

Mitochondria in Cell-Based Therapy for Stroke



Abstract

1. Introduction
2. Mitochondrial Impairment in the Oxidative Stress following Stroke and Reperfusion Injury
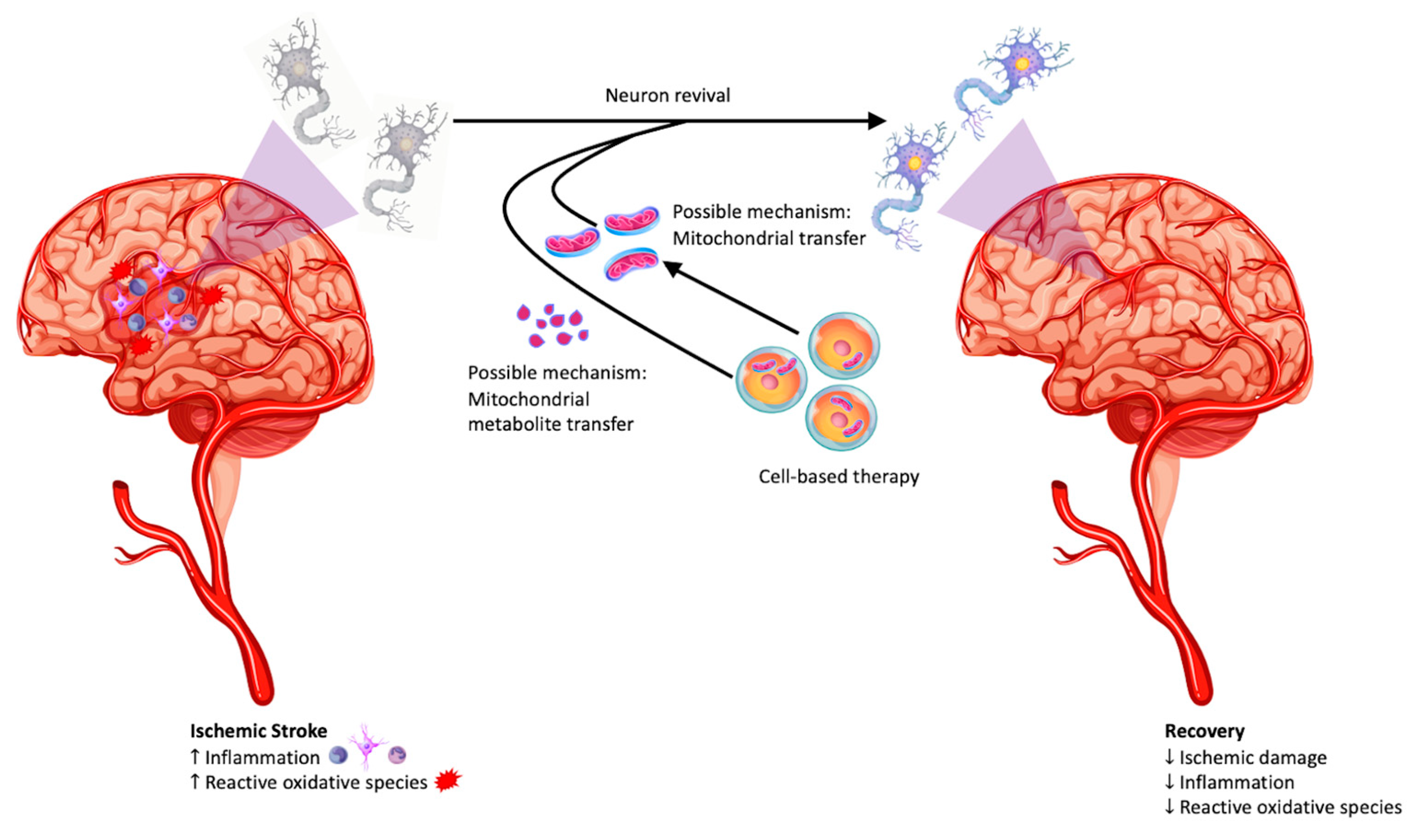
3. Repair of the Damaged Mitochondria in Stroke: Astrocytes-to Neurons Transfer of Mitochondria
4. Stem Cell-Neural Cell Crosstalk: Rescue of Mitochondria by Stem Cells
5. Non-Cell-Based Approaches to Mitochondrial Repair in Stroke
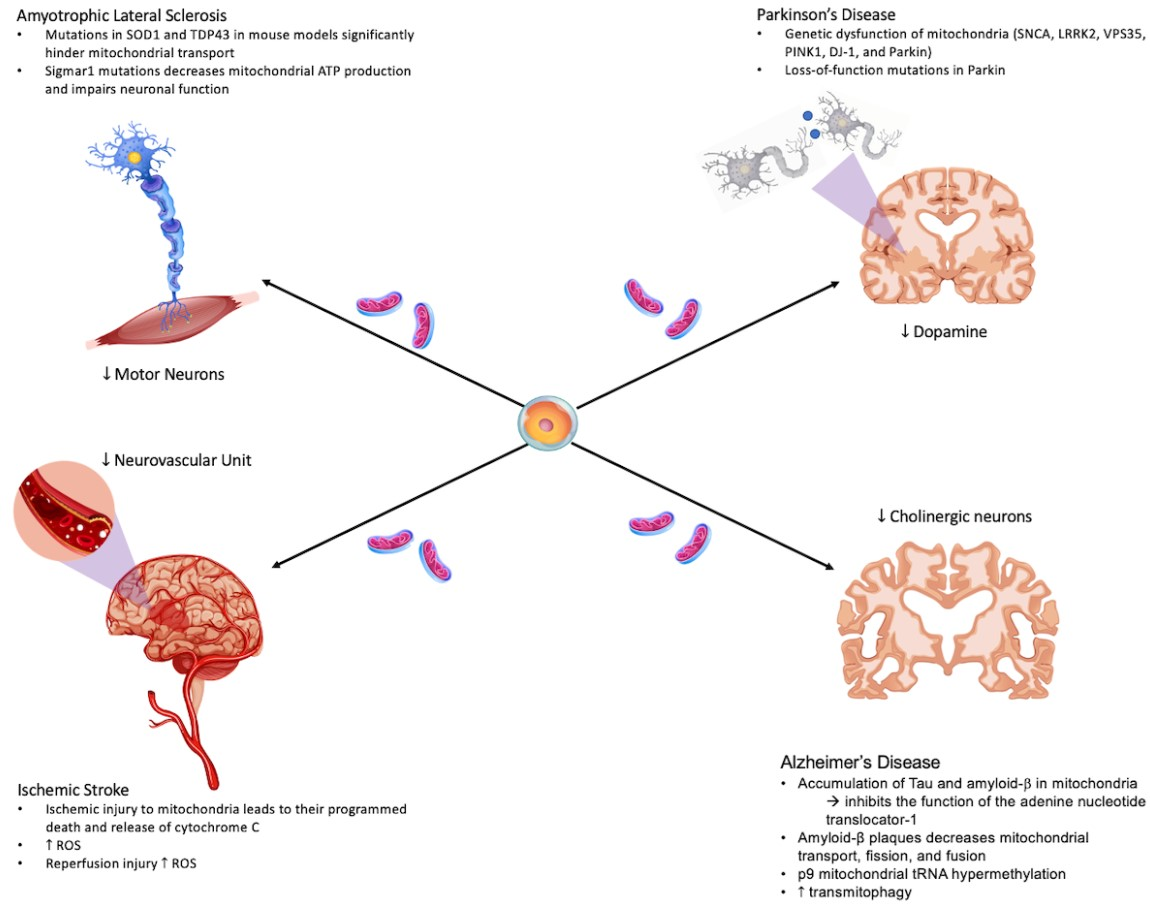
6. Mitochondrial Repair in Other Disorders of the Central Nervous System
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rangaraju, V.; Calloway, N.; Ryan, T.A. Activity-driven local ATP synthesis is required for synaptic function. Cell 2014, 156, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Dan Dunn, J.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox. Biol. 2015, 6, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Abate, M.; Festa, A.; Falco, M.; Lombardi, A.; Luce, A.; Grimaldi, A.; Zappavigna, S.; Sperlongano, P.; Irace, C.; Caraglia, M.; et al. Mitochondria as playmakers of apoptosis, autophagy and senescence. Semin. Cell Dev. Biol. 2020, 98, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Sagua, R.; Parra, V.; López-Crisosto, C.; Díaz, P.; Quest, A.F.; Lavandero, S. Calcium Transport and Signaling in Mitochondria. Compr. Physiol. 2017, 7, 623–634. [Google Scholar] [CrossRef]
- Cheng, A.; Hou, Y.; Mattson, M.P. Mitochondria and neuroplasticity. ASN Neuro. 2010, 2, e00045. [Google Scholar] [CrossRef]
- Marques-Aleixo, I.; Oliveira, P.J.; Moreira, P.I.; Magalhães, J.; Ascensão, A. Physical exercise as a possible strategy for brain protection: Evidence from mitochondrial-mediated mechanisms. Prog. Neurobiol. 2012, 99, 149–162. [Google Scholar] [CrossRef]
- Marques-Aleixo, I.; Santos-Alves, E.; Balça, M.M.; Rizo-Roca, D.; Moreira, P.I.; Oliveira, P.J.; Magalhães, J.; Ascensão, A. Physical exercise improves brain cortex and cerebellum mitochondrial bioenergetics and alters apoptotic, dynamic and auto(mito)phagy markers. Neuroscience 2015, 301, 480–495. [Google Scholar] [CrossRef]
- Fabricius, C.; Berthold, C.H.; Rydmark, M. Axoplasmic organelles at nodes of Ranvier. II. Occurrence and distribution in large myelinated spinal cord axons of the adult cat. J. Neurocytol. 1993, 22, 941–954. [Google Scholar] [CrossRef]
- Obashi, K.; Okabe, S. Regulation of mitochondrial dynamics and distribution by synapse position and neuronal activity in the axon. Eur. J. Neurosci. 2013, 38, 2350–2363. [Google Scholar] [CrossRef]
- Pilling, A.D.; Horiuchi, D.; Lively, C.M.; Saxton, W.M. Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol. Biol. Cell 2006, 17, 2057–2068. [Google Scholar] [CrossRef]
- Faits, M.C.; Zhang, C.; Soto, F.; Kerschensteiner, D. Dendritic mitochondria reach stable positions during circuit development. Elife 2016, 5, e11583. [Google Scholar] [CrossRef] [PubMed]
- Fukumitsu, K.; Hatsukano, T.; Yoshimura, A.; Heuser, J.; Fujishima, K.; Kengaku, M. Mitochondrial fission protein Drp1 regulates mitochondrial transport and dendritic arborization in cerebellar Purkinje cells. Mol. Cell Neurosci. 2016, 71, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Girard, M.; Larivière, R.; Parfitt, D.A.; Deane, E.C.; Gaudet, R.; Nossova, N.; Blondeau, F.; Prenosil, G.; Vermeulen, E.G.; Duchen, M.R.; et al. Mitochondrial dysfunction and Purkinje cell loss in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). Proc. Natl. Acad. Sci. USA 2012, 109, 1661–1666. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012, 2, 120080. [Google Scholar] [CrossRef]
- Rüb, C.; Wilkening, A.; Voos, W. Mitochondrial quality control by the Pink1/Parkin system. Cell Tissue Res. 2017, 367, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Nardin, A.; Schrepfer, E.; Ziviani, E. Counteracting PINK/Parkin Deficiency in the Activation of Mitophagy: A Potential Therapeutic Intervention for Parkinson’s Disease. Curr. Neuropharmacol. 2016, 14, 250–259. [Google Scholar] [CrossRef]
- Heo, J.M.; Ordureau, A.; Paulo, J.A.; Rinehart, J.; Harper, J.W. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cell. 2015, 60, 7–20. [Google Scholar] [CrossRef]
- Prakash, R.; Fauzia, E.; Siddiqui, A.J.; Yadav, S.K.; Kumari, N.; Singhai, A.; Khan, M.A.; Janowski, M.; Bhutia, S.K.; Raza, S.S. Oxidative Stress Enhances Autophagy-Mediated Death Of Stem Cells Through Erk1/2 Signaling Pathway-Implications For Neurotransplantations. Stem. Cell Rev. Rep. 2021, 17, 2347–2358. [Google Scholar] [CrossRef]
- Batlevi, Y.; La Spada, A.R. Mitochondrial autophagy in neural function, neurodegenerative disease, neuron cell death, and aging. Neurobiol. Dis. 2011, 43, 46–51. [Google Scholar] [CrossRef]
- Anthony, S.; Cabantan, D.; Monsour, M.; Borlongan, C.V. Neuroinflammation, Stem Cells, and Stroke. Stroke 2022, 53, 1460–1472. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis*. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [PubMed]
- Conte, F.; van Buuringen, N.; Voermans, N.C.; Lefeber, D.J. Galactose in human metabolism, glycosylation and congenital metabolic diseases: Time for a closer look. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129898. [Google Scholar] [CrossRef]
- Cadenas, E. Mitochondrial free radical production and cell signaling. Mol. Aspects Med. 2004, 25, 17–26. [Google Scholar] [CrossRef]
- Monsour, M.; Gorsky, A.; Nguyen, H.; Castelli, V.; Lee, J.Y.; Borlongan, C.V. Enhancing oxidative phosphorylation over glycolysis for energy production in cultured mesenchymal stem cells. Neuroreport 2022, 33, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.; Lockard, G.; Monsour, M.; Alayli, A.; Borlongan, C.V. The Role of Concomitant Nrf2 Targeting and Stem Cell Therapy in Cerebrovascular Disease. Antioxidants 2022, 11, 1447. [Google Scholar] [CrossRef] [PubMed]
- Jurcau, A.; Ardelean, A.I. Oxidative Stress in Ischemia/Reperfusion Injuries following Acute Ischemic Stroke. Biomedicines 2022, 10, 574. [Google Scholar] [CrossRef]
- Lu, M.; Guo, J.; Wu, B.; Zhou, Y.; Wu, M.; Farzaneh, M.; Khoshnam, S.E. Mesenchymal Stem Cell-Mediated Mitochondrial Transfer: A Therapeutic Approach for Ischemic Stroke. Transl. Stroke Res. 2021, 12, 212–229. [Google Scholar] [CrossRef]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef]
- Gorsky, A.; Monsour, M.; Nguyen, H.; Castelli, V.; Lee, J.-Y.; Borlongan, C.V. Metabolic Switching of Cultured Mesenchymal Stem Cells Creates Super Mitochondria in Rescuing Ischemic Neurons. NeuroMolecular Med. 2022. [Google Scholar] [CrossRef]
- Yan, W.; Diao, S.; Fan, Z. The role and mechanism of mitochondrial functions and energy metabolism in the function regulation of the mesenchymal stem cells. Stem. Cell Res. Ther. 2021, 12, 140. [Google Scholar] [CrossRef] [PubMed]
- Bastianetto, S.; Menard, C.; Quirion, R. Neuroprotective action of resveratrol. Biochim. Biophys. Acta 2015, 1852, 1195–1201. [Google Scholar] [CrossRef]
- Kahroba, H.; Ramezani, B.; Maadi, H.; Sadeghi, M.R.; Jaberie, H.; Ramezani, F. The role of Nrf2 in neural stem/progenitors cells: From maintaining stemness and self-renewal to promoting differentiation capability and facilitating therapeutic application in neurodegenerative disease. Ageing Res. Rev. 2021, 65, 101211. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Ding, Y.; Zhang, Y.; Tse, H.F.; Lian, Q. Paracrine mechanisms of mesenchymal stem cell-based therapy: Current status and perspectives. Cell Transplant. 2014, 23, 1045–1059. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.P.; Breakefield, X.O. Role of exosomes/microvesicles in the nervous system and use in emerging therapies. Front. Physiol. 2012, 3, 228. [Google Scholar] [CrossRef] [PubMed]
- Garbuzova-Davis, S.; Haller, E.; Lin, R.; Borlongan, C.V. Intravenously Transplanted Human Bone Marrow Endothelial Progenitor Cells Engraft Within Brain Capillaries, Preserve Mitochondrial Morphology, and Display Pinocytotic Activity Toward Blood-Brain Barrier Repair in Ischemic Stroke Rats. Stem. Cells 2017, 35, 1246–1258. [Google Scholar] [CrossRef]
- Babenko, V.A.; Silachev, D.N.; Popkov, V.A.; Zorova, L.D.; Pevzner, I.B.; Plotnikov, E.Y.; Sukhikh, G.T.; Zorov, D.B. Miro1 Enhances Mitochondria Transfer from Multipotent Mesenchymal Stem Cells (MMSC) to Neural Cells and Improves the Efficacy of Cell Recovery. Molecules 2018, 23, 687. [Google Scholar] [CrossRef]
- Liu, K.; Guo, L.; Zhou, Z.; Pan, M.; Yan, C. Mesenchymal stem cells transfer mitochondria into cerebral microvasculature and promote recovery from ischemic stroke. Microvasc. Res. 2019, 123, 74–80. [Google Scholar] [CrossRef]
- Pourmohammadi-Bejarpasi, Z.; Roushandeh, A.M.; Saberi, A.; Rostami, M.K.; Toosi, S.M.R.; Jahanian-Najafabadi, A.; Tomita, K.; Kuwahara, Y.; Sato, T.; Roudkenar, M.H. Mesenchymal stem cells-derived mitochondria transplantation mitigates I/R-induced injury, abolishes I/R-induced apoptosis, and restores motor function in acute ischemia stroke rat model. Brain Res. Bull. 2020, 165, 70–80. [Google Scholar] [CrossRef]
- Zhuo, Y.; Chen, W.; Li, W.; Huang, Y.; Duan, D.; Ge, L.; He, J.; Liu, J.; Hu, Z.; Lu, M. Ischemic-hypoxic preconditioning enhances the mitochondrial function recovery of transplanted olfactory mucosa mesenchymal stem cells via miR-181a signaling in ischemic stroke. Aging 2021, 13, 11234–11256. [Google Scholar] [CrossRef]
- Hosseini, L.; Karimipour, M.; Seyedaghamiri, F.; Abolhasanpour, N.; Sadigh-Eteghad, S.; Mahmoudi, J.; Farhoudi, M. Intranasal administration of mitochondria alleviated cognitive impairments and mitochondrial dysfunction in the photothrombotic model of mPFC stroke in mice. J. Stroke Cerebrovasc. Dis. 2022, 31, 106801. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Park, H.-H.; Choi, H.; Seo Kim, Y.; Yu, H.-J.; Lee, K.-Y.; Joo Lee, Y.; Hyun Kim, S.; Koh, S.-H. Coenzyme Q10 protects neural stem cells against hypoxia by enhancing survival signals. Brain Res. 2012, 1478, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, Y.; Tajiri, N.; Shojo, H.; Borlongan, C.V. Oxygen-glucose-deprived rat primary neural cells exhibit DJ-1 translocation into healthy mitochondria: A potent stroke therapeutic target. CNS Neurosci. Ther. 2014, 20, 275–281. [Google Scholar] [CrossRef]
- Hayakawa, K.; Chan, S.J.; Mandeville, E.T.; Park, J.H.; Bruzzese, M.; Montaner, J.; Arai, K.; Rosell, A.; Lo, E.H. Protective Effects of Endothelial Progenitor Cell-Derived Extracellular Mitochondria in Brain Endothelium. Stem. Cells 2018, 36, 1404–1410. [Google Scholar] [CrossRef]
- Gerdes, H.H.; Rustom, A.; Wang, X. Tunneling nanotubes, an emerging intercellular communication route in development. Mech. Dev. 2013, 130, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Torralba, D.; Baixauli, F.; Sanchez-Madrid, F. Mitochondria Know No Boundaries: Mechanisms and Functions of Intercellular Mitochondrial Transfer. Front. Cell Dev. Biol. 2016, 4, 107. [Google Scholar] [CrossRef]
- Las, G.; Shirihai, O.S. Miro1: New wheels for transferring mitochondria. Embo J. 2014, 33, 939–941. [Google Scholar] [CrossRef]
- Abdullahi, W.; Tripathi, D.; Ronaldson, P.T. Blood-brain barrier dysfunction in ischemic stroke: Targeting tight junctions and transporters for vascular protection. Am. J. Physiol. Cell Physiol. 2018, 315, C343–C356. [Google Scholar] [CrossRef]
- Monsour, M.; Borlongan, C.V. The central role of peripheral inflammation in ischemic stroke. J. Cereb. Blood Flow Metab. 2023, 0, 271678X221149509. [Google Scholar] [CrossRef]
- Clements, C.M.; McNally, R.S.; Conti, B.J.; Mak, T.W.; Ting, J.P. DJ-1, a cancer- and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc. Natl. Acad. Sci. USA 2006, 103, 15091–15096. [Google Scholar] [CrossRef]
- Fan, J.; Ren, H.; Jia, N.; Fei, E.; Zhou, T.; Jiang, P.; Wu, M.; Wang, G. DJ-1 decreases Bax expression through repressing p53 transcriptional activity. J. Biol. Chem. 2008, 283, 4022–4030. [Google Scholar] [CrossRef] [PubMed]
- Russo, E.; Nguyen, H.; Lippert, T.; Tuazon, J.; Borlongan, C.V.; Napoli, E. Mitochondrial targeting as a novel therapy for stroke. Brain Circ. 2018, 4, 84–94. [Google Scholar] [CrossRef]
- Borra, M.T.; Smith, B.C.; Denu, J.M. Mechanism of human SIRT1 activation by resveratrol. J. Biol. Chem. 2005, 280, 17187–17195. [Google Scholar] [CrossRef] [PubMed]
- Chong, Z.Z.; Shang, Y.C.; Wang, S.; Maiese, K. SIRT1: New avenues of discovery for disorders of oxidative stress. Expert. Opin. Ther. Targets 2012, 16, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; He, J.; Huang, Y.; Hu, Z. Resveratrol has an Overall Neuroprotective Role in Ischemic Stroke: A Meta-Analysis in Rodents. Front. Pharmacol. 2021, 12, 795409. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Li, W.; Poteet, E.C.; Xie, L.; Tan, C.; Yan, L.J.; Ju, X.; Liu, R.; Qian, H.; Marvin, M.A.; et al. Alternative mitochondrial electron transfer as a novel strategy for neuroprotection. J. Biol. Chem. 2011, 286, 16504–16515. [Google Scholar] [CrossRef]
- Huang, S.; Du, F.; Shih, Y.Y.; Shen, Q.; Gonzalez-Lima, F.; Duong, T.Q. Methylene blue potentiates stimulus-evoked fMRI responses and cerebral oxygen consumption during normoxia and hypoxia. Neuroimage 2013, 72, 237–242. [Google Scholar] [CrossRef]
- Enomoto, M.; Endo, A.; Yatsushige, H.; Fushimi, K.; Otomo, Y. Clinical Effects of Early Edaravone Use in Acute Ischemic Stroke Patients Treated by Endovascular Reperfusion Therapy. Stroke 2019, 50, 652–658. [Google Scholar] [CrossRef]
- Kimura, K.; Aoki, J.; Sakamoto, Y.; Kobayashi, K.; Sakai, K.; Inoue, T.; Iguchi, Y.; Shibazaki, K. Administration of edaravone, a free radical scavenger, during t-PA infusion can enhance early recanalization in acute stroke patients-a preliminary study. J. Neurol. Sci. 2012, 313, 132–136. [Google Scholar] [CrossRef]
- Tirilazad International Steering Committee. Tirilazad mesylate in acute ischemic stroke: A systematic review. Stroke 2000, 31, 2257–2265. [Google Scholar] [CrossRef]
- Davalos, A.; Alvarez-Sabin, J.; Castillo, J.; Diez-Tejedor, E.; Ferro, J.; Martinez-Vila, E.; Serena, J.; Segura, T.; Cruz, V.T.; Masjuan, J.; et al. Citicoline in the treatment of acute ischaemic stroke: An international, randomised, multicentre, placebo-controlled study (ICTUS trial). Lancet 2012, 380, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Diener, H.C.; Cortens, M.; Ford, G.; Grotta, J.; Hacke, W.; Kaste, M.; Koudstaal, P.J.; Wessel, T. Lubeluzole in acute ischemic stroke treatment: A double-blind study with an 8-hour inclusion window comparing a 10-mg daily dose of lubeluzole with placebo. Stroke 2000, 31, 2543–2551. [Google Scholar] [CrossRef] [PubMed]
- Gandolfo, C.; Sandercock, P.; Conti, M. Lubeluzole for acute ischaemic stroke. Cochrane Database Syst. Rev. 2002, CD001924. [Google Scholar] [CrossRef]
- Haley, E.C., Jr. High-dose tirilazad for acute stroke (RANTTAS II). RANTTAS II Investigators. Stroke 1998, 29, 1256–1257. [Google Scholar] [CrossRef]
- Koziol, J.A.; Feng, A.C. On the analysis and interpretation of outcome measures in stroke clinical trials: Lessons from the SAINT I study of NXY-059 for acute ischemic stroke. Stroke 2006, 37, 2644–2647. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Sano, K.; Takakura, K.; Saito, I.; Shinohara, Y.; Asano, T.; Yasuhara, H. Ebselen in acute ischemic stroke: A placebo-controlled, double-blind clinical trial. Ebselen Study Group. Stroke 1998, 29, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef]
- Bernard-Marissal, N.; Medard, J.J.; Azzedine, H.; Chrast, R. Dysfunction in endoplasmic reticulum-mitochondria crosstalk underlies SIGMAR1 loss of function mediated motor neuron degeneration. Brain 2015, 138, 875–890. [Google Scholar] [CrossRef]
- Chang, J.C.; Wu, S.L.; Liu, K.H.; Chen, Y.H.; Chuang, C.S.; Cheng, F.C.; Su, H.L.; Wei, Y.H.; Kuo, S.J.; Liu, C.S. Allogeneic/xenogeneic transplantation of peptide-labeled mitochondria in Parkinson’s disease: Restoration of mitochondria functions and attenuation of 6-hydroxydopamine-induced neurotoxicity. Transl. Res. 2016, 170, 40–56 e43. [Google Scholar] [CrossRef]
- Lee, M.; Ban, J.J.; Kim, K.Y.; Jeon, G.S.; Im, W.; Sung, J.J.; Kim, M. Adipose-derived stem cell exosomes alleviate pathology of amyotrophic lateral sclerosis in vitro. Biochem. Biophys. Res. Commun. 2016, 479, 434–439. [Google Scholar] [CrossRef]
- Shi, X.; Zhao, M.; Fu, C.; Fu, A. Intravenous administration of mitochondria for treating experimental Parkinson’s disease. Mitochondrion 2017, 34, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Nitzan, K.; Benhamron, S.; Valitsky, M.; Kesner, E.E.; Lichtenstein, M.; Ben-Zvi, A.; Ella, E.; Segalstein, Y.; Saada, A.; Lorberboum-Galski, H.; et al. Mitochondrial Transfer Ameliorates Cognitive Deficits, Neuronal Loss, and Gliosis in Alzheimer’s Disease Mice. J. Alzheimers Dis. 2019, 72, 587–604. [Google Scholar] [CrossRef]
- Cheng, X.Y.; Biswas, S.; Li, J.; Mao, C.J.; Chechneva, O.; Chen, J.; Li, K.; Li, J.; Zhang, J.R.; Liu, C.F.; et al. Human iPSCs derived astrocytes rescue rotenone-induced mitochondrial dysfunction and dopaminergic neurodegeneration in vitro by donating functional mitochondria. Transl. Neurodegener. 2020, 9, 13. [Google Scholar] [CrossRef]
- Verma, A.; Shteinfer-Kuzmine, A.; Kamenetsky, N.; Pittala, S.; Paul, A.; Nahon Crystal, E.; Ouro, A.; Chalifa-Caspi, V.; Pandey, S.K.; Monsengo, A.; et al. Targeting the overexpressed mitochondrial protein VDAC1 in a mouse model of Alzheimer’s disease protects against mitochondrial dysfunction and mitigates brain pathology. Transl. Neurodegener. 2022, 11, 58. [Google Scholar] [CrossRef] [PubMed]
- Calkins, M.J.; Manczak, M.; Mao, P.; Shirendeb, U.; Reddy, P.H. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2011, 20, 4515–4529. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Ohno, M. Mitochondrial dysfunction and accumulation of the β-secretase-cleaved C-terminal fragment of APP in Alzheimer’s disease transgenic mice. Neurobiol. Dis. 2012, 45, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Amadoro, G.; Corsetti, V.; Atlante, A.; Florenzano, F.; Capsoni, S.; Bussani, R.; Mercanti, D.; Calissano, P. Interaction between NH(2)-tau fragment and Aβ in Alzheimer’s disease mitochondria contributes to the synaptic deterioration. Neurobiol. Aging 2012, 33, 833.e1–833.e25. [Google Scholar] [CrossRef] [PubMed]
- Silzer, T.K.; Pathak, G.A.; Phillips, N.R. Mitochondrial tRNA methylation in Alzheimer’s disease and progressive supranuclear palsy. BMC Med. Genom. 2020, 13, 71. [Google Scholar] [CrossRef] [PubMed]
- Lampinen, R.; Belaya, I.; Saveleva, L.; Liddell, J.R.; Rait, D.; Huuskonen, M.T.; Giniatullina, R.; Sorvari, A.; Soppela, L.; Mikhailov, N.; et al. Neuron-astrocyte transmitophagy is altered in Alzheimer’s disease. Neurobiol. Dis. 2022, 170, 105753. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, P.J.; Sha, H.Y.; Ni, J.; Li, M.H.; Gu, G.J. Neural stem cell transplants improve cognitive function without altering amyloid pathology in an APP/PS1 double transgenic model of Alzheimer’s disease. Mol. Neurobiol. 2014, 50, 423–437. [Google Scholar] [CrossRef]
- Ager, R.R.; Davis, J.L.; Agazaryan, A.; Benavente, F.; Poon, W.W.; LaFerla, F.M.; Blurton-Jones, M. Human neural stem cells improve cognition and promote synaptic growth in two complementary transgenic models of Alzheimer’s disease and neuronal loss. Hippocampus 2015, 25, 813–826. [Google Scholar] [CrossRef]
- Yue, W.; Li, Y.; Zhang, T.; Jiang, M.; Qian, Y.; Zhang, M.; Sheng, N.; Feng, S.; Tang, K.; Yu, X.; et al. ESC-Derived Basal Forebrain Cholinergic Neurons Ameliorate the Cognitive Symptoms Associated with Alzheimer’s Disease in Mouse Models. Stem. Cell Reports 2015, 5, 776–790. [Google Scholar] [CrossRef] [PubMed]
- Lill, C.M. Genetics of Parkinson’s disease. Mol. Cell Probes. 2016, 30, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Paillusson, S.; Gomez-Suaga, P.; Stoica, R.; Little, D.; Gissen, P.; Devine, M.J.; Noble, W.; Hanger, D.P.; Miller, C.C.J. α-Synuclein binds to the ER-mitochondria tethering protein VAPB to disrupt Ca(2+) homeostasis and mitochondrial ATP production. Acta Neuropathol. 2017, 134, 129–149. [Google Scholar] [CrossRef] [PubMed]
- Yue, M.; Hinkle, K.M.; Davies, P.; Trushina, E.; Fiesel, F.C.; Christenson, T.A.; Schroeder, A.S.; Zhang, L.; Bowles, E.; Behrouz, B.; et al. Progressive dopaminergic alterations and mitochondrial abnormalities in LRRK2 G2019S knock-in mice. Neurobiol. Dis. 2015, 78, 172–195. [Google Scholar] [CrossRef]
- Ashrafi, G.; Schlehe, J.S.; LaVoie, M.J.; Schwarz, T.L. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J. Cell. Biol. 2014, 206, 655–670. [Google Scholar] [CrossRef]
- Stevens, D.A.; Lee, Y.; Kang, H.C.; Lee, B.D.; Lee, Y.I.; Bower, A.; Jiang, H.; Kang, S.U.; Andrabi, S.A.; Dawson, V.L.; et al. Parkin loss leads to PARIS-dependent declines in mitochondrial mass and respiration. Proc. Natl. Acad. Sci. USA 2015, 112, 11696–11701. [Google Scholar] [CrossRef]
- Prasuhn, J.; Davis, R.L.; Kumar, K.R. Targeting Mitochondrial Impairment in Parkinson’s Disease: Challenges and Opportunities. Front. Cell Dev. Biol. 2020, 8, 615461. [Google Scholar] [CrossRef]
- Fairley, L.H.; Grimm, A.; Eckert, A. Mitochondria Transfer in Brain Injury and Disease. Cells 2022, 11, 3603. [Google Scholar] [CrossRef]
- Grealish, S.; Diguet, E.; Kirkeby, A.; Mattsson, B.; Heuer, A.; Bramoulle, Y.; Van Camp, N.; Perrier, A.L.; Hantraye, P.; Bjorklund, A.; et al. Human ESC-derived dopamine neurons show similar preclinical efficacy and potency to fetal neurons when grafted in a rat model of Parkinson’s disease. Cell Stem. Cell 2014, 15, 653–665. [Google Scholar] [CrossRef]
- Hallett, P.J.; Deleidi, M.; Astradsson, A.; Smith, G.A.; Cooper, O.; Osborn, T.M.; Sundberg, M.; Moore, M.A.; Perez-Torres, E.; Brownell, A.L.; et al. Successful function of autologous iPSC-derived dopamine neurons following transplantation in a non-human primate model of Parkinson’s disease. Cell Stem. Cell 2015, 16, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Magrané, J.; Cortez, C.; Gan, W.B.; Manfredi, G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 2014, 23, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Tagashira, H.; Shinoda, Y.; Shioda, N.; Fukunaga, K. Methyl pyruvate rescues mitochondrial damage caused by SIGMAR1 mutation related to amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2014, 1840, 3320–3334. [Google Scholar] [CrossRef] [PubMed]
- Goutman, S.A.; Savelieff, M.G.; Sakowski, S.A.; Feldman, E.L. Stem cell treatments for amyotrophic lateral sclerosis: A critical overview of early phase trials. Expert Opin. Investig. Drugs 2019, 28, 525–543. [Google Scholar] [CrossRef]
- Chen, R.; Ende, N. The potential for the use of mononuclear cells from human umbilical cord blood in the treatment of amyotrophic lateral sclerosis in SOD1 mice. J. Med. 2000, 31, 21–30. [Google Scholar]
- Garbuzova-Davis, S.; Willing, A.E.; Zigova, T.; Saporta, S.; Justen, E.B.; Lane, J.C.; Hudson, J.E.; Chen, N.; Davis, C.D.; Sanberg, P.R. Intravenous administration of human umbilical cord blood cells in a mouse model of amyotrophic lateral sclerosis: Distribution, migration, and differentiation. J. Hematother. Stem. Cell. Res. 2003, 12, 255–270. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Sample | Cell Type | Route | Dosage | Outcome |
|---|---|---|---|---|
| MCAO rats | Human Bone Marrow Endothelial Progenitor Cells | Intravenous | 4 × 106 cells/mL | Endothelial cells, pericytes, and astrocytes demonstrate near normal morphology without perivascular edema. Mitochondrial morphology in endothelial cells and perivascular astrocytes shows near normal morphology and pinocytic vessels are observed in engrafted cells, which ameliorates post-stroke vasculature [36]. |
| MCAO rats | Miro1-overexpressing Multipotent MSCs | Intravenous | 3 × 106 cells/kg | Miro1, normally upregulated in the presence of ROS, promotes mitochondrial transfer to neural cells and reduces neurologic deficits after ischemic stroke [37]. |
| MCAO rats | MSCs | Intra-arterial | 5 × 105 cells | Mitochondrial transfer to injured cells of the cerebral microvasculature improves mitochondrial activity, upregulates angiogenesis, improves neurologic function, and decreases infarct volume [38]. |
| MCAO rats | hUC-MSC-derived mitochondria | Intraventricular | Isolate from 3 × 107 cells | Transplanted mitochondria improve ischemic injury exemplified through inhibition of apoptosis, decreased astrogliosis, microglial downregulation, reduced infarct size, and enhanced preservation of motor function [39]. |
| MCAO rats | Ischemic-hypoxic preconditioned olfactory mucosa MSCs | Intravenous | 1 × 106 cells | Mitochondrial function is preserved through upregulation of downstream target genes (GRP78 and Bcl-2) by miR-181a and the presence of ROS is significantly reduced. Apoptosis and pyroptosis of neurons are inhibited [40]. |
| Photothrombotic mPFC stroke mice | BM-MSC-derived mitochondria | Intranasal | 12 μL | Mitochondrial transplant significantly reduced the presence of ROS in the mPFC following ischemia. Transplant also ameliorates memory impairment, upregulates ATP generation, improves mitochondrial membrane potential, and upregulates expression of synaptic markers (GAP-43 and PSD-95) [41]. |
| Model of Injury | Cell Type | Outcome |
|---|---|---|
| Hypoxia-reperfusion | Rat Neural Stem Cells | Coenzyme Q10 achieves an antioxidant effect through interaction with the electron transport chain, increasing expression of survival proteins (pAkt, pGSK3-β, and Bcl-2) and reducing levels of cleaved caspase-3 [42]. |
| OGD | Primary Rat Neural Cells | DJ-1, a protein involved in neuroprotection through regulation of oxidative stress, translocated to mitochondria and enhanced both cell viability mitochondrial activity while reducing ROS concentrations [43]. |
| OGD | Primary Rat Neural Cells | Ischemic conditions promote the uptake of astrocyte-released mitochondrial particles by adjacent neurons, which increases survival signaling [29]. |
| OGD | Human Endothelial Progenitor Cell-derived Extracellular Mitochondria | Incorporation of extracellular mitochondria promotes angiogenesis, decreases BBB permeability, and increases expression of TOM40, mtDNA copy number, and intracellular ATP [44]. |
| Metabolic Switching Paradigm, OGD | Human MSCs, Primary Rat Neurons | Metabolic switching in MSCs yields greater energy generation, respiratory capacity, and ATP production. Co-culture with ODG neurons enhances cellular metabolism, decreases mitochondrial ROS mRNA, and overall improves cell viability [30]. |
| Metabolic Switching Paradigm | Human MSCs | Metabolic switching in MSCs results in mitochondria with enhanced capability for oxidative phosphorylation [25]. |
| Sample | Treatment | Route | Dosage | Outcome |
|---|---|---|---|---|
| Sigmar1(-/-) mice | BAPTA-AM, a selective intracellular calcium chelator or an endoplasmic reticulum stress inhibitor salubrinal | - | - | Restoration of calcium homeostasis and endoplasmic reticulum stability recovered mitochondrial movement and morphology, ultimately reducing motor neuron degeneration [68]. |
| 6-hydroxydopamine-induced selective parkinsonian rats | PC12 cell- and Human Osteosarcoma cybrid-derived mitochondria | Intracranial | 1.05 μg | Peptide-mediated allogeneic mitochondrial delivery maintains mitochondrial function in the setting of oxidative stress and apoptotic death. Motor activity is improved up to three months following transplantation, and dopaminergic neuron loss is reduced [69]. |
| G93A ALS mice neurons | ADSC-derived exosomes | - | 200 μg/mL | Transplanted exosomes reduce aggregation of superoxide dismutase 1 and normalize mitochondrial phospho-CREB/CREB ratio and PGC-1α expression [70]. |
| MPTP-HCL-induced parkinsonian mice | HepG2-derived mitochondria | Intravenous | 0.5 mg/kg | Transplanted mitochondria distributed to the brain, liver, kidney, muscle, and heart. PD progression is halted through increased electron transport chain activity, reduced levels of reactive oxygen species, and prevention of apoptosis [71]. |
| Aβ-ICV Alzheimer’s Disease mice | HeLa cell-derived mitochondria | Intravenous | 200 μg | Treated mice show enhanced cognitive performance, reduced loss of neurons, and reduced hippocampal gliosis. Mitochondrial function is further ameliorated peripherally in organs such as the liver [72]. |
| Rotenone-induced Parkinson’s neurons | iPSC- and hESC-derived DA neurons and astrocytes | - | - | iPSC-derived astrocytes and astrocytic conditioned media reduce degeneration of dopaminergic neurons and reverse axonal pruning through mitochondrial transfer [73]. |
| 5 x Familial Alzheimer’s Disease mice | VBIT-4 (Voltage-Dependent anion channel-1 inhibitor) | PO (in drinking water) | 20 mg/kg | VBIT-4 reduces neuronal cell death, downregulates neuroinflammation, and ameliorates metabolic dysfunction, leading to improved cognitive outcomes in behavioral assessments [74]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monsour, M.; Gordon, J.; Lockard, G.; Alayli, A.; Borlongan, C.V. Mitochondria in Cell-Based Therapy for Stroke. Antioxidants 2023, 12, 178. https://doi.org/10.3390/antiox12010178
Monsour M, Gordon J, Lockard G, Alayli A, Borlongan CV. Mitochondria in Cell-Based Therapy for Stroke. Antioxidants. 2023; 12(1):178. https://doi.org/10.3390/antiox12010178
Chicago/Turabian StyleMonsour, Molly, Jonah Gordon, Gavin Lockard, Adam Alayli, and Cesar V. Borlongan. 2023. "Mitochondria in Cell-Based Therapy for Stroke" Antioxidants 12, no. 1: 178. https://doi.org/10.3390/antiox12010178
APA StyleMonsour, M., Gordon, J., Lockard, G., Alayli, A., & Borlongan, C. V. (2023). Mitochondria in Cell-Based Therapy for Stroke. Antioxidants, 12(1), 178. https://doi.org/10.3390/antiox12010178