
Inflammation: Roles in Skeletal Muscle Atrophy

 ,
,
Abstract
:1. Introduction
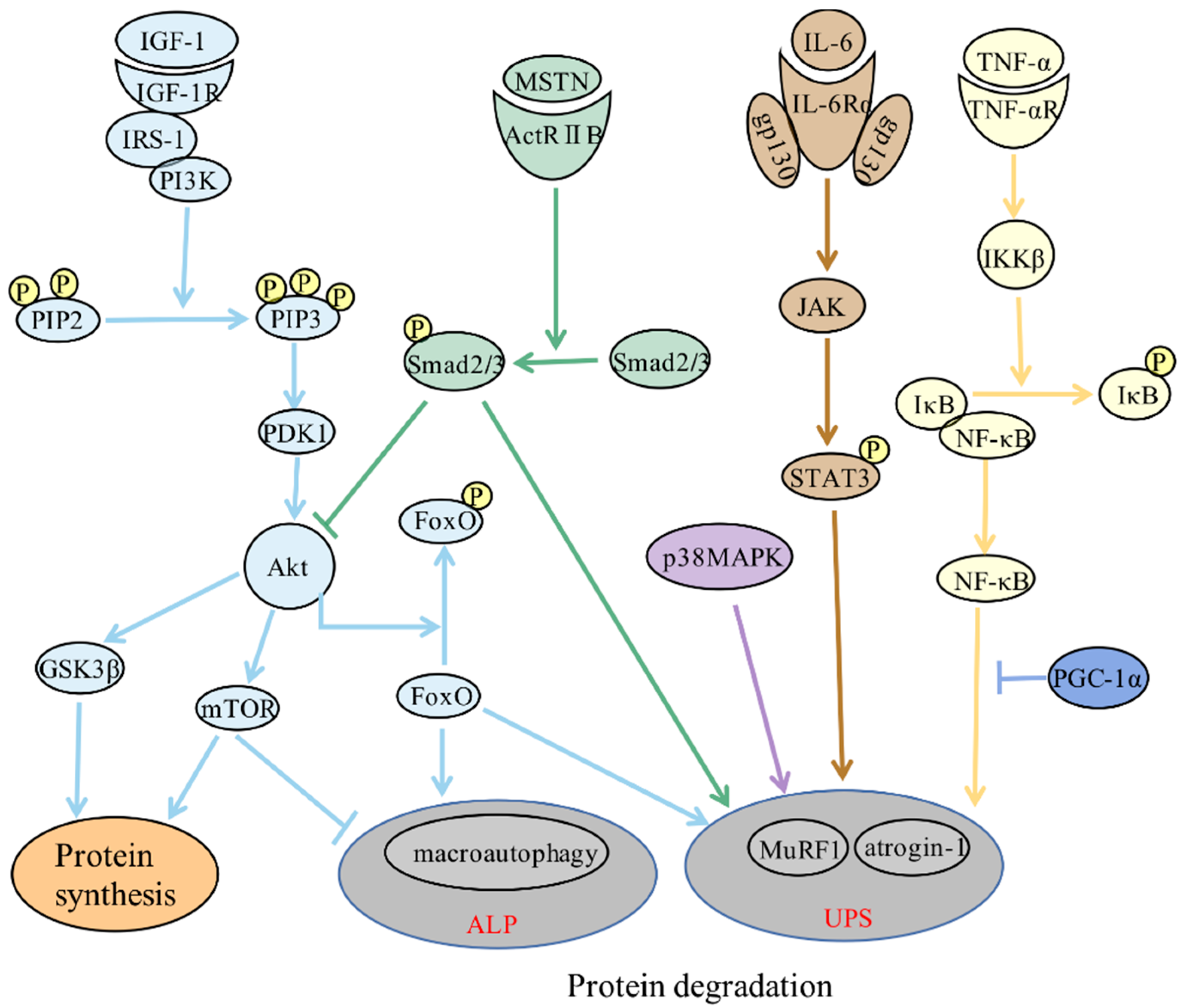
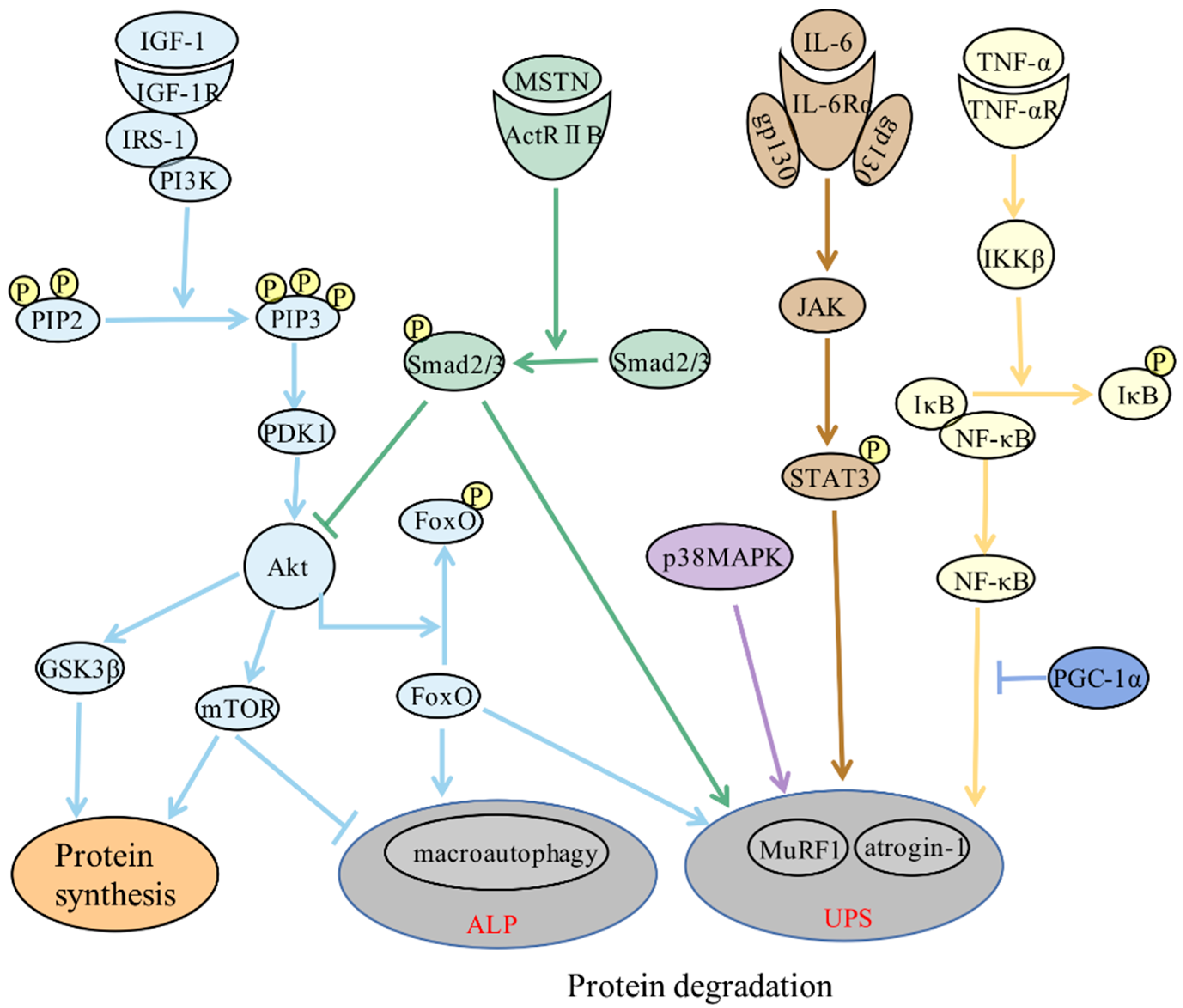
2. Molecular Mechanisms of Skeletal Muscle Atrophy
3. Role of Inflammation in Skeletal Muscle Atrophy
3.1. Direct Effect: Inflammatory Signaling Pathways Directly Regulate Skeletal Muscle Protein Metabolism
3.2. Indirect Effects
3.2.1. Inflammation Is Involved in Skeletal Muscle Atrophy via the Hypothalamic-Pituitary-Adrenal Axis
3.2.2. Inflammation Is Involved in Skeletal Muscle Atrophy through Fat Metabolism Controls
4. Relationship between Inflammation-Related Diseases and Skeletal Muscle Atrophy
4.1. Cachexia and Skeletal Muscle Atrophy
4.2. Sepsis and Skeletal Muscle Atrophy
4.3. Type 2 Diabetes Mellitus and Skeletal Muscle Atrophy
4.4. Obesity and Skeletal Muscle Atrophy
4.5. COPD and Skeletal Muscle Atrophy
4.6. CKD and Skeletal Muscle Atrophy
4.7. Nerve Injury and Skeletal Muscle Atrophy
5. Treatment of Skeletal Muscle Atrophy with Anti-Inflammatory Strategies
6. Prospects
Funding
Acknowledgments
Conflicts of Interest
References
- Yin, L.; Li, N.; Jia, W.; Wang, N.; Liang, M.; Yang, X.; Du, G. Skeletal muscle atrophy: From mechanisms to treatments. Pharmacol. Res. 2021, 172, 105807. [Google Scholar] [CrossRef]
- Wang, W.; Li, M.; Chen, Z.; Xu, L.; Chang, M.; Wang, K.; Deng, C.; Gu, Y.; Zhou, S.; Shen, Y.; et al. Biogenesis and function of extracellular vesicles in pathophysiological processes of skeletal muscle atrophy. Biochem. Pharmacol. 2022, 198, 114954. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Gong, Y.; Qiu, J.; Chen, Y.; Ding, F.; Zhao, Q. TRAF6 inhibition rescues dexamethasone-induced muscle atrophy. Int. J. Mol. Sci. 2014, 15, 11126–11141. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Fang, Q.; Ma, W.; Zhang, Q.; Qiu, J.; Gu, X.; Yang, H.; Sun, H. Skeletal Muscle Atrophy Was Alleviated by Salidroside Through Suppressing Oxidative Stress and Inflammation During Denervation. Front. Pharmacol. 2019, 10, 997. [Google Scholar] [CrossRef] [PubMed]
- Haberecht-Muller, S.; Kruger, E.; Fielitz, J. Out of Control: The Role of the Ubiquitin Proteasome System in Skeletal Muscle during Inflammation. Biomolecules 2021, 11, 1327. [Google Scholar] [CrossRef]
- Dolly, A.; Dumas, J.F.; Servais, S. Cancer cachexia and skeletal muscle atrophy in clinical studies: What do we really know? J. Cachexia Sarcopenia Muscle 2020, 11, 1413–1428. [Google Scholar] [CrossRef]
- Madaro, L.; Passafaro, M.; Sala, D.; Etxaniz, U.; Lugarini, F.; Proietti, D.; Alfonsi, M.V.; Nicoletti, C.; Gatto, S.; De Bardi, M.; et al. Denervation-activated STAT3-IL-6 signalling in fibro-adipogenic progenitors promotes myofibres atrophy and fibrosis. Nat. Cell Biol. 2018, 20, 917–927. [Google Scholar] [CrossRef]
- Wu, C.; Tang, L.; Ni, X.; Xu, T.; Fang, Q.; Xu, L.; Ma, W.; Yang, X.; Sun, H. Salidroside Attenuates Denervation-Induced Skeletal Muscle Atrophy Through Negative Regulation of Pro-inflammatory Cytokine. Front. Physiol. 2019, 10, 665. [Google Scholar] [CrossRef]
- Shen, Y.; Zhang, R.; Xu, L.; Wan, Q.; Zhu, J.; Gu, J.; Huang, Z.; Ma, W.; Shen, M.; Ding, F.; et al. Microarray Analysis of Gene Expression Provides New Insights Into Denervation-Induced Skeletal Muscle Atrophy. Front. Physiol. 2019, 10, 1298. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, X.; Ji, Y.; Zhang, L.; Wang, W.; Shen, Y.; Sun, H. A narrative review of the role of m6A in oxidative stress and inflammation. Biotarget 2021, 5, 1. [Google Scholar] [CrossRef]
- Ma, W.; Xu, T.; Wang, Y.; Wu, C.; Wang, L.; Yang, X.; Sun, H. The role of inflammatory factors in skeletal muscle injury. Biotarget 2018, 2, 7. [Google Scholar]
- Huang, Z.; Zhong, L.; Zhu, J.; Xu, H.; Ma, W.; Zhang, L.; Shen, Y.; Law, B.Y.; Ding, F.; Gu, X.; et al. Inhibition of IL-6/JAK/STAT3 pathway rescues denervation-induced skeletal muscle atrophy. Ann. Transl. Med. 2020, 8, 1681. [Google Scholar] [CrossRef]
- Wan, Q.; Zhang, L.; Huang, Z.; Zhang, H.; Gu, J.; Xu, H.; Yang, X.; Shen, Y.; Law, B.Y.; Zhu, J.; et al. Aspirin alleviates denervation-induced muscle atrophy via regulating the Sirt1/PGC-1alpha axis and STAT3 signaling. Ann. Transl. Med. 2020, 8, 1524. [Google Scholar] [CrossRef]
- Sun, H.; Sun, J.; Li, M.; Qian, L.; Zhang, L.; Huang, Z.; Shen, Y.; Law, B.Y.; Liu, L.; Gu, X. Transcriptome Analysis of Immune Receptor Activation and Energy Metabolism Reduction as the Underlying Mechanisms in Interleukin-6-Induced Skeletal Muscle Atrophy. Front. Immunol. 2021, 12, 730070. [Google Scholar] [CrossRef]
- Jackman, R.W.; Cornwell, E.W.; Wu, C.L.; Kandarian, S.C. Nuclear factor-kappaB signalling and transcriptional regulation in skeletal muscle atrophy. Exp. Physiol. 2013, 98, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhu, J.; Sun, J.; Ma, W.; Wang, L.; Zhang, Q.; Sun, H. Effect of mammalian target of rapamycin signaling pathway on nerve regeneration. Biotarget 2018, 2, 18. [Google Scholar]
- Webster, J.M.; Kempen, L.; Hardy, R.S.; Langen, R.C.J. Inflammation and Skeletal Muscle Wasting During Cachexia. Front. Physiol. 2020, 11, 597675. [Google Scholar] [CrossRef]
- Ma, W.; Cai, Y.; Shen, Y.; Chen, X.; Zhang, L.; Ji, Y.; Chen, Z.; Zhu, J.; Yang, X.; Sun, H. HDAC4 Knockdown Alleviates Denervation-Induced Muscle Atrophy by Inhibiting Myogenin-Dependent Atrogene Activation. Front. Cell Neurosci. 2021, 15, 663384. [Google Scholar] [CrossRef]
- Wang, W.; Shen, D.; Zhang, L.; Ji, Y.; Xu, L.; Chen, Z.; Shen, Y.; Gong, L.; Zhang, Q.; Shen, M.; et al. SKP-SC-EVs Mitigate Denervated Muscle Atrophy by Inhibiting Oxidative Stress and Inflammation and Improving Microcirculation. Antioxidants 2021, 11, 66. [Google Scholar] [CrossRef]
- Yoshida, T.; Delafontaine, P. Mechanisms of IGF-1-Mediated Regulation of Skeletal Muscle Hypertrophy and Atrophy. Cells 2020, 9, 1970. [Google Scholar] [CrossRef]
- Lee, Y.S.; Huynh, T.V.; Lee, S.J. Paracrine and endocrine modes of myostatin action. J. Appl. Physiol. 2016, 120, 592–598. [Google Scholar] [CrossRef] [PubMed]
- Dumont, N.A.; Bentzinger, C.F.; Sincennes, M.C.; Rudnicki, M.A. Satellite Cells and Skeletal Muscle Regeneration. Compr. Physiol. 2015, 5, 1027–1059. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Cho, J.; Song, E.J. Ubiquitin–proteasome system (UPS) as a target for anticancer treatment. Arch. Pharmacal Res. 2020, 43, 1144–1161. [Google Scholar] [CrossRef]
- Khalil, R. Ubiquitin-Proteasome Pathway and Muscle Atrophy. Adv. Exp. Med. Biol. 2018, 1088, 235–248. [Google Scholar] [CrossRef]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef]
- Kitajima, Y.; Yoshioka, K.; Suzuki, N. The ubiquitin-proteasome system in regulation of the skeletal muscle homeostasis and atrophy: From basic science to disorders. J. Physiol. Sci. JPS 2020, 70, 40. [Google Scholar] [CrossRef]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef]
- Nakao, R.; Hirasaka, K.; Goto, J.; Ishidoh, K.; Yamada, C.; Ohno, A.; Okumura, Y.; Nonaka, I.; Yasutomo, K.; Baldwin, K.M.; et al. Ubiquitin ligase Cbl-b is a negative regulator for insulin-like growth factor 1 signaling during muscle atrophy caused by unloading. Mol. Cell. Biol. 2009, 29, 4798–4811. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Grumati, P.; Coletto, L.; Sabatelli, P.; Cescon, M.; Angelin, A.; Bertaggia, E.; Blaauw, B.; Urciuolo, A.; Tiepolo, T.; Merlini, L.; et al. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat. Med. 2010, 16, 1313–1320. [Google Scholar] [CrossRef]
- Shen, Y.; Zhang, Q.; Huang, Z.; Zhu, J.; Qiu, J.; Ma, W.; Yang, X.; Ding, F.; Sun, H. Isoquercitrin Delays Denervated Soleus Muscle Atrophy by Inhibiting Oxidative Stress and Inflammation. Front. Physiol. 2020, 11, 988. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.E.; Hayashi, Y.K.; Bonne, G.; Arimura, T.; Noguchi, S.; Nonaka, I.; Nishino, I. Autophagic degradation of nuclear components in mammalian cells. Autophagy 2009, 5, 795–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raben, N.; Hill, V.; Shea, L.; Takikita, S.; Baum, R.; Mizushima, N.; Ralston, E.; Plotz, P. Suppression of autophagy in skeletal muscle uncovers the accumulation of ubiquitinated proteins and their potential role in muscle damage in Pompe disease. Hum. Mol. Genet. 2008, 17, 3897–3908. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Phogat, J.; Yadav, A.; Dabur, R. The dependency of autophagy and ubiquitin proteasome system during skeletal muscle atrophy. Biophys. Rev. 2021, 13, 203–219. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef]
- Spinozzi, S.; Albini, S.; Best, H.; Richard, I. Calpains for dummies: What you need to know about the calpain family. Biochim. Biophys. Acta Proteins Proteom. 2021, 1869, 140616. [Google Scholar] [CrossRef]
- Ono, Y.; Ojima, K.; Shinkai-Ouchi, F.; Hata, S.; Sorimachi, H. An eccentric calpain, CAPN3/p94/calpain-3. Biochimie 2016, 122, 169–187. [Google Scholar] [CrossRef]
- Hyatt, H.W.; Powers, S.K. The Role of Calpains in Skeletal Muscle Remodeling with Exercise and Inactivity-induced Atrophy. Int. J. Sports Med. 2020, 41, 994–1008. [Google Scholar] [CrossRef]
- Goll, D.E.; Neti, G.; Mares, S.W.; Thompson, V.F. Myofibrillar protein turnover: The proteasome and the calpains. J. Anim. Sci. 2008, 86, E19–E35. [Google Scholar] [CrossRef]
- Goll, D.E.; Thompson, V.F.; Li, H.; Wei, W.; Cong, J. The calpain system. Physiol. Rev. 2003, 83, 731–801. [Google Scholar] [CrossRef] [PubMed]
- Polster, B.M.; Basañez, G.; Etxebarria, A.; Hardwick, J.M.; Nicholls, D.G. Calpain I induces cleavage and release of apoptosis-inducing factor from isolated mitochondria. J. Biol. Chem. 2005, 280, 6447–6454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, T.J.; Han, L.H.; Cong, R.S.; Liang, J. Caspase family proteases and apoptosis. Acta Biochim. Biophys. Sin. 2005, 37, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Fernando, P.; Kelly, J.F.; Balazsi, K.; Slack, R.S.; Megeney, L.A. Caspase 3 activity is required for skeletal muscle differentiation. Proc. Natl. Acad. Sci. USA 2002, 99, 11025–11030. [Google Scholar] [CrossRef]
- Boonstra, K.; Bloemberg, D.; Quadrilatero, J. Caspase-2 is required for skeletal muscle differentiation and myogenesis. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 95–104. [Google Scholar] [CrossRef]
- Du, J.; Wang, X.; Miereles, C.; Bailey, J.L.; Debigare, R.; Zheng, B.; Price, S.R.; Mitch, W.E. Activation of caspase-3 is an initial step triggering accelerated muscle proteolysis in catabolic conditions. J. Clin. Investig. 2004, 113, 115–123. [Google Scholar] [CrossRef]
- Plant, P.J.; Bain, J.R.; Correa, J.E.; Woo, M.; Batt, J. Absence of caspase-3 protects against denervation-induced skeletal muscle atrophy. J. Appl. Physiol. 2009, 107, 224–234. [Google Scholar] [CrossRef]
- Siu, P.M.; Alway, S.E. Mitochondria-associated apoptotic signalling in denervated rat skeletal muscle. J. Physiol. 2005, 565, 309–323. [Google Scholar] [CrossRef]
- Shimizu, N.; Yoshikawa, N.; Ito, N.; Maruyama, T.; Suzuki, Y.; Takeda, S.; Nakae, J.; Tagata, Y.; Nishitani, S.; Takehana, K.; et al. Crosstalk between glucocorticoid receptor and nutritional sensor mTOR in skeletal muscle. Cell Metab. 2011, 13, 170–182. [Google Scholar] [CrossRef]
- Grüner, S.; Peter, D.; Weber, R.; Wohlbold, L.; Chung, M.Y.; Weichenrieder, O.; Valkov, E.; Igreja, C.; Izaurralde, E. The Structures of eIF4E-eIF4G Complexes Reveal an Extended Interface to Regulate Translation Initiation. Mol. Cell 2016, 64, 467–479. [Google Scholar] [CrossRef]
- Risson, V.; Mazelin, L.; Roceri, M.; Sanchez, H.; Moncollin, V.; Corneloup, C.; Richard-Bulteau, H.; Vignaud, A.; Baas, D.; Defour, A.; et al. Muscle inactivation of mTOR causes metabolic and dystrophin defects leading to severe myopathy. J. Cell Biol. 2009, 187, 859–874. [Google Scholar] [CrossRef] [PubMed]
- Lokireddy, S.; Mouly, V.; Butler-Browne, G.; Gluckman, P.D.; Sharma, M.; Kambadur, R.; McFarlane, C. Myostatin promotes the wasting of human myoblast cultures through promoting ubiquitin-proteasome pathway-mediated loss of sarcomeric proteins. Am. J. Physiol. Cell Physiol. 2011, 301, C1316–C1324. [Google Scholar] [CrossRef] [Green Version]
- Gilson, H.; Schakman, O.; Combaret, L.; Lause, P.; Grobet, L.; Attaix, D.; Ketelslegers, J.M.; Thissen, J.P. Myostatin gene deletion prevents glucocorticoid-induced muscle atrophy. Endocrinology 2007, 148, 452–460. [Google Scholar] [CrossRef]
- Verzola, D.; Barisione, C.; Picciotto, D.; Garibotto, G.; Koppe, L. Emerging role of myostatin and its inhibition in the setting of chronic kidney disease. Kidney Int. 2019, 95, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Dyar, K.A.; Ciciliot, S.; Blaauw, B.; Sandri, M. Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. 2013, 280, 4294–4314. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Rajan, V.; Lin, E.; Hu, Z.; Han, H.Q.; Zhou, X.; Song, Y.; Min, H.; Wang, X.; Du, J.; et al. Pharmacological inhibition of myostatin suppresses systemic inflammation and muscle atrophy in mice with chronic kidney disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2011, 25, 1653–1663. [Google Scholar] [CrossRef]
- Philip, B.; Lu, Z.; Gao, Y. Regulation of GDF-8 signaling by the p38 MAPK. Cell. Signal. 2005, 17, 365–375. [Google Scholar] [CrossRef]
- Workeneh, B.T.; Rondon-Berrios, H.; Zhang, L.; Hu, Z.; Ayehu, G.; Ferrando, A.; Kopple, J.D.; Wang, H.; Storer, T.; Fournier, M.; et al. Development of a diagnostic method for detecting increased muscle protein degradation in patients with catabolic conditions. J. Am. Soc. Nephrol. JASN 2006, 17, 3233–3239. [Google Scholar] [CrossRef]
- Bentzinger, C.F.; Wang, Y.X.; Dumont, N.A.; Rudnicki, M.A. Cellular dynamics in the muscle satellite cell niche. EMBO Rep. 2013, 14, 1062–1072. [Google Scholar] [CrossRef]
- Chen, S.E.; Jin, B.; Li, Y.P. TNF-alpha regulates myogenesis and muscle regeneration by activating p38 MAPK. Am. J. Physiol. Cell Physiol. 2007, 292, C1660–C1671. [Google Scholar] [CrossRef]
- Alter, J.; Rozentzweig, D.; Bengal, E. Inhibition of myoblast differentiation by tumor necrosis factor alpha is mediated by c-Jun N-terminal kinase 1 and leukemia inhibitory factor. J. Biol. Chem. 2008, 283, 23224–23234. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Huang, D.; Saederup, N.; Charo, I.F.; Ransohoff, R.M.; Zhou, L. Macrophages recruited via CCR2 produce insulin-like growth factor-1 to repair acute skeletal muscle injury. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2011, 25, 358–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horsley, V.; Jansen, K.M.; Mills, S.T.; Pavlath, G.K. IL-4 acts as a myoblast recruitment factor during mammalian muscle growth. Cell 2003, 113, 483–494. [Google Scholar] [CrossRef]
- Fang, W.Y.; Tseng, Y.T.; Lee, T.Y.; Fu, Y.C.; Chang, W.H.; Lo, W.W.; Lin, C.L.; Lo, Y.C. Triptolide prevents LPS-induced skeletal muscle atrophy via inhibiting NF-kappaB/TNF-alpha and regulating protein synthesis/degradation pathway. Br. J. Pharmacol. 2021, 178, 2998–3016. [Google Scholar] [CrossRef]
- Zanders, L.; Kny, M.; Hahn, A.; Schmidt, S.; Wundersitz, S.; Todiras, M.; Lahmann, I.; Bandyopadhyay, A.; Wollersheim, T.; Kaderali, L.; et al. Sepsis induces interleukin 6, gp130/JAK2/STAT3, and muscle wasting. J. Cachexia Sarcopenia Muscle 2022, 13, 713–727. [Google Scholar] [CrossRef]
- Ueno, M.; Maeshige, N.; Hirayama, Y.; Yamaguchi, A.; Ma, X.; Uemura, M.; Kondo, H.; Fujino, H. Pulsed ultrasound prevents lipopolysaccharide-induced muscle atrophy through inhibiting p38 MAPK phosphorylation in C2C12 myotubes. Biochem. Biophys. Res. Commun. 2021, 570, 184–190. [Google Scholar] [CrossRef]
- Thoma, A.; Lightfoot, A.P. NF-kB and Inflammatory Cytokine Signalling: Role in Skeletal Muscle Atrophy. Adv. Exp. Med. Biol. 2018, 1088, 267–279. [Google Scholar] [CrossRef]
- Bakkar, N.; Guttridge, D.C. NF-kappaB signaling: A tale of two pathways in skeletal myogenesis. Physiol. Rev. 2010, 90, 495–511. [Google Scholar] [CrossRef]
- Mukund, K.; Subramaniam, S. Skeletal muscle: A review of molecular structure and function, in health and disease. Wiley Interdiscip. Rev. Syst. Biol. Med. 2020, 12, e1462. [Google Scholar] [CrossRef]
- Suntar, I.; Sureda, A.; Belwal, T.; Sanches Silva, A.; Vacca, R.A.; Tewari, D.; Sobarzo-Sanchez, E.; Nabavi, S.F.; Shirooie, S.; Dehpour, A.R.; et al. Natural products, PGC-1 alpha, and Duchenne muscular dystrophy. Acta Pharm. Sin. B 2020, 10, 734–745. [Google Scholar] [CrossRef]
- Schindler, C.; Levy, D.E.; Decker, T. JAK-STAT signaling: From interferons to cytokines. J. Biol. Chem. 2007, 282, 20059–20063. [Google Scholar] [CrossRef]
- Baldini, C.; Moriconi, F.R.; Galimberti, S.; Libby, P.; De Caterina, R. The JAK-STAT pathway: An emerging target for cardiovascular disease in rheumatoid arthritis and myeloproliferative neoplasms. Eur. Heart J. 2021, 42, 4389–4400. [Google Scholar] [CrossRef] [PubMed]
- Belizario, J.E.; Fontes-Oliveira, C.C.; Borges, J.P.; Kashiabara, J.A.; Vannier, E. Skeletal muscle wasting and renewal: A pivotal role of myokine IL-6. Springerplus 2016, 5, 619. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.A.; Jones, S.A. IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 2015, 16, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Silva, K.A.; Dong, J.; Dong, Y.; Dong, Y.; Schor, N.; Tweardy, D.J.; Zhang, L.; Mitch, W.E. Inhibition of Stat3 activation suppresses caspase-3 and the ubiquitin-proteasome system, leading to preservation of muscle mass in cancer cachexia. J. Biol. Chem. 2015, 290, 11177–11187. [Google Scholar] [CrossRef]
- Zhang, L.; Pan, J.; Dong, Y.; Tweardy, D.J.; Dong, Y.; Garibotto, G.; Mitch, W.E. Stat3 activation links a C/EBPδ to myostatin pathway to stimulate loss of muscle mass. Cell Metab. 2013, 18, 368–379. [Google Scholar] [CrossRef]
- Yoon, S.; Woo, S.U.; Kang, J.H.; Kim, K.; Shin, H.J.; Gwak, H.S.; Park, S.; Chwae, Y.J. NF-κB and STAT3 cooperatively induce IL6 in starved cancer cells. Oncogene 2012, 31, 3467–3481. [Google Scholar] [CrossRef]
- Babon, J.J.; Kershaw, N.J.; Murphy, J.M.; Varghese, L.N.; Laktyushin, A.; Young, S.N.; Lucet, I.S.; Norton, R.S.; Nicola, N.A. Suppression of cytokine signaling by SOCS3: Characterization of the mode of inhibition and the basis of its specificity. Immunity 2012, 36, 239–250. [Google Scholar] [CrossRef]
- Muniyappa, H.; Das, K.C. Activation of c-Jun N-terminal kinase (JNK) by widely used specific p38 MAPK inhibitors SB202190 and SB203580: A MLK-3-MKK7-dependent mechanism. Cell. Signal. 2008, 20, 675–683. [Google Scholar] [CrossRef]
- Wagner, E.F.; Nebreda, A.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef]
- Yuasa, K.; Okubo, K.; Yoda, M.; Otsu, K.; Ishii, Y.; Nakamura, M.; Itoh, Y.; Horiuchi, K. Targeted ablation of p38alpha MAPK suppresses denervation-induced muscle atrophy. Sci. Rep. 2018, 8, 9037. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, A.; Thakur, N.; Grimsby, S.; Marcusson, A.; von Bulow, V.; Schuster, N.; Zhang, S.; Heldin, C.H.; Landström, M. The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat. Cell Biol. 2008, 10, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Zhu, J.; Zhang, R.; Liang, W.; Ma, W.; Zhang, Q.; Huang, Z.; Ding, F.; Sun, H. miR-125b-5p targeting TRAF6 relieves skeletal muscle atrophy induced by fasting or denervation. Ann. Transl. Med. 2019, 7, 456. [Google Scholar] [CrossRef] [PubMed]
- Tomida, T.; Adachi-Akahane, S. Roles of p38 MAPK signaling in the skeletal muscle formation, regeneration, and pathology. Nihon Yakurigaku Zasshi Folia Pharmacol. Jpn. 2020, 155, 241–247. [Google Scholar] [CrossRef]
- Engeland, W.C.; Yoder, J.M.; Karsten, C.A.; Kofuji, P. Phase-Dependent Shifting of the Adrenal Clock by Acute Stress-Induced ACTH. Front. Endocrinol. 2016, 7, 81. [Google Scholar] [CrossRef]
- Silverman, M.N.; Sternberg, E.M. Glucocorticoid regulation of inflammation and its functional correlates: From HPA axis to glucocorticoid receptor dysfunction. Ann. N. Y. Acad. Sci. 2012, 1261, 55–63. [Google Scholar] [CrossRef]
- Ruggiero, C.; Lalli, E. Impact of ACTH Signaling on Transcriptional Regulation of Steroidogenic Genes. Front. Endocrinol. 2016, 7, 24. [Google Scholar] [CrossRef]
- Shimizu, N.; Maruyama, T.; Yoshikawa, N.; Matsumiya, R.; Ma, Y.; Ito, N.; Tasaka, Y.; Kuribara-Souta, A.; Miyata, K.; Oike, Y.; et al. A muscle-liver-fat signalling axis is essential for central control of adaptive adipose remodelling. Nat. Commun. 2015, 6, 6693. [Google Scholar] [CrossRef]
- Meijsing, S.H.; Pufall, M.A.; So, A.Y.; Bates, D.L.; Chen, L.; Yamamoto, K.R. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science 2009, 324, 407–410. [Google Scholar] [CrossRef]
- Menconi, M.; Fareed, M.; O’Neal, P.; Poylin, V.; Wei, W.; Hasselgren, P.O. Role of glucocorticoids in the molecular regulation of muscle wasting. Crit. Care Med. 2007, 35, S602–S608. [Google Scholar] [CrossRef]
- Braun, T.P.; Zhu, X.; Szumowski, M.; Scott, G.D.; Grossberg, A.J.; Levasseur, P.R.; Graham, K.; Khan, S.; Damaraju, S.; Colmers, W.F.; et al. Central nervous system inflammation induces muscle atrophy via activation of the hypothalamic-pituitary-adrenal axis. J. Exp. Med. 2011, 208, 2449–2463. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Cánoves, P.; Scheele, C.; Pedersen, B.K.; Serrano, A.L. Interleukin-6 myokine signaling in skeletal muscle: A double-edged sword? FEBS J. 2013, 280, 4131–4148. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Yang, X.; Wu, C.; Qiu, J.; Fang, Q.; Wang, L.; Yu, S.; Sun, H. Pyrroloquinoline quinone attenuates cachexia-induced muscle atrophy via suppression of reactive oxygen species. J. Thorac. Dis. 2018, 10, 2752–2759. [Google Scholar] [CrossRef]
- Flores, E.A.; Bistrian, B.R.; Pomposelli, J.J.; Dinarello, C.A.; Blackburn, G.L.; Istfan, N.W. Infusion of tumor necrosis factor/cachectin promotes muscle catabolism in the rat. A synergistic effect with interleukin 1. J. Clin. Investig. 1989, 83, 1614–1622. [Google Scholar] [CrossRef] [PubMed]
- Finn, P.F.; Dice, J.F. Proteolytic and lipolytic responses to starvation. Nutrition 2006, 22, 830–844. [Google Scholar] [CrossRef]
- Argilés, J.M.; Busquets, S.; López-Soriano, F.J. Metabolic interrelationships between liver and skeletal muscle in pathological states. Life Sci. 2001, 69, 1345–1361. [Google Scholar] [CrossRef]
- Flint, T.R.; Janowitz, T.; Connell, C.M.; Roberts, E.W.; Denton, A.E.; Coll, A.P.; Jodrell, D.I.; Fearon, D.T. Tumor-Induced IL-6 Reprograms Host Metabolism to Suppress Anti-tumor Immunity. Cell Metab. 2016, 24, 672–684. [Google Scholar] [CrossRef]
- Goncalves, M.D.; Hwang, S.K.; Pauli, C.; Murphy, C.J.; Cheng, Z.; Hopkins, B.D.; Wu, D.; Loughran, R.M.; Emerling, B.M.; Zhang, G.; et al. Fenofibrate prevents skeletal muscle loss in mice with lung cancer. Proc. Natl. Acad. Sci. USA 2018, 115, E743–E752. [Google Scholar] [CrossRef]
- Martín, A.I.; Priego, T.; Moreno-Ruperez, Á.; González-Hedström, D.; Granado, M.; López-Calderón, A. IGF-1 and IGFBP-3 in Inflammatory Cachexia. Int. J. Mol. Sci. 2021, 22, 9469. [Google Scholar] [CrossRef]
- Spate, U.; Schulze, P.C. Proinflammatory cytokines and skeletal muscle. Curr. Opin. Clin. Nutr. Metab. Care 2004, 7, 265–269. [Google Scholar] [CrossRef]
- Grounds, M.D.; Radley, H.G.; Gebski, B.L.; Bogoyevitch, M.A.; Shavlakadze, T. Implications of cross-talk between tumour necrosis factor and insulin-like growth factor-1 signalling in skeletal muscle. Clin. Exp. Pharmacol. Physiol. 2008, 35, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Thibaut, M.M.; Sboarina, M.; Roumain, M.; Pötgens, S.A.; Neyrinck, A.M.; Destrée, F.; Gillard, J.; Leclercq, I.A.; Dachy, G.; Demoulin, J.B.; et al. Inflammation-induced cholestasis in cancer cachexia. J. Cachexia Sarcopenia Muscle 2021, 12, 70–90. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.J.; Patel, B.M. TNF-α and cancer cachexia: Molecular insights and clinical implications. Life Sci. 2017, 170, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Laird, B.J.; McMillan, D.; Skipworth, R.J.E.; Fallon, M.T.; Paval, D.R.; McNeish, I.; Gallagher, I.J. The Emerging Role of Interleukin 1beta (IL-1beta) in Cancer Cachexia. Inflammation 2021, 44, 1223–1228. [Google Scholar] [CrossRef]
- Bolton, C.F. Neuromuscular manifestations of critical illness. Muscle Nerve 2005, 32, 140–163. [Google Scholar] [CrossRef]
- Cao, Y.Y.; Wang, Z.; Yu, T.; Zhang, Y.; Wang, Z.H.; Lu, Z.M.; Lu, W.H.; Yu, J.B. Sepsis induces muscle atrophy by inhibiting proliferation and promoting apoptosis via PLK1-AKT signalling. J. Cell. Mol. Med. 2021, 25, 9724–9739. [Google Scholar] [CrossRef]
- Leduc-Gaudet, J.P.; Mayaki, D.; Reynaud, O.; Broering, F.E.; Chaffer, T.J.; Hussain, S.N.A.; Gouspillou, G. Parkin Overexpression Attenuates Sepsis-Induced Muscle Wasting. Cells 2020, 9, 1454. [Google Scholar] [CrossRef]
- Owen, A.M.; Patel, S.P.; Smith, J.D.; Balasuriya, B.K.; Mori, S.F.; Hawk, G.S.; Stromberg, A.J.; Kuriyama, N.; Kaneki, M.; Rabchevsky, A.G.; et al. Chronic muscle weakness and mitochondrial dysfunction in the absence of sustained atrophy in a preclinical sepsis model. eLife 2019, 8, e49920. [Google Scholar] [CrossRef]
- Yin, D.; Lin, D.; Xie, Y.; Gong, A.; Jiang, P.; Wu, J. Neuregulin-1β Alleviates Sepsis-Induced Skeletal Muscle Atrophy by Inhibiting Autophagy via AKT/mTOR Signaling Pathway in Rats. Shock 2022, 57, 397–407. [Google Scholar] [CrossRef]
- Stana, F.; Vujovic, M.; Mayaki, D.; Leduc-Gaudet, J.P.; Leblanc, P.; Huck, L.; Hussain, S.N.A. Differential Regulation of the Autophagy and Proteasome Pathways in Skeletal Muscles in Sepsis. Crit. Care Med. 2017, 45, e971–e979. [Google Scholar] [CrossRef]
- Russell, R.C.; Yuan, H.X.; Guan, K.L. Autophagy regulation by nutrient signaling. Cell Res. 2014, 24, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Trejo-Solís, C.; Serrano-Garcia, N.; Escamilla-Ramírez, Á.; Castillo-Rodríguez, R.A.; Jimenez-Farfan, D.; Palencia, G.; Calvillo, M.; Alvarez-Lemus, M.A.; Flores-Nájera, A.; Cruz-Salgado, A.; et al. Autophagic and Apoptotic Pathways as Targets for Chemotherapy in Glioblastoma. Int. J. Mol. Sci. 2018, 19, 3773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yung, J.H.M.; Giacca, A. Role of c-Jun N-terminal Kinase (JNK) in Obesity and Type 2 Diabetes. Cells 2020, 9, 706. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Li, M.; Wang, K.; Qi, G.; Liu, H.; Wang, W.; Ji, Y.; Chang, M.; Deng, C.; Xu, F.; et al. Diabetic Muscular Atrophy: Molecular Mechanisms and Promising Therapies. Front. Endocrinol. 2022, 13, 917113. [Google Scholar] [CrossRef]
- Bassil, M.S.; Gougeon, R. Muscle protein anabolism in type 2 diabetes. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 83–88. [Google Scholar] [CrossRef]
- Andreasen, A.S.; Kelly, M.; Berg, R.M.; Møller, K.; Pedersen, B.K. Type 2 diabetes is associated with altered NF-κB DNA binding activity, JNK phosphorylation, and AMPK phosphorylation in skeletal muscle after LPS. PLoS ONE 2011, 6, e23999. [Google Scholar] [CrossRef]
- Kim, J.H.; Bachmann, R.A.; Chen, J. Interleukin-6 and insulin resistance. Vitam. Horm. 2009, 80, 613–633. [Google Scholar] [CrossRef]
- Scheele, C.; Nielsen, S.; Kelly, M.; Broholm, C.; Nielsen, A.R.; Taudorf, S.; Pedersen, M.; Fischer, C.P.; Pedersen, B.K. Satellite cells derived from obese humans with type 2 diabetes and differentiated into myocytes in vitro exhibit abnormal response to IL-6. PLoS ONE 2012, 7, e39657. [Google Scholar] [CrossRef]
- D’Souza, D.M.; Trajcevski, K.E.; Al-Sajee, D.; Wang, D.C.; Thomas, M.; Anderson, J.E.; Hawke, T.J. Diet-induced obesity impairs muscle satellite cell activation and muscle repair through alterations in hepatocyte growth factor signaling. Physiol. Rep. 2015, 3, e12506. [Google Scholar] [CrossRef]
- Van Gaal, L.F.; Mertens, I.L.; De Block, C.E. Mechanisms linking obesity with cardiovascular disease. Nature 2006, 444, 875–880. [Google Scholar] [CrossRef]
- Longo, M.; Zatterale, F.; Naderi, J.; Parrillo, L.; Formisano, P.; Raciti, G.A.; Beguinot, F.; Miele, C. Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications. Int. J. Mol. Sci. 2019, 20, 2358. [Google Scholar] [CrossRef] [PubMed]
- Yuen, D.Y.; Dwyer, R.M.; Matthews, V.B.; Zhang, L.; Drew, B.G.; Neill, B.; Kingwell, B.A.; Clark, M.G.; Rattigan, S.; Febbraio, M.A. Interleukin-6 attenuates insulin-mediated increases in endothelial cell signaling but augments skeletal muscle insulin action via differential effects on tumor necrosis factor-alpha expression. Diabetes 2009, 58, 1086–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engin, A. The Pathogenesis of Obesity-Associated Adipose Tissue Inflammation. Adv. Exp. Med. Biol. 2017, 960, 221–245. [Google Scholar] [CrossRef] [PubMed]
- Rogero, M.M.; Calder, P.C. Obesity, Inflammation, Toll-Like Receptor 4 and Fatty Acids. Nutrients 2018, 10, 432. [Google Scholar] [CrossRef]
- Ju, S.; Lee, S.J.; Park, M.J.; Cho, Y.J.; Jeong, Y.Y.; Jeon, K.N.; Bae, K.; Lee, J.D.; Kim, H.C. Clinical importance of cross-sectional area of intercostal muscles in patients with chronic obstructive pulmonary disease. Clin. Respir. J. 2018, 12, 939–947. [Google Scholar] [CrossRef]
- Tanimura, K.; Sato, S.; Fuseya, Y.; Hasegawa, K.; Uemasu, K.; Sato, A.; Oguma, T.; Hirai, T.; Mishima, M.; Muro, S. Quantitative Assessment of Erector Spinae Muscles in Patients with Chronic Obstructive Pulmonary Disease. Novel Chest Computed Tomography-derived Index for Prognosis. Ann. Am. Thorac. Soc. 2016, 13, 334–341. [Google Scholar] [CrossRef]
- Barreiro, E.; Jaitovich, A. Muscle atrophy in chronic obstructive pulmonary disease: Molecular basis and potential therapeutic targets. J. Thorac. Dis. 2018, 10, S1415–S1424. [Google Scholar] [CrossRef]
- Plant, P.J.; Brooks, D.; Faughnan, M.; Bayley, T.; Bain, J.; Singer, L.; Correa, J.; Pearce, D.; Binnie, M.; Batt, J. Cellular markers of muscle atrophy in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2010, 42, 461–471. [Google Scholar] [CrossRef]
- Zhang, L.; Sun, Y. Muscle-Bone Crosstalk in Chronic Obstructive Pulmonary Disease. Front. Endocrinol. 2021, 12, 724911. [Google Scholar] [CrossRef]
- Lee, S.J. Regulation of muscle mass by myostatin. Annu. Rev. Cell Dev. Biol. 2004, 20, 61–86. [Google Scholar] [CrossRef]
- Langley, B.; Thomas, M.; Bishop, A.; Sharma, M.; Gilmour, S.; Kambadur, R. Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J. Biol. Chem. 2002, 277, 49831–49840. [Google Scholar] [CrossRef] [PubMed]
- Sriram, S.; Subramanian, S.; Juvvuna, P.K.; Ge, X.; Lokireddy, S.; McFarlane, C.D.; Wahli, W.; Kambadur, R.; Sharma, M. Myostatin augments muscle-specific ring finger protein-1 expression through an NF-kB independent mechanism in SMAD3 null muscle. Mol. Endocrinol. 2014, 28, 317–330. [Google Scholar] [CrossRef]
- Gu, L.; Wang, Z.; Zhang, Y.; Zhu, N.; Li, J.; Yang, M.; Wang, L.; Rong, S. TLR13 contributes to skeletal muscle atrophy by increasing insulin resistance in chronic kidney disease. Cell Prolif. 2022, 55, e13181. [Google Scholar] [CrossRef] [PubMed]
- Kooman, J.P.; Dekker, M.J.; Usvyat, L.A.; Kotanko, P.; van der Sande, F.M.; Schalkwijk, C.G.; Shiels, P.G.; Stenvinkel, P. Inflammation and premature aging in advanced chronic kidney disease. Am. J. Physiology. Ren. Physiol. 2017, 313, F938–F950. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, Z.; Wang, Y.; Tweardy, D.J.; Mitch, W.E. Stat3 activation induces insulin resistance via a muscle-specific E3 ubiquitin ligase Fbxo40. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E625–E635. [Google Scholar] [CrossRef]
- Wu, J.; Dong, J.; Verzola, D.; Hruska, K.; Garibotto, G.; Hu, Z.; Mitch, W.E.; Thomas, S.S. Signal regulatory protein alpha initiates cachexia through muscle to adipose tissue crosstalk. J. Cachexia Sarcopenia Muscle 2019, 10, 1210–1227. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.; Kny, M.; Pablo-Tortola, C.; Todiras, M.; Willenbrock, M.; Schmidt, S.; Schmoeckel, K.; Jorde, I.; Nowak, M.; Jarosch, E.; et al. Serum amyloid A1 mediates myotube atrophy via Toll-like receptors. J. Cachexia Sarcopenia Muscle 2020, 11, 103–119. [Google Scholar] [CrossRef]
- Gu, L.J.; Zhang, Y.Y.; Wang, L.; Wang, J.L.; Wang, X.; Yuan, W.J. Changes of insulin-like growth factor 1 axis in chronic kidney disease and its role in skeletal muscle atrophy. Zhonghua Yi Xue Za Zhi 2018, 98, 749–754. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Yang, M.; Bao, J.F.; Gu, L.J.; Yu, H.L.; Yuan, W.J. Phosphate stimulates myotube atrophy through autophagy activation: Evidence of hyperphosphatemia contributing to skeletal muscle wasting in chronic kidney disease. BMC Nephrol. 2018, 19, 45. [Google Scholar] [CrossRef]
- Gu, X. Biodegradable Materials and the Tissue Engineering of Nerves. Engineering 2021, 7, 1700–1703. [Google Scholar] [CrossRef]
- Qiu, J.; Yang, X.; Wang, L.; Zhang, Q.; Ma, W.; Huang, Z.; Bao, Y.; Zhong, L.; Sun, H.; Ding, F. Isoquercitrin promotes peripheral nerve regeneration through inhibiting oxidative stress following sciatic crush injury in mice. Ann. Transl. Med. 2019, 7, 680. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Fang, Q.; Xu, T.; Wu, C.; Xu, L.; Wang, L.; Yang, X.; Yu, S.; Zhang, Q.; Ding, F.; et al. Mechanistic Role of Reactive Oxygen Species and Therapeutic Potential of Antioxidants in Denervation- or Fasting-Induced Skeletal Muscle Atrophy. Front. Physiol. 2018, 9, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, W.; Zhang, R.; Huang, Z.; Zhang, Q.; Xie, X.; Yang, X.; Zhang, Q.; Liu, H.; Ding, F.; Zhu, J.; et al. PQQ ameliorates skeletal muscle atrophy, mitophagy and fiber type transition induced by denervation via inhibition of the inflammatory signaling pathways. Ann. Transl. Med. 2019, 7, 440. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.; Pomerantz, J.H. The Role of Muscle Stem Cells in Regeneration and Recovery after Denervation: A Review. Plast. Reconstr. Surg. 2019, 143, 779–788. [Google Scholar] [CrossRef]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef]
- Wan, B.; Feng, P.; Guan, Z.; Sheng, L.; Liu, Z.; Hua, Y. A severe mouse model of spinal muscular atrophy develops early systemic inflammation. Hum. Mol. Genet. 2018, 27, 4061–4076. [Google Scholar] [CrossRef]
- Yang, X.; Ji, Y.; Wang, W.; Zhang, L.; Chen, Z.; Yu, M.; Shen, Y.; Ding, F.; Gu, X.; Sun, H. Amyotrophic Lateral Sclerosis: Molecular Mechanisms, Biomarkers, and Therapeutic Strategies. Antioxidants 2021, 10, 1012. [Google Scholar] [CrossRef]
- D’Ambrosi, N.; Cozzolino, M.; Carrì, M.T. Neuroinflammation in Amyotrophic Lateral Sclerosis: Role of Redox (dys)Regulation. Antioxid Redox Signal. 2018, 29, 15–36. [Google Scholar] [CrossRef]
- Sun, H.; Li, M.; Ji, Y.; Zhu, J.; Chen, Z.; Zhang, L.; Deng, C.; Cheng, Q.; Wang, W.; Shen, Y.; et al. Identification of Regulatory Factors and Prognostic Markers in Amyotrophic Lateral Sclerosis. Antioxidants 2022, 11, 303. [Google Scholar] [CrossRef]
- Källstig, E.; McCabe, B.D.; Schneider, B.L. The Links between ALS and NF-κB. Int. J. Mol. Sci. 2021, 22, 3875. [Google Scholar] [CrossRef]
- Cao, R.Y.; Li, J.; Dai, Q.; Li, Q.; Yang, J. Muscle Atrophy: Present and Future. Adv. Exp. Med. Biol. 2018, 1088, 605–624. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Jang, M.; Park, R.; Jo, D.; Choi, I.; Choe, J.; Oh, W.K.; Park, J. Conessine Treatment Reduces Dexamethasone-Induced Muscle Atrophy by Regulating MuRF1 and Atrogin-1 Expression. J. Microbiol. Biotechnol. 2018, 28, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Heo, J.W.; Kim, A.R.; Kweon, M.; Nam, S.; Lim, J.S.; Sung, M.K.; Kim, S.E.; Ryu, J.H. Z-ajoene from Crushed Garlic Alleviates Cancer-Induced Skeletal Muscle Atrophy. Nutrients 2019, 11, 2724. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Hu, R.; You, H.; Li, J.; Liu, Y.; Li, Q.; Wu, X.; Huang, J.; Cai, X.; Wang, M.; et al. Formononetin ameliorates muscle atrophy by regulating myostatin-mediated PI3K/Akt/FoxO3a pathway and satellite cell function in chronic kidney disease. J. Cell. Mol. Med. 2021, 25, 1493–1506. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.B.; Lee, J.S.; Moon, S.O.; Lee, H.D.; Yoon, Y.S.; Son, C.G. A standardized herbal combination of Astragalus membranaceus and Paeonia japonica, protects against muscle atrophy in a C26 colon cancer cachexia mouse model. J. Ethnopharmacol. 2021, 267, 113470. [Google Scholar] [CrossRef]
- Zhang, D.; Cao, L.; Wang, Z.; Feng, H.; Cai, X.; Xu, M.; Li, M.; Yu, N.; Yin, Y.; Wang, W.; et al. Salidroside mitigates skeletal muscle atrophy in rats with cigarette smoke-induced COPD by up-regulating myogenin and down-regulating myostatin expression. Biosci. Rep. 2019, 39, 440. [Google Scholar] [CrossRef]
- Chen, C.; Ju, R.; Zhu, L.; Li, J.; Chen, W.; Zhang, D.C.; Ye, C.Y.; Guo, L. Carboxyamidotriazole alleviates muscle atrophy in tumor-bearing mice by inhibiting NF-κB and activating SIRT1. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2017, 390, 423–433. [Google Scholar] [CrossRef]
- Han, Y.; Lee, H.; Li, H.; Ryu, J.H. Corylifol A from Psoralea corylifolia L. Enhances Myogenesis and Alleviates Muscle Atrophy. Int. J. Mol. Sci. 2020, 21, 1571. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, S.W.; Kim, J.H.; Kim, H.J.; Um, J.; Jung, D.W.; Williams, D.R. Lithium Chloride Protects against Sepsis-Induced Skeletal Muscle Atrophy and Cancer Cachexia. Cells 2021, 10, 1017. [Google Scholar] [CrossRef]
- Oh, S.; Choi, C.H.; Lee, B.J.; Park, J.H.; Son, K.H.; Byun, K. Fermented Oyster Extract Attenuated Dexamethasone-Induced Muscle Atrophy by Decreasing Oxidative Stress. Molecules 2021, 26, 7128. [Google Scholar] [CrossRef]
- Tseng, Y.T.; Chang, W.H.; Lin, C.C.; Chang, F.R.; Wu, P.C.; Lo, Y.C. Protective effects of Liuwei dihuang water extracts on diabetic muscle atrophy. Phytomed. Int. J. Phytother. Phytopharm. 2019, 53, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Chi, M.Y.; Zhang, H.; Wang, Y.X.; Sun, X.P.; Yang, Q.J.; Guo, C. Silibinin Alleviates Muscle Atrophy Caused by Oxidative Stress Induced by Cisplatin through ERK/FoxO and JNK/FoxO Pathways. Oxidative Med. Cell. Longev. 2022, 2022, 5694223. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.C.; Liu, H.M.; Lee, M.C.; Leu, Y.L.; Chiang, W.H.; Chang, H.H.; Lee, T.Y. Phytochemical-rich herbal formula ATG-125 protects against sucrose-induced gastrocnemius muscle atrophy by rescuing Akt signaling and improving mitochondrial dysfunction in young adult mice. Mol. Med. Rep. 2022, 25, 57. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, C.; Lee, H.; Hwang, J.K. Inhibitory Effects of Standardized Leonurus japonicus Extract and Its Bioactive Leonurine on TNF-α-Induced Muscle Atrophy in L6 Myotubes. J. Microbiol. Biotechnol. 2020, 30, 1896–1904. [Google Scholar] [CrossRef]
- Lee, S.J.; Im, M.; Park, S.K.; Kim, J.Y.; So, E.Y.; Liang, O.D.; Kang, J.S.; Bae, G.U. BST204, a Rg3 and Rh2 Enriched Ginseng Extract, Upregulates Myotube Formation and Mitochondrial Function in TNF-α-Induced Atrophic Myotubes. Am. J. Chin. Med. 2020, 48, 631–650. [Google Scholar] [CrossRef]
- Liu, L.; Liu, X.; Bai, Y.; Tang, N.; Li, J.; Zhang, Y.; Wu, J.; Wang, X.; Wei, J. Neuregulin-1β modulates myogenesis in septic mouse serum-treated C2C12 myotubes in vitro through PPARγ/NF-κB signaling. Mol. Biol. Rep. 2018, 45, 1611–1619. [Google Scholar] [CrossRef]
- Sun, L.J.; Sun, Y.N.; Chen, S.J.; Liu, S.; Jiang, G.R. Resveratrol attenuates skeletal muscle atrophy induced by chronic kidney disease via MuRF1 signaling pathway. Biochem. Biophys. Res. Commun. 2017, 487, 83–89. [Google Scholar] [CrossRef]
- Bai, C.H.; Alizargar, J.; Peng, C.Y.; Wu, J.P. Combination of exercise training and resveratrol attenuates obese sarcopenia in skeletal muscle atrophy. Chin. J. Physiol. 2020, 63, 101–112. [Google Scholar] [CrossRef]
- Shadfar, S.; Couch, M.E.; McKinney, K.A.; Weinstein, L.J.; Yin, X.; Rodriguez, J.E.; Guttridge, D.C.; Willis, M. Oral resveratrol therapy inhibits cancer-induced skeletal muscle and cardiac atrophy in vivo. Nutr. Cancer 2011, 63, 749–762. [Google Scholar] [CrossRef]
- Lu, S.; Li, Y.; Shen, Q.; Zhang, W.; Gu, X.; Ma, M.; Li, Y.; Zhang, L.; Liu, X.; Zhang, X. Carnosol and its analogues attenuate muscle atrophy and fat lipolysis induced by cancer cachexia. J. Cachexia Sarcopenia Muscle 2021, 12, 779–795. [Google Scholar] [CrossRef]
- Li, C.; Deng, Z.; Zheng, G.; Xie, T.; Wei, X.; Huo, Z.; Bai, J. Histone Deacetylase 2 Suppresses Skeletal Muscle Atrophy and Senescence via NF-κB Signaling Pathway in Cigarette Smoke-Induced Mice with Emphysema. Int. J. Chronic Obstr. Pulm. Dis. 2021, 16, 1661–1675. [Google Scholar] [CrossRef]
- Dai, J.; Xiang, Y.; Fu, D.; Xu, L.; Jiang, J.; Xu, J. Ficus carica L. Attenuates Denervated Skeletal Muscle Atrophy via PPARα/NF-κB Pathway. Front. Physiol. 2020, 11, 580223. [Google Scholar] [CrossRef] [PubMed]
- Lala-Tabbert, N.; Lejmi-Mrad, R.; Timusk, K.; Fukano, M.; Holbrook, J.; St-Jean, M.; LaCasse, E.C.; Korneluk, R.G. Targeted ablation of the cellular inhibitor of apoptosis 1 (cIAP1) attenuates denervation-induced skeletal muscle atrophy. Skelet. Muscle 2019, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Choi, Y.H.; Nam, T.J. Pyropia yezoensis protein protects against TNF-α-induced myotube atrophy in C2C12 myotubes via the NF-κB signaling pathway. Mol. Med. Rep. 2021, 24, 486. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Choi, J.W.; Choi, Y.H.; Nam, T.J. Pyropia yezoensis Protein Prevents Dexamethasone-Induced Myotube Atrophy in C2C12 Myotubes. Mar. Drugs 2018, 16, 497. [Google Scholar] [CrossRef]
- Zhou, L.; Huang, Y.; Xie, H.; Mei, X. Buyang Huanwu Tang improves denervation-dependent muscle atrophy by increasing ANGPTL4, and increases NF-κB and MURF1 levels. Mol. Med. Rep. 2018, 17, 3674–3680. [Google Scholar] [CrossRef]
- Yoshioka, Y.; Kubota, Y.; Samukawa, Y.; Yamashita, Y.; Ashida, H. Glabridin inhibits dexamethasone-induced muscle atrophy. Arch. Biochem. Biophys. 2019, 664, 157–166. [Google Scholar] [CrossRef]
- Chen, L.; Xu, W.; Yang, Q.; Zhang, H.; Wan, L.; Xin, B.; Zhang, J.; Guo, C. Imperatorin alleviates cancer cachexia and prevents muscle wasting via directly inhibiting STAT3. Pharmacol. Res. 2020, 158, 104871. [Google Scholar] [CrossRef]
- Chen, L.; Yang, Q.; Zhang, H.; Wan, L.; Xin, B.; Cao, Y.; Zhang, J.; Guo, C. Cryptotanshinone prevents muscle wasting in CT26-induced cancer cachexia through inhibiting STAT3 signaling pathway. J. Ethnopharmacol. 2020, 260, 113066. [Google Scholar] [CrossRef]
- Shen, Q.; Kuang, J.X.; Miao, C.X.; Zhang, W.L.; Li, Y.W.; Zhang, X.W.; Liu, X. Alantolactone ameliorates cancer cachexia-associated muscle atrophy mainly by inhibiting the STAT3 signaling pathway. Phytomed. Int. J. Phytother. Phytopharm. 2022, 95, 153858. [Google Scholar] [CrossRef]
- Dutt, V.; Saini, V.; Gupta, P.; Kaur, N.; Bala, M.; Gujar, R.; Grewal, A.; Gupta, S.; Dua, A.; Mittal, A. S-allyl cysteine inhibits TNFα-induced skeletal muscle wasting through suppressing proteolysis and expression of inflammatory molecules. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Zhu, M.; Lu, Z.; Zhang, Y.; Li, L.; Li, N.; Yin, L.; Wang, H.; Song, W.; Xu, H. L-carnitine ameliorates the muscle wasting of cancer cachexia through the AKT/FOXO3a/MaFbx axis. Nutr. Metab. 2021, 18, 98. [Google Scholar] [CrossRef]
- Kou, X.; Li, J.; Liu, X.; Yang, X.; Fan, J.; Chen, N. Ampelopsin attenuates the atrophy of skeletal muscle from d-gal-induced aging rats through activating AMPK/SIRT1/PGC-1α signaling cascade. Biomed. Pharmacother. 2017, 90, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Perumal, E.; Bi, X.; Wang, Y.; Ding, W. Potential mechanisms of uremic muscle wasting and the protective role of the mitochondria-targeted antioxidant Mito-TEMPO. Int. Urol. Nephrol. 2020, 52, 1551–1561. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Han, W.; Wang, C.; Sui, D.; Bian, J.; Bo, L.; Deng, X. Upregulation of Heme Oxygenase-1 by Hemin Alleviates Sepsis-Induced Muscle Wasting in Mice. Oxidative Med. Cell. Longev. 2018, 2018, 8927104. [Google Scholar] [CrossRef]
- Yeo, D.; Kang, C.; Zhang, T.; Ji, L.L. Avenanthramides attenuate inflammation and atrophy in muscle cells. J. Sport Health Sci. 2019, 8, 189–195. [Google Scholar] [CrossRef]

{kind=link}
| Drug/Compound | Targeted Signaling Pathway | Function | References |
|---|---|---|---|
| Conessine: a steroidal alkaloid, a potent histamine H3 antagonist | Inhibits NF-κB pathway; inhibits UPS. | Conessine reduces dexamethasone-induced muscle atrophy. | [152] |
| Salidroside: a glucoside of tyrosol found mostly in the roots of Rhodiola spp. | Inhibits inflammatory response; mitigates oxidative stress; inhibits UPS and mitophagy. | Salidroside alleviates denervation-induced muscle atrophy by suppressing oxidative stress and inflammation. | [4,156] |
| Carboxyamidotriazole: a noncytotoxic chemotherapy agent, shows anti-inflammatory features | Inhibits inflammation; inhibits NF-κB pathway. | Carboxyamidotriazole can alleviate cancer cachexia-induced muscle wasting. | [157] |
| Corylifol A: isolated from P. corylifolia | Activates p38MAPK pathway; protects PI3K/Akt pathway. | Corylifol A alleviates muscle atrophy through activating myoblast differentiation and suppressing muscle degradation. | [158] |
| Lithium chloride | Inhibits inflammatory response; enhances myogenic differentiation; inhibits UPS. | Lithium chloride exerts therapeutic effects on inflammation-mediated skeletal muscle wasting, such as sepsis-induced muscle atrophy and cancer cachexia. | [159] |
| Fermented oyster extracts (FO): rich in γ-aminobutyric acid (GABA) and lactate | Inhibits inflammatory response; inhibits oxidative stress; inhibits NF-κB pathway; enhances IGF-1/Akt pathway; inhibits UPS. | Fermented oyster extract attenuated dexamethasone-induced muscle atrophy by decreasing oxidative stress and inflammation. | [160] |
| Liuwei dihuang water extracts: a Chinese herbal medicine composed of Rehmanniae Radix Praeparata, Corni Sarcocarpium, Dioscoreae Rhizoma, Alismatis Rhizoma, Moutan Radicis Cortex and Poria | Attenuates oxidative damage; enhances IGF-1/Akt pathway; inhibits UPS. | Liuwei dihuang water extracts attenuated diabetic muscle atrophy. | [161] |
| Silibinin (SLI) | Alleviates oxidative stress; inhibits UPS and MSTN; regulates MAPK pathway. | SLI can reduce DDP-induced skeletal muscle atrophy by reducing oxidative stress and regulating ERK/FoxO and JNK/FoxO pathways. | [162] |
| ATG-125: herbal formula ATG-125, a phytochemical-rich formula | Inhibits chronic inflammation; improves mitochondrial dysfunction; enhances IGF/Akt-mTOR pathway. | ATG-125, as an integrator of protein synthesis and degradative pathways, prevented muscle wasting. | [163] |
| Leonurus japonicus extract (LJE) | Alleviates inflammatory responses; inhibits NF-κB pathway; inhibits UPS; activates PI3K/Akt pathway. | LJE and leonurine inhibits skeletal muscle atrophy by activating the PI3K/Akt pathway and reducing inflammatory responses. | [164] |
| Formononetin (FMN): a major bioactive isoflavone compound in Astragalus membranaceus | Inhibits inflammation; inhibits UPS and MSTN; enhances PI3K/Akt/FoxO3a pathway. | Formononetin ameliorates muscle atrophy by regulating myostatin-mediated PI3K/Akt/FoxO3a pathway and satellite cell function in chronic kidney disease. | [154] |
| BST204: a Rg3 and Rh2 enriched ginseng extract | Inhibits inflammation; reduces oxidative damage; inhibits UPS; activates IGF-1/Akt pathway. | BST204 upregulates myotube formation and mitochondrial function in TNF-α-induced atrophic myotubes. | [165] |
| Neuregulin-1β (NRG-1β) | Inhibits NF-κB pathway. | Neuregulin-1β modulates myogenesis in septic mouse serum-treated C2C12 myotubes in vitro through PPARγ/NF-κB signaling. | [166] |
| Resveratrol: a polyphenol that is abundant in grape skin and seeds | Inhibits NF-κB pathway; activates IGF-1/Akt pathway; inhibits UPS and ALP; increases mitochondrial biogenesis. | Resveratrol attenuates skeletal muscle atrophy induced by chronic kidney disease, TNF-α, cancer, and obese muscle atrophy. | [167,168,169] |
| Carnosol: a bioactive diterpene compound present in Lamiaceae spp. | Inhibits NF-κB and AMPK pathway; activates Akt pathway; inhibits proteolysis. | Carnosol and its analogues attenuate muscle atrophy and fat lipolysis induced by cancer cachexia. | [170] |
| Histone deacetylase 2 | Inhibits NF-κB pathway. | Histone deacetylase 2 suppresses skeletal muscle atrophy and senescence in cigarette-smoke-induced mice with emphysema. | [171] |
| Ficus carica L.: a flowering plant that contains flavonoids, psoralen, and bergapten | Inhibits the inflammatory response; inhibits NF-κB pathway. | Ficus carica L. attenuates denervated skeletal muscle atrophy via PPARα/NF-κB pathway. | [172] |
| Targeted ablation of cellular inhibitor of apoptosis 1 (cIAP1) | Inhibits NF-κB pathway; inhibits UPS. | Targeted ablation of cIAP1 attenuates denervation-induced skeletal muscle atrophy. | [173] |
| P. yezoensis protein (PYCP): from red algae Pyropia yezoensis | Inhibits NF-κB pathway; inhibits UPS and ALP; antioxidant activity. | PYCP protects against TNF-α-induced myotube atrophy in C2C12 myotubes via the NF-κB signaling pathway. | [174,175] |
| Astragalus membranaceus and Paeonia japonica (APX) | Inhibits inflammatory cytokines; inhibits NF-κB pathway; inhibits p38. | APX protects against muscle atrophy in a C26 colon cancer cachexia mouse model. | [155] |
| Buyang Huanwu Tang (BYHWT): a compound traditional Chinese herbal medicine | Inhibits inflammatory response; inhibits NF-κB pathway. | BYHWT improves denervation-dependent muscle atrophy by decreasing NF-κB and MuRF1 levels. | [176] |
| Pyrroloquinoline quinone (PQQ) | Inhibits inflammation; inhibits oxidative stress; inhibits UPS and mitophagy. | PQQ ameliorates skeletal muscle atrophy induced by denervation via inhibition of the inflammatory signaling pathways. | [93,143] |
| Ajoene: a sulfur compound found in crushed garlic | Inhibits JAK/STAT3 and NF-κB pathway; inhibits SMADs/FoxO pathway; inhibits UPS. | Ajoene extract and Z-ajoene can attenuate skeletal muscle atrophy induced by cancer cachexia through suppressing inflammatory responses. | [153] |
| Glabridin: a prenylated isoflavone | Inhibits phosphorylation of p38; inhibits FoxO3a. | Glabridin inhibits dexamethasone-induced muscle atrophy. | [177] |
| Imperatorin (IMP): a main bioactive component of Angelica dahurica Radix | Inhibits JAK/STAT3 pathway; inhibits UPS. | Imperatorin alleviates cancer cachexia and prevents muscle wasting via directly inhibiting STAT3. | [178] |
| Cryptotanshinone: a major lipophilic compound extracted from the root of Danshen | Inhibits JAK/STAT3 pathway; inhibits UPS. | Cryptotanshinone prevents muscle wasting in CT26-induced cancer cachexia through inhibiting STAT3 signaling pathway. | [179] |
| Isoquercitrin: a biologically active flavonoid with antioxidative and anti-inflammatory properties | Inhibits inflammatory response; inhibits oxidative stress; inhibits UPS and mitophagy. | Isoquercitrin delays denervated muscle atrophy by inhibiting oxidative stress and inflammation. | [31] |
| Alantolactone: a sesquiterpene lactone isolated from Inula helenium | Inhibits inflammatory response; inhibits STAT3 pathway. | Alantolactone ameliorates cancer cachexia-associated muscle atrophy mainly by inhibiting the STAT3 signaling pathway. | [180] |
| S-allyl cysteine: an active component of garlic (Allium sativum) | Inhibits inflammatory response; inhibits NF-κB pathway; inhibits UPS. | S-allyl cysteine inhibits TNFα-induced skeletal muscle wasting through suppressing the expression of inflammatory molecules. | [181] |
| L-carnitine: antioxidant and anti-inflammatory properties | Reduces the levels of IL-1 and IL-6; inhibits UPS; activates AKT/FOXO3a pathway. | L-carnitine ameliorates the muscle wasting of cancer cachexia. | [182] |
| Ampelopsin: a natural flavonoid with antioxidant and anti-inflammatory properties | Anti-inflammatory function; anti-oxidative function; inhibits UPS; up-regulates AMPK/SIRT1 pathway. | Ampelopsin attenuates the atrophy of skeletal muscle from d-gal-induced aging rats. | [183] |
| Aspirin | Inhibits inflammatory response; inhibits JAK2/STAT3 pathway; inhibits UPS. | Aspirin alleviates denervation-induced muscle atrophy via regulating the Sirt1/PGC-1α axis and STAT3 signaling. | [13] |
| Mito-TEMPO: a mitochondria-targeted antioxidant | Inhibits inflammatory response; ameliorates mitochondrial dysfunction. | Mito-TEMPO improved muscle wasting in CKD mice possibly through inhibiting inflammation and oxidative stress. | [184] |
| Hemin: an inducer of heme oxygenase-1 (HO-1) | Inhibits proinflammatory cytokine; inhibits ROS; inhibits UPS. | Upregulation of heme oxygenase-1 by hemin alleviates sepsis-induced muscle wasting in mice. | [185] |
| Avenanthramides (AVAs) | Inhibits inflammatory response; inhibits NF-κB pathway; mitigates oxidative stress; inhibits UPS. | AVAs can reduce proinflammatory cytokine and ROS production and ameliorate TNF-α-induced myotube atrophy in muscle cells. | [186] |
| SKP-SC-EVs: skin-derived precursors pre-differentiated Schwann cells’ extracellular vesicles | Inhibits inflammation; inhibits oxidative stress; inhibits UPS; inhibits mitophagy. | SKP-SC-EVs mitigate denervated muscle atrophy by inhibiting oxidative stress and inflammation. | [19] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, Y.; Li, M.; Chang, M.; Liu, R.; Qiu, J.; Wang, K.; Deng, C.; Shen, Y.; Zhu, J.; Wang, W.; et al. Inflammation: Roles in Skeletal Muscle Atrophy. Antioxidants 2022, 11, 1686. https://doi.org/10.3390/antiox11091686
Ji Y, Li M, Chang M, Liu R, Qiu J, Wang K, Deng C, Shen Y, Zhu J, Wang W, et al. Inflammation: Roles in Skeletal Muscle Atrophy. Antioxidants. 2022; 11(9):1686. https://doi.org/10.3390/antiox11091686
Chicago/Turabian StyleJi, Yanan, Ming Li, Mengyuan Chang, Ruiqi Liu, Jiayi Qiu, Kexin Wang, Chunyan Deng, Yuntian Shen, Jianwei Zhu, Wei Wang, and et al. 2022. "Inflammation: Roles in Skeletal Muscle Atrophy" Antioxidants 11, no. 9: 1686. https://doi.org/10.3390/antiox11091686
APA StyleJi, Y., Li, M., Chang, M., Liu, R., Qiu, J., Wang, K., Deng, C., Shen, Y., Zhu, J., Wang, W., Xu, L., & Sun, H. (2022). Inflammation: Roles in Skeletal Muscle Atrophy. Antioxidants, 11(9), 1686. https://doi.org/10.3390/antiox11091686