
Integrin-Linked Kinase Expression in Human Valve Endothelial Cells Plays a Protective Role in Calcific Aortic Valve Disease
 , , , , ,
, , , , ,  ,
,  and
and 
Abstract
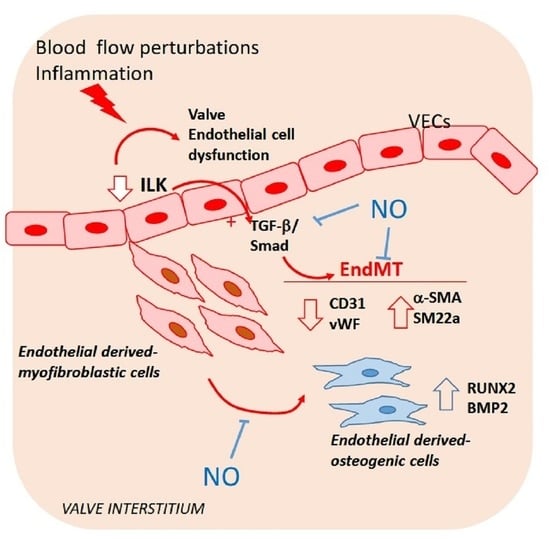
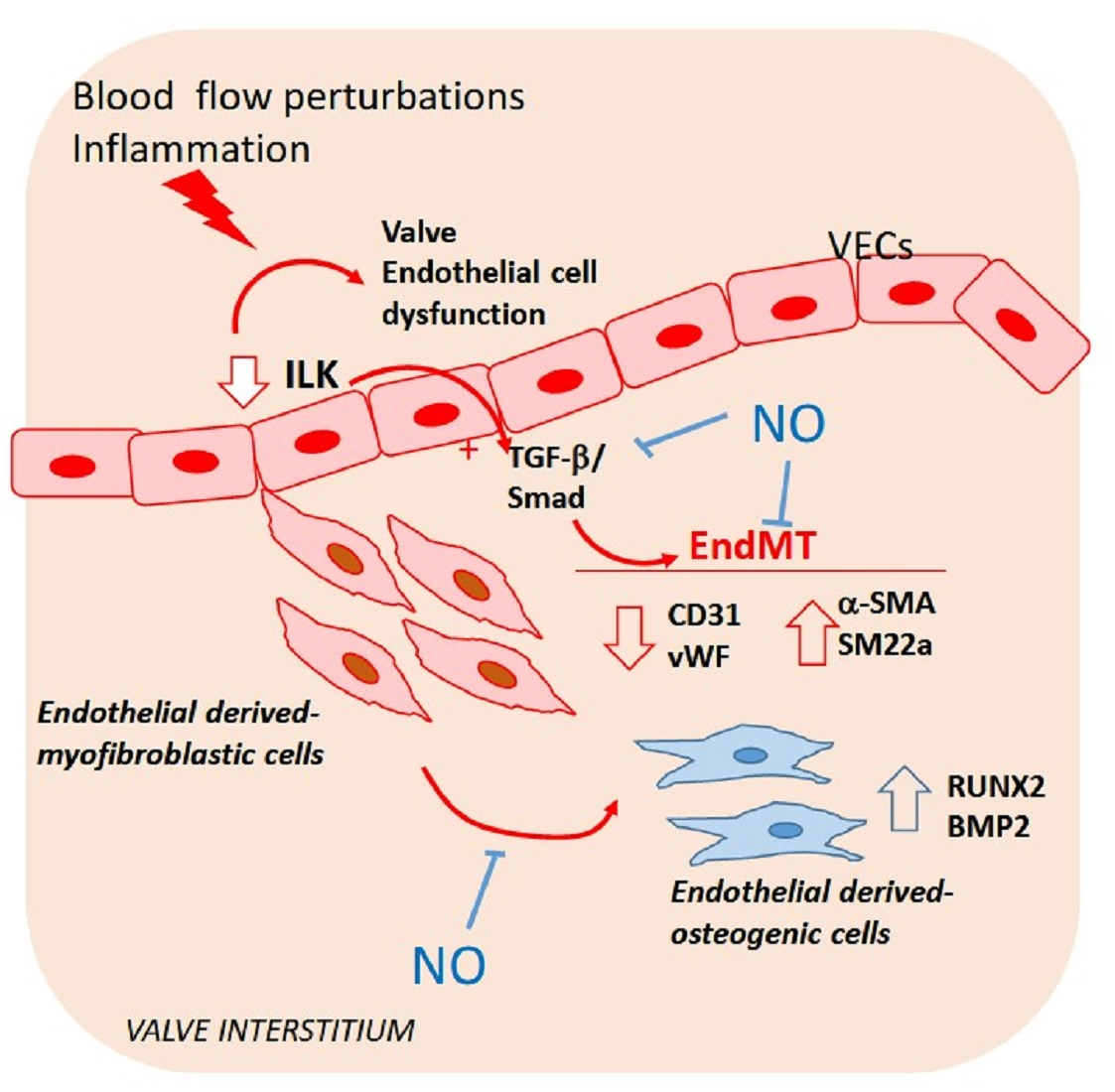
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Human Calcified Aortic Valves
2.2. Animals
2.3. Histological Detection of Calcium Deposits
2.4. Immunohistochemistry
2.5. Confocal Microscopy
2.6. Human Valve Endothelial Cell (hVEC) Isolation
2.7. Immunoblot
2.8. Cell Transfection
2.9. Cell Treatments
2.10. Pro-Osteogenic Culture Medium
2.11. Nitric Oxide and Superoxide Anion Production
2.12. Statistical Analysis
3. Results
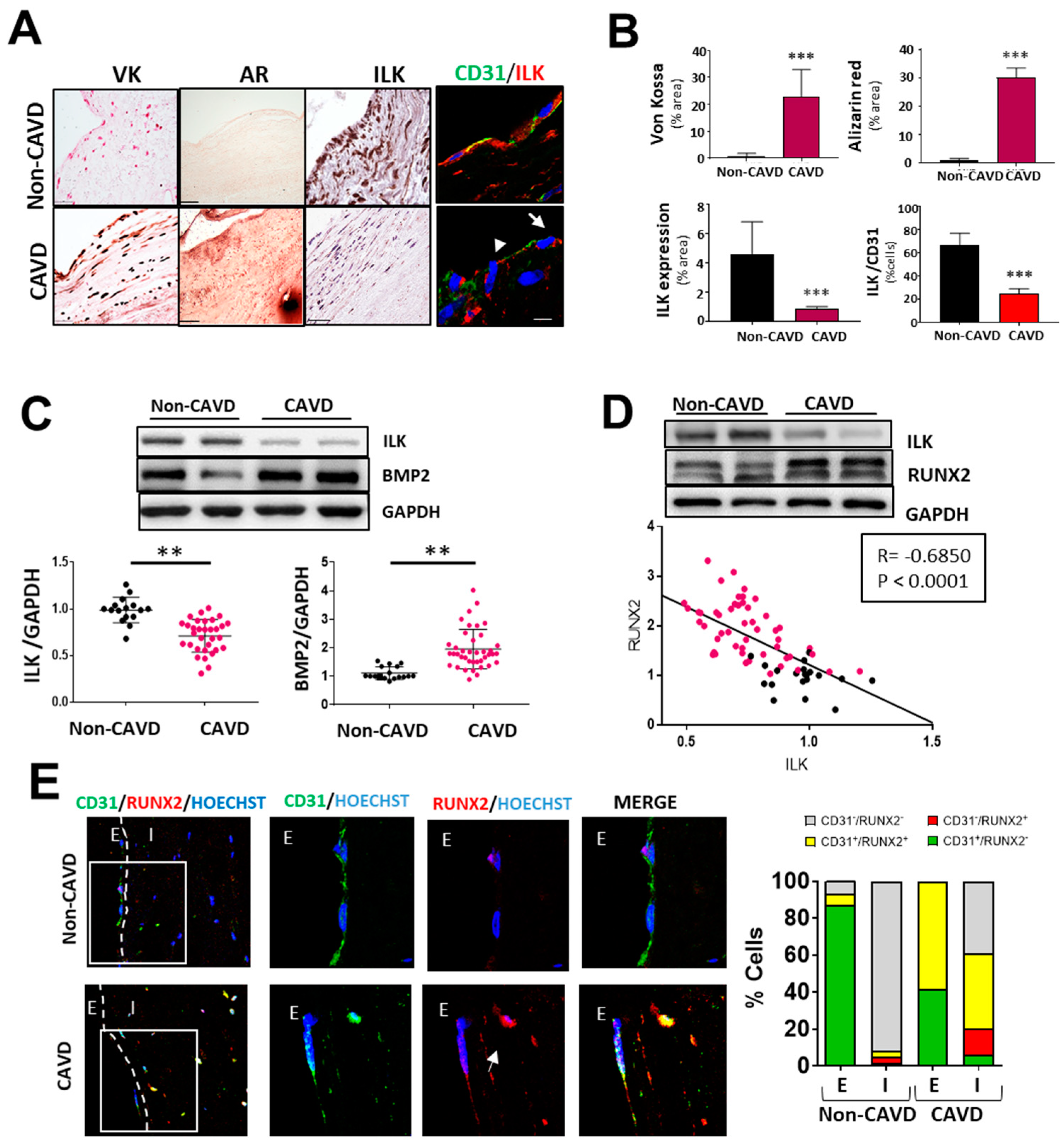
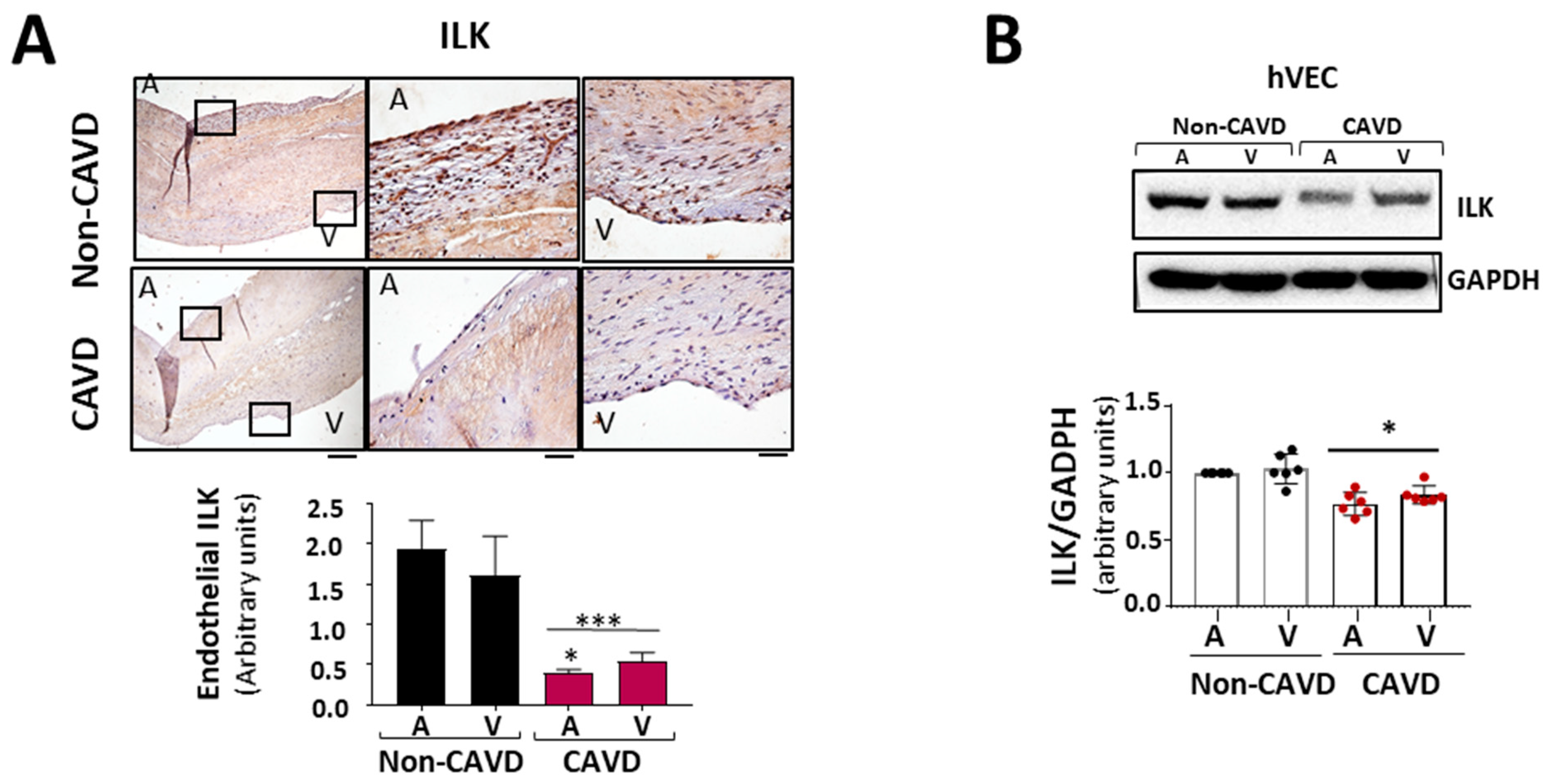
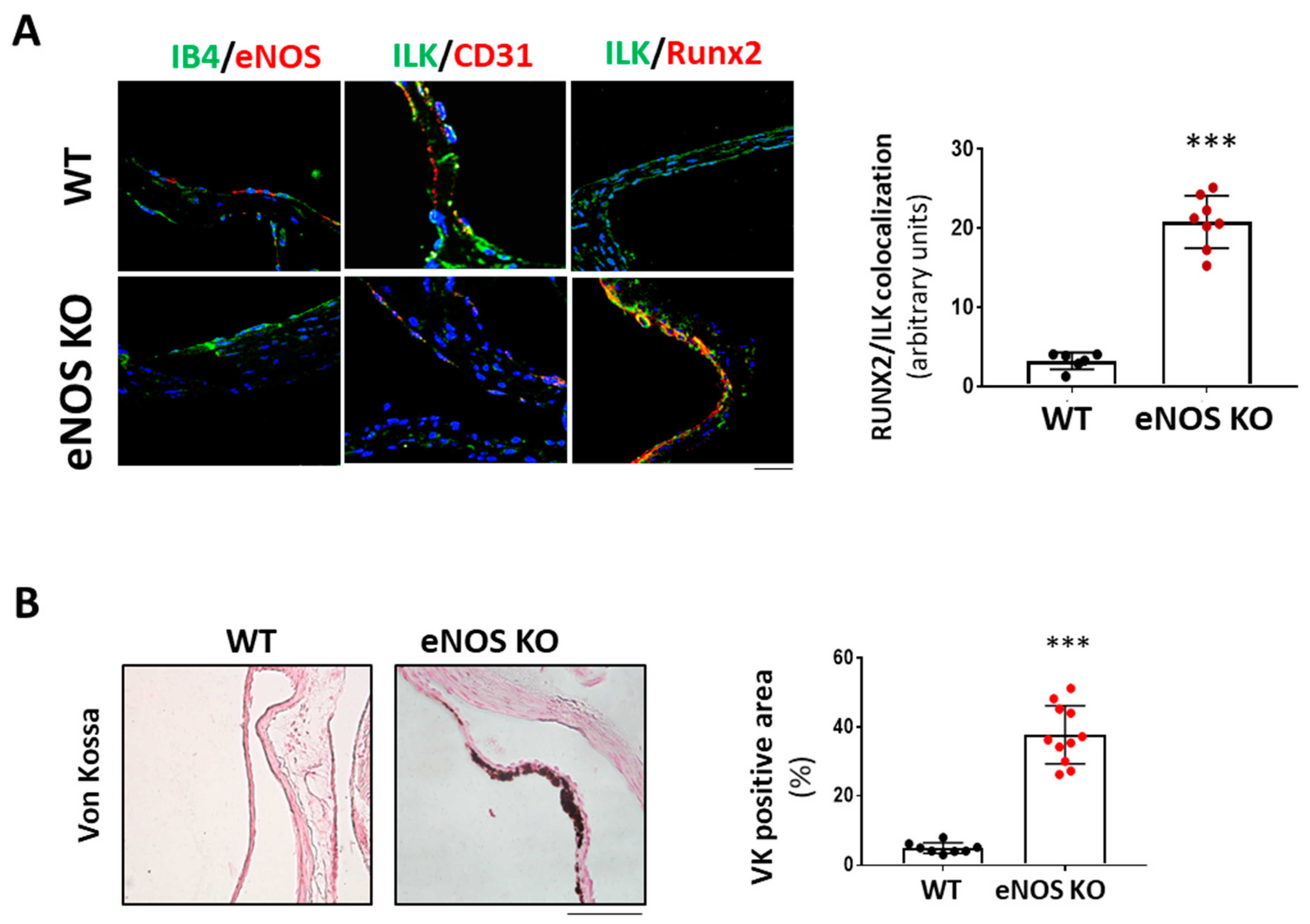
3.1. ILK Decrease in Aortic Endothelial Cells of Calcific Aortic stenosis Patients
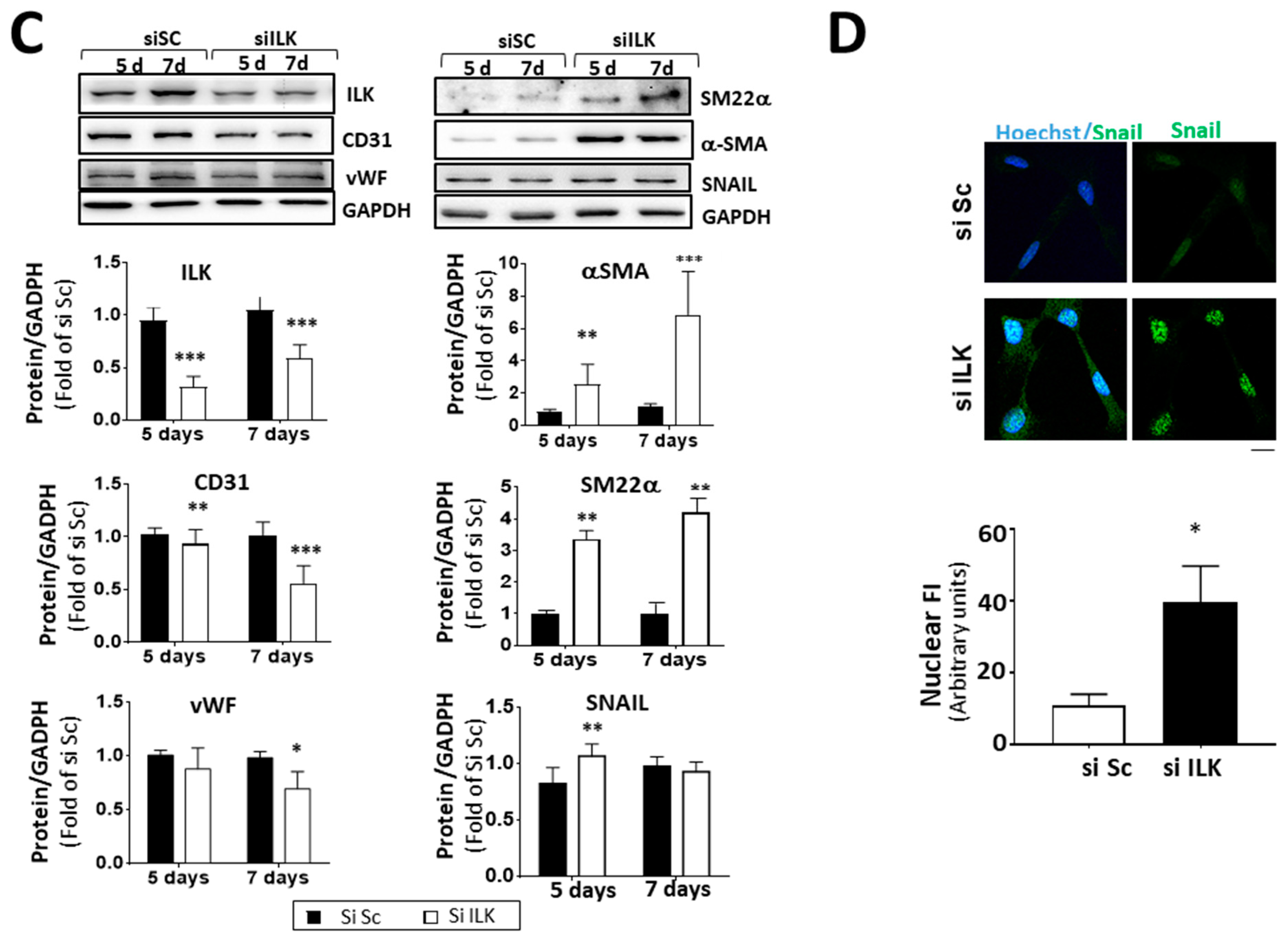
3.2. ILK Prevents EndMT and the Osteogenic Phenotype Switch in hVECs
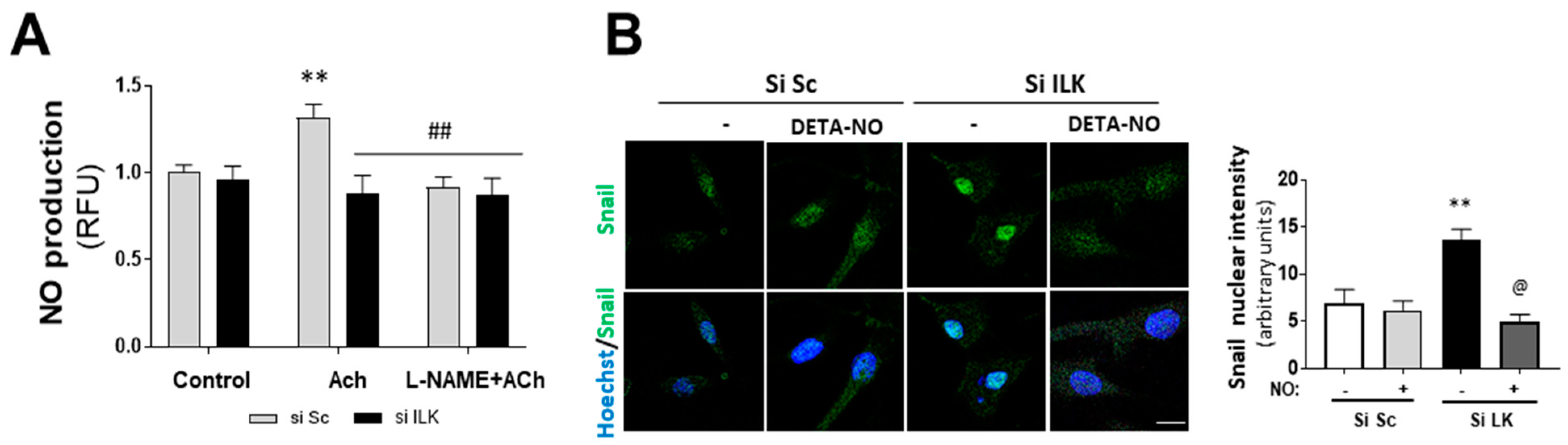
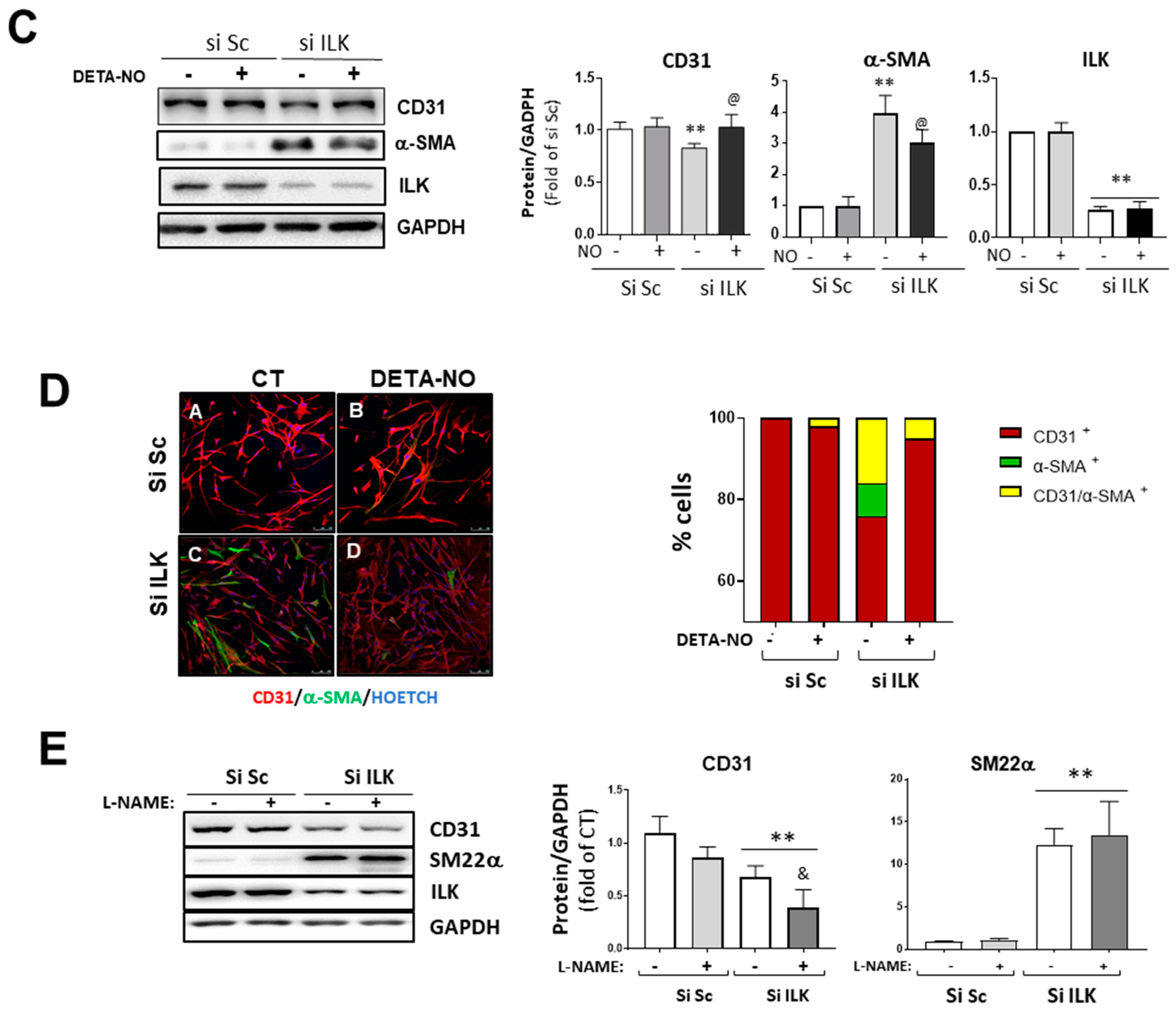
3.3. Nitric Oxide (NO) Prevents Osteogenic Differentiation Induced by ILK Silencing in hVECs
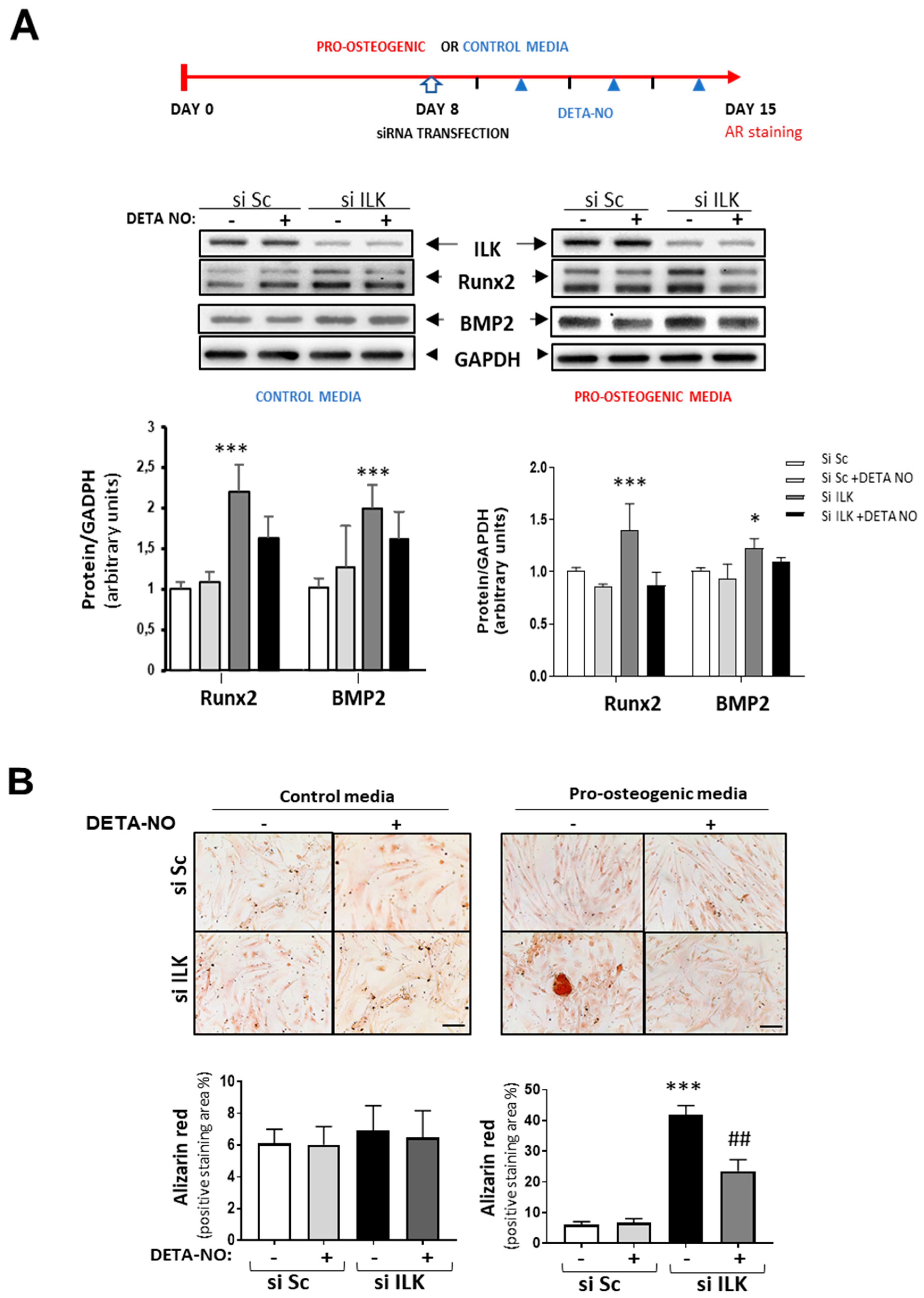
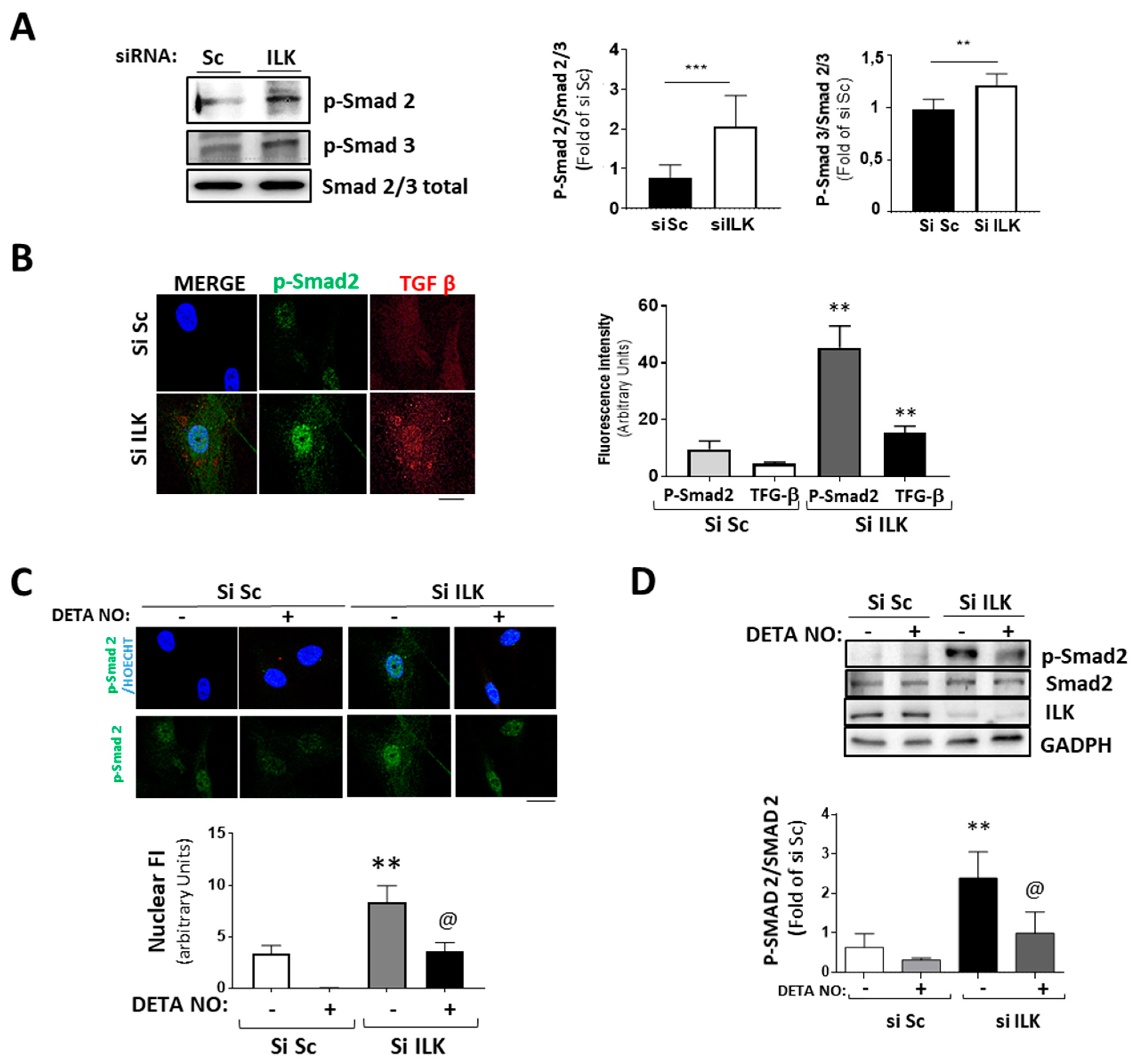
3.4. NO Prevents ILK Silencing Effects on Valve Calcification through the TGF-β/Smad2/3 Signaling Pathway
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rutkovskiy, A.; Malashicheva, A.; Sullivan, G.; Bogdanova, M.; Kostareva, A.; Stensløkken, K.O.; Fiane, A.; Vaage, J. Valve Interstitial Cells: The Key to Understanding the Pathophysiology of Heart Valve Calcification. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Yadgir, S.; Johnson, C.O.; Aboyans, V.; Adebayo, O.M.; Adedoyin, R.A.; Afarideh, M.; Alahdab, F.; Alashi, A.; Alipour, V.; Arabloo, J.; et al. Global, Regional, and National Burden of Calcific Aortic Valve and Degenerative Mitral Valve Diseases, 1990–2017. Circulation 2020, 141, 1670–1680. [Google Scholar] [CrossRef] [PubMed]
- Goody, P.R.; Hosen, M.R.; Christmann, D.; Niepmann, S.T.; Zietzer, A.; Adam, M.; Bönner, F.; Zimmer, S.; Nickenig, G.; Jansen, F. Aortic Valve Stenosis: From Basic Mechanisms to Novel Therapeutic Targets. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 885–900. [Google Scholar] [CrossRef] [PubMed]
- Lindman, B.R.; Sukul, D.; Dweck, M.R.; Madhavan, M.V.; Arsenault, B.J.; Coylewright, M.; Merryman, W.D.; Newby, D.E.; Lewis, J.; Harrell, F.E.; et al. Evaluating Medical Therapy for Calcific Aortic Stenosis: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2021, 78, 2354–2376. [Google Scholar] [CrossRef]
- Ma, X.; Zhao, D.; Yuan, P.; Li, J.; Yun, Y.; Cui, Y.; Zhang, T.; Ma, J.; Sun, L.; Ma, H.; et al. Endothelial-to-Mesenchymal Transition in Calcific Aortic Valve Disease. Acta Cardiol. Sin. 2020, 36, 183–194. [Google Scholar] [CrossRef]
- Hjortnaes, J.; Shapero, K.; Goettsch, C.; Hutcheson, J.D.; Keegan, J.; Kluin, J.; Mayer, J.E.; Bischoff, J.; Aikawa, E. Valvular Interstitial Cells Suppress Calcification of Valvular Endothelial Cells. Atherosclerosis 2015, 242, 251. [Google Scholar] [CrossRef]
- Mahler, G.J.; Farrar, E.J.; Butcher, J.T. Inflammatory Cytokines Promote Mesenchymal Transformation in Embryonic and Adult Valve Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 121. [Google Scholar] [CrossRef]
- Hannigan, G.E.; Coles, J.G.; Dedhar, S. Integrin-Linked Kinase at the Heart of Cardiac Contractility, Repair, and Disease. Circ. Res. 2007, 100, 1408–1414. [Google Scholar] [CrossRef]
- Górska, A.; Mazur, A.J. Integrin-Linked Kinase (ILK): The Known vs. the Unknown and Perspectives. Cell. Mol. Life Sci. 2022, 79, 100. [Google Scholar] [CrossRef]
- Herranz, B.; Marquez, S.; Guijarro, B.; Aracil, E.; Aicart-Ramos, C.; Rodriguez-Crespo, I.; Rodríguez-Puyol, M.; Zaragoza, C.; Saura, M. Integrin-Linked Kinase Regulates Vasomotor Function by Preventing Endothelial Nitric Oxide Synthase Uncoupling: Role in Atherosclerosis. Circ. Res. 2012, 110, 439–449. [Google Scholar] [CrossRef] [Green Version]
- Reventun, P.; Alique, M.; Cuadrado, I.; Márquez, S.; Toro, R.; Zaragoza, C.; Saura, M. INOS-Derived Nitric Oxide Induces Integrin-Linked Kinase Endocytic Lysosome-Mediated Degradation in the Vascular Endothelium. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1272–1281. [Google Scholar] [CrossRef]
- Cuadrado, I.; Castejon, B.; Martin, A.M.; Saura, M.; Reventun-Torralba, P.; Zamorano, J.L.; Zaragoza, C. Nitric Oxide Induces Cardiac Protection by Preventing Extracellular Matrix Degradation through the Complex Caveolin-3/Emmprin in Cardiac Myocytes. PLoS ONE 2016, 11, e0162912. [Google Scholar] [CrossRef]
- Ramos-Vara, J.A. Technical Aspects of Immunohistochemistry. Vet. Pathol. 2005, 42, 405–426. [Google Scholar] [CrossRef]
- Liu, M.M.; Flanagan, T.C.; Lu, C.C.; French, A.T.; Argyle, D.J.; Corcoran, B.M. Culture and Characterisation of Canine Mitral Valve Interstitial and Endothelial Cells. Vet. J. 2015, 204, 32–39. [Google Scholar] [CrossRef]
- Hanna, L.; Armour, C.; Xu, X.Y.; Gibbs, R. The Haemodynamic and Pathophysiological Mechanisms of Calcific Aortic Valve Disease. Biomedicines 2022, 10, 1317. [Google Scholar] [CrossRef]
- Kovacic, J.C.; Dimmeler, S.; Harvey, R.P.; Finkel, T.; Aikawa, E.; Krenning, G.; Baker, A.H. Endothelial to Mesenchymal Transition in Cardiovascular Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 190–209. [Google Scholar] [CrossRef]
- Majumdar, U.; Manivannan, S.; Basu, M.; Ueyama, Y.; Blaser, M.C.; Cameron, E.; McDermott, M.R.; Lincoln, J.; Cole, S.E.; Wood, S.; et al. Nitric Oxide Prevents Aortic Valve Calcification by S-Nitrosylation of USP9X to Activate NOTCH Signaling. Sci. Adv. 2021, 7, eabe3706. [Google Scholar] [CrossRef]
- Bosse, K.; Hans, C.P.; Zhao, N.; Koenig, S.N.; Huang, N.; Guggilam, A.; LaHaye, S.; Tao, G.; Lucchesi, P.A.; Lincoln, J.; et al. Endothelial Nitric Oxide Signaling Regulates Notch1 in Aortic Valve Disease. J. Mol. Cell. Cardiol. 2013, 60, 27. [Google Scholar] [CrossRef]
- Chen, G.; Deng, C.; Li, Y.P. TGF-β and BMP Signaling in Osteoblast Differentiation and Bone Formation. Int. J. Biol. Sci. 2012, 8, 272. [Google Scholar] [CrossRef]
- Yang, X.; Meng, X.; Su, X.; Mauchley, D.C.; Ao, L.; Cleveland, J.C.; Fullerton, D.A. Bone Morphogenic Protein 2 Induces Runx2 and Osteopontin Expression in Human Aortic Valve Interstitial Cells: Role of Smad1 and Extracellular Signal-Regulated Kinase 1/2. J. Thorac. Cardiovasc. Surg. 2009, 138, 1008–1015. [Google Scholar] [CrossRef] [Green Version]
- Song, R.; Fullerton, D.A.; Ao, L.; Zhao, K.S.; Meng, X. An Epigenetic Regulatory Loop Controls Pro-Osteogenic Activation by TGF-Β1 or Bone Morphogenetic Protein 2 in Human Aortic Valve Interstitial Cells. J. Biol. Chem. 2017, 292, 8657. [Google Scholar] [CrossRef]
- Groppe, J.; Greenwald, J.; Wiater, E.; Rodriguez-Leon, J.; Economides, A.N.; Kwiatkowski, W.; Affolter, M.; Vale, W.W.; Izpisua Belmonte, J.C.; Choe, S. Structural Basis of BMP Signalling Inhibition by the Cystine Knot Protein Noggin. Nature 2002, 420, 636–642. [Google Scholar] [CrossRef]
- Paranya, G.; Vineberg, S.; Dvorin, S.; Kaushal, S.; Roth, S.J.; Rabkin, E.; Schoen, F.J.; Bischoff, J. Aortic Valve Endothelial Cells Undergo Transforming Growth Factor-β-Mediated and Non-Transforming Growth Factor-β-Mediated Transdifferentiation in Vitro. Am. J. Pathol. 2001, 159, 1335. [Google Scholar] [CrossRef]
- Saura, M.; Zaragoza, C.; Herranz, B.; Griera, M.; Diez-Marqués, L.; Rodriguez-Puyol, D.; Rodriguez-Puyol, M. Nitric Oxide Regulates Transforming Growth Factor-Beta Signaling in Endothelial Cells. Circ. Res. 2005, 97, 1115–1123. [Google Scholar] [CrossRef]
- Gomel, M.A.; Lee, R.; Grande-Allen, K.J. Comparing the Role of Mechanical Forces in Vascular and Valvular Calcification Progression. Front. Cardiovasc. Med. 2018, 5, 197. [Google Scholar] [CrossRef]
- Butcher, J.T.; Nerem, R.M. Valvular Endothelial Cells Regulate the Phenotype of Interstitial Cells in Co-Culture: Effects of Steady Shear Stress. Tissue Eng. 2006, 12, 905–915. [Google Scholar] [CrossRef]
- Gould, S.T.; Matherly, E.E.; Smith, J.N.; Heistad, D.D.; Anseth, K.S. The Role of Valvular Endothelial Cell Paracrine Signaling and Matrix Elasticity on Valvular Interstitial Cell Activation. Biomaterials 2014, 35, 3596–3606. [Google Scholar] [CrossRef]
- Kraler, S.; Blaser, M.C.; Aikawa, E.; Camici, G.G.; Lüscher, T.F. Calcific Aortic Valve Disease: From Molecular and Cellular Mechanisms to Medical Therapy. Eur. Heart J. 2022, 43, 683–697. [Google Scholar] [CrossRef]
- Balachandran, K.; Alford, P.W.; Wylie-Sears, J.; Goss, J.A.; Grosberg, A.; Bischoff, J.; Aikawa, E.; Levine, R.A.; Parker, K.K. Cyclic Strain Induces Dual-Mode Endothelialmesenchymal Transformation of the Cardiac Valve. Proc. Natl. Acad. Sci. USA 2011, 108, 19943–19948. [Google Scholar] [CrossRef]
- Wylie-Sears, J.; Aikawa, E.; Levine, R.A.; Yang, J.H.; Bischoff, J. Mitral Valve Endothelial Cells with Osteogenic Differentiation Potential. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 598–607. [Google Scholar] [CrossRef] [Green Version]
- El-Hoss, J.; Arabian, A.; Dedhar, S.; St-Arnaud, R. Inactivation of the Integrin-Linked Kinase (ILK) in Osteoblasts Increases Mineralization. Gene 2014, 533, 246–252. [Google Scholar] [CrossRef]
- Rustad, K.C.; Wong, V.W.; Gurtner, G.C. The Role of Focal Adhesion Complexes in Fibroblast Mechanotransduction during Scar Formation. Differentiation 2013, 86, 87–91. [Google Scholar] [CrossRef]
- Yamamoto-Fukuda, T.; Akiyama, N.; Kojima, H. L1CAM-ILK-YAP Mechanotransduction Drives Proliferative Activity of Epithelial Cells in Middle Ear Cholesteatoma. Am. J. Pathol. 2020, 190, 1667–1679. [Google Scholar] [CrossRef]
- McDonald, P.C.; Fielding, A.B.; Dedhar, S. Integrin-Linked Kinase--Essential Roles in Physiology and Cancer Biology. J. Cell Sci. 2008, 121, 3121–3132. [Google Scholar] [CrossRef]
- Hannigan, G.E.; Leung-Hagesteijn, C.; Fitz-Gibbon, L.; Coppolino, M.G.; Radeva, G.; Filmus, J.; Bell, J.C.; Dedhar, S. Regulation of Cell Adhesion and Anchorage-Dependent Growth by a New Beta 1-Integrin-Linked Protein Kinase. Nature 1996, 379, 91–96. [Google Scholar] [CrossRef]
- Sánchez-Esteban, S.; Cook, A.; Reventún, P.; Zaragoza, C.; Zamorano, J.L.; Saura, M. Aging-Related ILK Levels Are Associated with Calcified Aortic Valve and Circulating MiR 199-3p Levels. Rev. Española Cardiol. 2022, 75, 88–91. [Google Scholar] [CrossRef]
- van Driel, B.O.; Schuldt, M.; Algül, S.; Levin, E.; Güclü, A.; Germans, T.; van Rossum, A.C.; Pei, J.; Harakalova, M.; Baas, A.; et al. Metabolomics in Severe Aortic Stenosis Reveals Intermediates of Nitric Oxide Synthesis as Most Distinctive Markers. Int. J. Mol. Sci. 2021, 22, 3569. [Google Scholar] [CrossRef]
- Liu, Z.; Dong, N.; Hui, H.; Wang, Y.; Liu, F.; Xu, L.; Liu, M.; Rao, Z.; Yuan, Z.; Shang, Y.; et al. Endothelial Cell-Derived Tetrahydrobiopterin Prevents Aortic Valve Calcification. Eur. Heart J. 2022, 43, 1652–1664. [Google Scholar] [CrossRef]
- Gomez-Stallons, M.V.; Wirrig-Schwendeman, E.E.; Hassel, K.R.; Conway, S.J.; Yutzey, K.E. Bone Morphogenetic Protein Signaling Is Required for Aortic Valve Calcification. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1398–1405. [Google Scholar] [CrossRef]
- Hiepen, C.; Mendez, P.L.; Knaus, P. It Takes Two to Tango: Endothelial TGFβ/BMP Signaling Crosstalk with Mechanobiology. Cells 2020, 9, 1965. [Google Scholar] [CrossRef]
- Xian, S.; Chen, A.; Wu, X.; Lu, C.; Wu, Y.; Huang, F.; Zeng, Z. Activation of Activin/Smad2 and 3 Signaling Pathway and the Potential Involvement of Endothelial-mesenchymal Transition in the Valvular Damage Due to Rheumatic Heart Disease. Mol. Med. Rep. 2021, 23, 10. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.M.T.; Billiar, K.L. Investigating the Role of Substrate Stiffness in the Persistence of Valvular Interstitial Cell Activation. J. Biomed. Mater. Res. A 2012, 100, 2474–2482. [Google Scholar] [CrossRef] [PubMed]
- Serrano, I.; Mcdonald, P.C.; Lock, F.E.; Dedhar, S. Role of the Integrin-Linked Kinase (ILK)/Rictor Complex in TGFβ-1-Induced Epithelial-Mesenchymal Transition (EMT). Oncogene 2013, 32, 50–60. [Google Scholar] [CrossRef]
- Chen, D.; Zhang, Y.; Zhang, X.; Li, J.; Han, B.; Liu, S.; Wang, L.; Ling, Y.; Mao, S.; Wang, X. Overexpression of Integrin-Linked Kinase Correlates with Malignant Phenotype in Non-Small Cell Lung Cancer and Promotes Lung Cancer Cell Invasion and Migration via Regulating Epithelial-Mesenchymal Transition (EMT)-Related Genes. Acta Histochem. 2013, 115, 128–136. [Google Scholar] [CrossRef]
- Gil, D.; Ciołczyk-Wierzbicka, D.; Dulińska-Litewka, J.; Laidler, P. Integrin-Linked Kinase Regulates Cadherin Switch in Bladder Cancer. Tumour Biol. 2016, 37, 15185–15191. [Google Scholar] [CrossRef]
- Jia, Y.Y.; Yu, Y.; Li, H.J. POSTN Promotes Proliferation and Epithelial-Mesenchymal Transition in Renal Cell Carcinoma through ILK/AKT/MTOR Pathway. J. Cancer 2021, 12, 4183–4195. [Google Scholar] [CrossRef]
- Qi, F.H.; Cai, P.P.; Liu, X.; Si, G.M. Adenovirus-Mediated P311 Ameliorates Renal Fibrosis through Inhibition of Epithelial-Mesenchymal Transition via TGF-Β1-Smad-ILK Pathway in Unilateral Ureteral Obstruction Rats. Int. J. Mol. Med. 2018, 41, 3015–3023. [Google Scholar] [CrossRef]
- Chou, C.C.; Chuang, H.C.; Salunke, S.B.; Kulp, S.K.; Chen, C.S. A Novel HIF-1α-Integrin-Linked Kinase Regulatory Loop That Facilitates Hypoxia-Induced HIF-1α Expression and Epithelial-Mesenchymal Transition in Cancer Cells. Oncotarget 2015, 6, 8271–8285. [Google Scholar] [CrossRef]
- Huang, Q.; Gan, Y.; Yu, Z.; Wu, H.; Zhong, Z. Endothelial to Mesenchymal Transition: An Insight in Atherosclerosis. Front. Cardiovasc. Med. 2021, 8, 734550. [Google Scholar] [CrossRef]
- Khan, K.; Yu, B.; Kiwan, C.; Shalal, Y.; Filimon, S.; Cipro, M.; Shum-Tim, D.; Cecere, R.; Schwertani, A. The Role of Wnt/β-Catenin Pathway Mediators in Aortic Valve Stenosis. Front. Cell Dev. Biol. 2020, 8, 862. [Google Scholar] [CrossRef]
- Zheng, R.; Zhu, P.; Gu, J.; Ni, B.; Sun, H.; He, K.; Bian, J.; Shao, Y.; Du, J. Transcription Factor Sp2 Promotes TGFB-Mediated Interstitial Cell Osteogenic Differentiation in Bicuspid Aortic Valves through a SMAD-Dependent Pathway. Exp. Cell Res. 2022, 411, 112972. [Google Scholar] [CrossRef]
- Li, J.; Ge, L.; Zhao, Y.; Zhai, Y.; Rao, N.; Yuan, X.; Yang, J.; Li, J.; Yu, S. TGF-Β2 and TGF-Β1 Differentially Regulate the Odontogenic and Osteogenic Differentiation of Mesenchymal Stem Cells. Arch. Oral Biol. 2022, 135, 105357. [Google Scholar] [CrossRef]
- Bruderer, M.; Richards, R.G.; Alini, M.; Stoddart, M.J. Role and Regulation of RUNX2 in Osteogenesis. Eur. Cell. Mater. 2014, 28, 269–286. [Google Scholar] [CrossRef]
- Ankeny, R.F.; Thourani, V.H.; Weiss, D.; Vega, J.D.; Taylor, W.R.; Nerem, R.M.; Jo, H. Preferential Activation of SMAD1/5/8 on the Fibrosa Endothelium in Calcified Human Aortic Valves--Association with Low BMP Antagonists and SMAD6. PLoS ONE 2011, 6, e20969. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Esteban, S.; Castro-Pinto, M.; Cook-Calvete, A.; Reventún, P.; Delgado-Marín, M.; Benito-Manzanaro, L.; Hernandez, I.; López-Menendez, J.; Zamorano, J.L.; Zaragoza, C.; et al. Integrin-Linked Kinase Expression in Human Valve Endothelial Cells Plays a Protective Role in Calcific Aortic Valve Disease. Antioxidants 2022, 11, 1736. https://doi.org/10.3390/antiox11091736
Sánchez-Esteban S, Castro-Pinto M, Cook-Calvete A, Reventún P, Delgado-Marín M, Benito-Manzanaro L, Hernandez I, López-Menendez J, Zamorano JL, Zaragoza C, et al. Integrin-Linked Kinase Expression in Human Valve Endothelial Cells Plays a Protective Role in Calcific Aortic Valve Disease. Antioxidants. 2022; 11(9):1736. https://doi.org/10.3390/antiox11091736
Chicago/Turabian StyleSánchez-Esteban, Sandra, Mercedes Castro-Pinto, Alberto Cook-Calvete, Paula Reventún, María Delgado-Marín, Lucía Benito-Manzanaro, Ignacio Hernandez, José López-Menendez, José Luis Zamorano, Carlos Zaragoza, and et al. 2022. "Integrin-Linked Kinase Expression in Human Valve Endothelial Cells Plays a Protective Role in Calcific Aortic Valve Disease" Antioxidants 11, no. 9: 1736. https://doi.org/10.3390/antiox11091736
APA StyleSánchez-Esteban, S., Castro-Pinto, M., Cook-Calvete, A., Reventún, P., Delgado-Marín, M., Benito-Manzanaro, L., Hernandez, I., López-Menendez, J., Zamorano, J. L., Zaragoza, C., & Saura, M. (2022). Integrin-Linked Kinase Expression in Human Valve Endothelial Cells Plays a Protective Role in Calcific Aortic Valve Disease. Antioxidants, 11(9), 1736. https://doi.org/10.3390/antiox11091736