lyu, X. Research progress of itaconate on the regulation of macrophage inflammation. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 2021, 33, 1388–1392. [Google Scholar] [CrossRef]
- Mora, A.L.; Rojas, M.; Pardo, A.; Selman, M. Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease. Nat. Rev. Drug Discov. 2017, 16, 810. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; Pardo, A. Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. an integral model. Am. J. Respir. Crit. Care Med. 2014, 189, 1161–1172. [Google Scholar] [CrossRef]
- Loomis-King, H.; Flaherty, K.R.; Moore, B.B. Pathogenesis, current treatments and future directions for idiopathic pulmonary fibrosis. Curr. Opin. Pharmacol. 2013, 13, 377–385. [Google Scholar] [CrossRef]
- Saito, S.; Zhuang, Y.; Suzuki, T.; Ota, Y.; Bateman, M.E.; Alkhatib, A.L.; Morris, G.F.; Lasky, J.A. HDAC8 inhibition ameliorates pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L175–L186. [Google Scholar] [CrossRef]
- Aziz, R.K.; Kulkarni, A.A.; Thatcher, T.H.; Olsen, K.C.; Maggirwar, S.B.; Phipps, R.P.; Sime, P.J. PPAR-γ Ligands Repress TGFβ-Induced Myofibroblast Differentiation by Targeting the PI3K/Akt Pathway: Implications for Therapy of Fibrosis. PLoS ONE 2011, 6, e15909. [Google Scholar] [CrossRef]
- Fernandez, I.E.; Eickelberg, O. The impact of TGF-beta on lung fibrosis: From targeting to biomarkers. Proc. Am. Thorac. Soc. 2012, 9, 111–116. [Google Scholar] [CrossRef]
- Han, Y.; Gu, X.; Yang, C.; Ji, H.; Lan, Y.; Bi, Y.; Si, R.; Qu, J.; Cheng, M.; Gao, J. Protective effect of dimethyl itaconate against fibroblast-myofibroblast differentiation during pulmonary fibrosis by inhibiting TXNIP. J. Cell. Physiol. 2021, 236, 7734–7744. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.S.; Liu, C.; Lv, X.X.; Cui, B.; Yan, J.; Li, Y.X.; Li, K.; Hua, F.; Zhang, X.W.; Yu, J.J.; et al. The chemokine CCL1 triggers an AMFR-SPRY1 pathway that promotes differentiation of lung fibroblasts into myofibroblasts and drives pulmonary fibrosis. Immunity 2021, 54, 2042–2056.e48. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, J.; Hu, Y.; Pan, T.; Xu, Y.; Yu, J.; Xiong, W.; Zhou, Q.; Wang, Y. Local administration of liposomal-based Srpx2 gene therapy reverses pulmonary fibrosis by blockading fibroblast-to-myofibroblast transition. Theranostics 2021, 11, 7110–7125. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Prunotto, M.; Desmouliere, A.; Varga, J.; De Wever, O.; Mareel, M.; Gabbiani, G. Recent developments in myofibroblast biology: Paradigms for connective tissue remodeling. Am. J. Pathol. 2012, 180, 1340–1355. [Google Scholar] [CrossRef] [PubMed]
- Tsubouchi, K.; Araya, J.; Minagawa, S.; Hara, H.; Ichikawa, A.; Saito, N.; Kadota, T.; Sato, N.; Yoshida, M.; Kurita, Y.; et al. Azithromycin attenuates myofibroblast differentiation and lung fibrosis development through proteasomal degradation of NOX4. Autophagy 2017, 13, 1420–1434. [Google Scholar] [CrossRef]
- El Agha, E.; Kramann, R.; Schneider, R.K.; Li, X.; Seeger, W.; Humphreys, B.D.; Bellusci, S. Mesenchymal Stem Cells in Fibrotic Disease. Cell Stem Cell 2017, 21, 166–177. [Google Scholar] [CrossRef]
- Artaud-Macari, E.; Goven, D.; Brayer, S.; Hamimi, A.; Besnard, V.; Marchal-Somme, J.; Ali, Z.E.; Crestani, B.; Kerdine-Romer, S.; Boutten, A.; et al. Nuclear factor erythroid 2-related factor 2 nuclear translocation induces myofibroblastic dedifferentiation in idiopathic pulmonary fibrosis. Antioxid. Redox Signal. 2013, 18, 66–79. [Google Scholar] [CrossRef]
- Zhang, Z.; Qu, J.; Zheng, C.; Zhang, P.; Zhou, W.; Cui, W.; Mo, X.; Li, L.; Xu, L.; Gao, J. Nrf2 antioxidant pathway suppresses Numb-mediated epithelial-mesenchymal transition during pulmonary fibrosis. Cell Death Dis. 2018, 9, 83. [Google Scholar] [CrossRef]
- Bartis, D.; Mise, N.; Mahida, R.Y.; Eickelberg, O.; Thickett, D.R. Epithelial-mesenchymal transition in lung development and disease: Does it exist and is it important? Thorax 2014, 69, 760–765. [Google Scholar] [CrossRef]
- Barkauskas, C.E.; Noble, P.W. Cellular mechanisms of tissue fibrosis. 7. New insights into the cellular mechanisms of pulmonary fibrosis. Am. J. Physiol. Cell Physiol. 2014, 306, C987–C996. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Liu, C.; Zhou, D.; Zhang, L. TGF-beta/SMAD Pathway and Its Regulation in Hepatic Fibrosis. J. Histochem. Cytochem. 2016, 64, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhang, T.; Shan, S.; Wang, S.; Bian, W.; Ren, T.; Yang, D. MiR-124 regulates transforming growth factor-beta1 induced differentiation of lung resident mesenchymal stem cells to myofibroblast by repressing Wnt/beta-catenin signaling. Dev. Biol. 2019, 449, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Mo, X.; Cui, W.; Zhang, Z.; Li, D.; Li, L.; Xu, L.; Yao, H.; Gao, J. Nrf2 inhibits epithelial-mesenchymal transition by suppressing snail expression during pulmonary fibrosis. Sci. Rep. 2016, 6, 38646. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Wu, X.; Wang, Y.; Ji, S.; Zhang, W.; Kang, J.; Li, J.; Fei, G. Melatonin prevents LPS-induced epithelial-mesenchymal transition in human alveolar epithelial cells via the GSK-3β/Nrf2 pathway. Biomed. Pharmacother. 2020, 132, 110827. [Google Scholar] [CrossRef]
- Ebrahimpour, A.; Wang, M.; Li, L.; Jegga, A.G.; Bonnen, M.D.; Eissa, N.T.; Raghu, G.; Jyothula, S.; Kheradmand, F.; Hanania, N.A.; et al. Esomeprazole attenuates inflammatory and fibrotic response in lung cells through the MAPK/Nrf2/HO1 pathway. J. Inflamm. 2021, 18, 17. [Google Scholar] [CrossRef]
- Richter, K.; Kietzmann, T. Reactive oxygen species and fibrosis: Further evidence of a significant liaison. Cell Tissue Res. 2016, 365, 591–605. [Google Scholar] [CrossRef]
- Huang, L.S.; Jiang, P.; Feghali-Bostwick, C.; Reddy, S.P.; Garcia, J.G.N.; Natarajan, V. Lysocardiolipin acyltransferase regulates TGF-beta mediated lung fibroblast differentiation. Free Radic. Biol. Med. 2017, 112, 162–173. [Google Scholar] [CrossRef]
- Hecker, L.; Vittal, R.; Jones, T.; Jagirdar, R.; Luckhardt, T.R.; Horowitz, J.C.; Pennathur, S.; Martinez, F.J.; Thannickal, V.J. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 2009, 15, 1077–1081. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Däbritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457.e13–470.e13. [Google Scholar] [CrossRef]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Peace, C.G.; O’Neill, L.A. The role of itaconate in host defense and inflammation. J. Clin. Investig. 2022, 132, e148548. [Google Scholar] [CrossRef] [PubMed]
- Ogger, P.P.; Albers, G.J.; Hewitt, R.J.; O’Sullivan, B.J.; Powell, J.E.; Calamita, E.; Ghai, P.; Walker, S.A.; McErlean, P.; Saunders, P.; et al. Itaconate controls the severity of pulmonary fibrosis. Sci. Immunol. 2020, 5, eaan2946. [Google Scholar] [CrossRef]
- Sehgal, S.N. Rapamune (RAPA, rapamycin, sirolimus): Mechanism of action immunosuppressive effect results from blockade of signal transduction and inhibition of cell cycle progression. Clin. Biochem. 1998, 31, 335–340. [Google Scholar] [CrossRef]
- Korfhagen, T.R.; Le Cras, T.D.; Davidson, C.R.; Schmidt, S.M.; Ikegami, M.; Whitsett, J.A.; Hardie, W.D. Rapamycin Prevents Transforming Growth Factor-α–Induced Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2009, 41, 562–572. [Google Scholar] [CrossRef]
- Shao, X.; Li, M.; Luo, C.; Wang, Y.-Y.; Lu, Y.-Y.; Feng, S.; Li, H.; Lang, X.-B.; Wang, Y.-C.; Lin, C.; et al. Effects of rapamycin against paraquat-induced pulmonary fibrosis in mice. J. Zhejiang Univ. Sci. B 2015, 16, 52–61. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Tai, W.; Qu, X.; Wu, W.; Li, Z.; Deng, S.; Vongphouttha, C.; Dong, Z. Rapamycin protects against paraquat-induced pulmonary fibrosis: Activation of Nrf2 signaling pathway. Biochem. Biophys. Res. Commun. 2017, 490, 535–540. [Google Scholar] [CrossRef]
- Yan, B.; Ma, Z.; Shi, S.; Hu, Y.; Ma, T.; Rong, G.; Yang, J. Sulforaphane prevents bleomycininduced pulmonary fibrosis in mice by inhibiting oxidative stress via nuclear factor erythroid 2related factor2 activation. Mol. Med. Rep. 2017, 15, 4005–4014. [Google Scholar] [CrossRef]
- Dong, H.; Yang, J.; Wang, Y.; Jiang, Y.; Chen, J.; Zhang, W.; Lu, Y.; Chen, L.; Chen, Y. Polysaccharide SAFP from Sarcodon aspratus attenuates oxidative stress-induced cell damage and bleomycin-induced pulmonary fibrosis. Int. J. Biol. Macromol. 2020, 164, 1215–1236. [Google Scholar] [CrossRef]
- Cui, Y.; Xin, H.; Tao, Y.; Mei, L.; Wang, Z. Arenaria kansuensis attenuates pulmonary fibrosis in mice via the activation of Nrf2 pathway and the inhibition of NF-kB/TGF-beta1/Smad2/3 pathway. Phytother. Res. 2021, 35, 974–986. [Google Scholar] [CrossRef]
- Veith, C.; Drent, M.; Bast, A.; van Schooten, F.; Boots, A. The disturbed redox-balance in pulmonary fibrosis is modulated by the plant flavonoid quercetin. Toxicol. Appl. Pharmacol. 2017, 336, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Wen, L.; Shi, Q.; Gao, F.; Huang, B.; Wang, C. Chelerythrine Ameliorates Pulmonary Fibrosis via Activating the Nrf2/ARE Signaling Pathway. Cell Biochem. Biophys. 2021, 79, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Zhou, T.; Zhao, F.; Xiong, D.; He, B.; Hua, Q.; Lin, M.; Deng, L.; Sang, X.; Xie, W.; et al. p62-Nrf2 Regulatory Loop Mediates the Anti-Pulmonary Fibrosis Effect of Bergenin. Antioxidants 2022, 11, 307. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Li, J.; Zhao, P.; Li, Y.; Li, M.; Feng, S.; Qin, Y.; Tian, Y.; Zhou, T. A Chinese Herbal Formula Ameliorates Pulmonary Fibrosis by Inhibiting Oxidative Stress via Upregulating Nrf2. Front. Pharmacol. 2018, 9, 628. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wu, Z.; Liu, Y.; Zhan, Z.; Yang, L.; Wang, C.; Jiang, Q.; Ran, H.; Li, P.; Wang, Z. ROS-responsive liposomes as an inhaled drug delivery nanoplatform for idiopathic pulmonary fibrosis treatment via Nrf2 signaling. J. Nanobiotechnol. 2022, 20, 213. [Google Scholar] [CrossRef]
- Shen, H.; Wu, N.; Wang, Y.; Zhao, H.; Zhang, L.; Li, T.; Zhao, M. Chloroquine attenuates paraquat-induced lung injury in mice by altering inflammation, oxidative stress and fibrosis. Int. Immunopharmacol. 2017, 46, 16–22. [Google Scholar] [CrossRef]
- Wang, L.; Li, S.; Yao, Y.; Yin, W.; Ye, T. The role of natural products in the prevention and treatment of pulmonary fibrosis: A review. Food Funct. 2021, 12, 990–1007. [Google Scholar] [CrossRef]
- Mansour, H.M.; Rhee, Y.-S.; Wu, X. Nanomedicine in pulmonary delivery. Int. J. Nanomed. 2009, 4, 299–319. [Google Scholar] [CrossRef]
- Alipour, S.; Montaseri, H.; Tafaghodi, M. Preparation and characterization of biodegradable paclitaxel loaded alginate microparticles for pulmonary delivery. Colloids Surf. B Biointerfaces 2010, 81, 521–529. [Google Scholar] [CrossRef]
- Ahmad, J.; Akhter, S.; Rizwanullah, M.; Amin, S.; Rahman, M.; Ahmad, M.Z.; Rizvi, M.A.; Kamal, M.A.; Ahmad, F.J. Nanotechnology-based inhalation treatments for lung cancer: State of the art. Nanotechnol. Sci. Appl. 2015, 8, 55–66. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Wei, J.; Deng, H.; Zheng, L.; Yang, H.; Lv, X. The Role of Nrf2 in Pulmonary Fibrosis: Molecular Mechanisms and Treatment Approaches. Antioxidants 2022, 11, 1685. https://doi.org/10.3390/antiox11091685
Wang Y, Wei J, Deng H, Zheng L, Yang H, Lv X. The Role of Nrf2 in Pulmonary Fibrosis: Molecular Mechanisms and Treatment Approaches. Antioxidants. 2022; 11(9):1685. https://doi.org/10.3390/antiox11091685
Chicago/Turabian StyleWang, Yu, Juan Wei, Huimin Deng, Li Zheng, Hao Yang, and Xin Lv. 2022. "The Role of Nrf2 in Pulmonary Fibrosis: Molecular Mechanisms and Treatment Approaches" Antioxidants 11, no. 9: 1685. https://doi.org/10.3390/antiox11091685
APA StyleWang, Y., Wei, J., Deng, H., Zheng, L., Yang, H., & Lv, X. (2022). The Role of Nrf2 in Pulmonary Fibrosis: Molecular Mechanisms and Treatment Approaches. Antioxidants, 11(9), 1685. https://doi.org/10.3390/antiox11091685