1. Introduction
Repetitive exposure of the skin to ultraviolet (UV) radiation causes an imbalance in the redox state of the cellular environment, leading to oxidative stress, the starting point of skin photodamage, which in turn provokes photoaging and photocarcinogenesis. Photoaging leads to profound changes in the composition and architecture of the dermal extracellular matrix [
1]. Additionally, photocarcinogenesis involves multiple steps of biochemical reactions, including tumor initiation, tumor promotion, and tumor progression, eventually causing skin cancer [
2]. Moreover, UV radiation leads to high-intensity inflammatory responses in acute exposure, causing sunburn, and in chronic exposure, leads to low-intensity inflammatory reactions, which are closely related to photoaging, immunosuppression, photocarcinogenesis and skin cancer development comprised of melanoma and non-melanoma types [
3,
4]. Among the possible mechanisms that explain the damage caused by UV radiation to the skin is the intense generation of reactive oxygen species (ROS), and the inability to neutralize them by the endogenous antioxidant defense mechanism, which leads to oxidative stress followed by direct oxidative damage to cellular macromolecules including lipids, proteins, and DNA [
5]. As a long-term consequence, a variety of pathological processes are aggravated or caused by exposure to solar radiation [
6,
7].
The prevention and treatment of injuries induced by exposure to UV radiation are of great importance for promoting the health of populations that are more frequently exposed to solar radiation. In addition, it is known that life expectancy is longer compared to past decades [
8]. As the damage caused by UV exposure is cumulative, increased longevity favors the emergence of photoinduced chronic changes, such as the intensification of photoaging and the increased incidence of skin cancers, especially in elderly populations, reflecting the long latency between exposure to the carcinogen and cancer formation [
9]. The therapeutic modalities for the management of photodamage include the topical administration of sunscreens and antioxidants, which reduce the oxidative injuries of the integumentary tissue cells, compromising keratinocytes and fibroblasts. In this context, studies demonstrate the beneficial properties of phenolic compounds as preventive agents against photodamage in skin cells [
10], combating oxidative damage to macromolecules and decreasing the prevalence of inflammatory processes [
11].
A recent study of our research group evidenced that tannic acid (TA), a phenolic compound, exhibited high antioxidant capacity and that TA treatment in UVB-irradiated fibroblasts prevented cellular redox imbalance, oxidative damage to cellular macromolecules, and photoaging [
12]. In another study from our group [
13], the photoprotective potential of cerium oxide nanoparticles (CNPs) was evaluated. CNP treatment was able to combat cellular photodamage in UVB-irradiated fibroblasts by decreasing ROS generation and increasing the activities of endogenous antioxidant enzymes. There has been an increase in interest in the application of CNPs as antioxidants for a variety of treatments related to oxidative stress disorders, due to their mimetic antioxidant superoxide dismutase (SOD) and catalase (CAT) activity and their direct ROS scavenging effect in cell and animal models. The antioxidant effect of CNPs depends on their physicochemical properties, as their nanoscale size offers the thermodynamic efficiency of the redox cycling between cerium 3+ (Ce
3+) and cerium 4+ (Ce
4+) ions on the nanoparticle surface, and the ability to absorb and release oxygen [
14].
To date, to the best of our knowledge, few studies have been reported the effects of cerium oxide nanoparticles against UV radiation-induced oxidative photodamage in cells. For instance, zholobak et al. [
15] reported that CNPs prevented the decrease in cell viability of mouse fibroblasts (L929) and fibroblast-like cells of African Green monkey (VERO) exposed to UV. Fujita and Kamada [
16] demonstrated that CNPs decreased oxidized DNA under UV light. Zholobak et al. [
17] reported that panthenol-stabilized CNPs protect human epidermoid cancer cells (Hep-2) and the diploid epithelial swine testicular cell line (ST-cells) from ROS and UV irradiation. Additionally, Ribeiro et al. [
18] showed that CNPs with a higher Ce
3+ content decreased UVA-induced L929 fibroblast death by regulating protein kinases 1 and 2 (ERK 1/2). Therefore, the present study proposed to synthesize a novel delivery system of TA conjugated with CNPs (CNP-TA) and to investigate the photoprotective potential of this nanoparticulate system, using as in vitro model L929 fibroblasts exposed to UVB, with emphasis on combating photodamage processes.
2. Materials and Methods
2.1. Materials
All chemicals and solvents used were of analytical grade. Tannic acid (purity 95%) was purchased from ACROS organics (Geel, Belgium, Europe). Bradford reagent was acquired from Bio-Rad (Hercules, CA, USA). Acridine orange, catalase, cerium nitrate hexahydrate (purity 99.999%), dimethyl sulfoxide (DMSO), ethyl diamine, HBSS (Hank’s balanced salt solution), hydrogen peroxide (99.9%), ortho-phthalaldehyde, protease inhibitor cocktail, reduced glutathione (GSH), and the chemical cross-linkers of 1, 1′-carbonyldiimidazole (CDI) were obtained from Sigma-Aldrich (St. Louis, MO, USA). DPPP (diphenyl-1-pyrenylphosphine) and H2DCF-DA (2′,7′-dichlorodihydrofluorescein diacetate) were purchased from Molecular Probes (Eugene, OR, USA). DMEM (Dulbecco’s modified Eagle’s medium) and fetal bovine serum were purchased from Gibco (Carlsbad, CA, USA). Neutral red (3-amino-7-dimethylamino-2-methylphenazine hydrochloride) was obtained from Inlab (São Paulo, Brazil). HRP-conjugated goat anti-rabbit IgG secondary antibody, mouse anti-COX-2, mouse anti-MMP-1, mouse anti-β-actin and ECL reagent were purchased from Santa Cruz Biotechnology (Paso Robles, CA, USA).
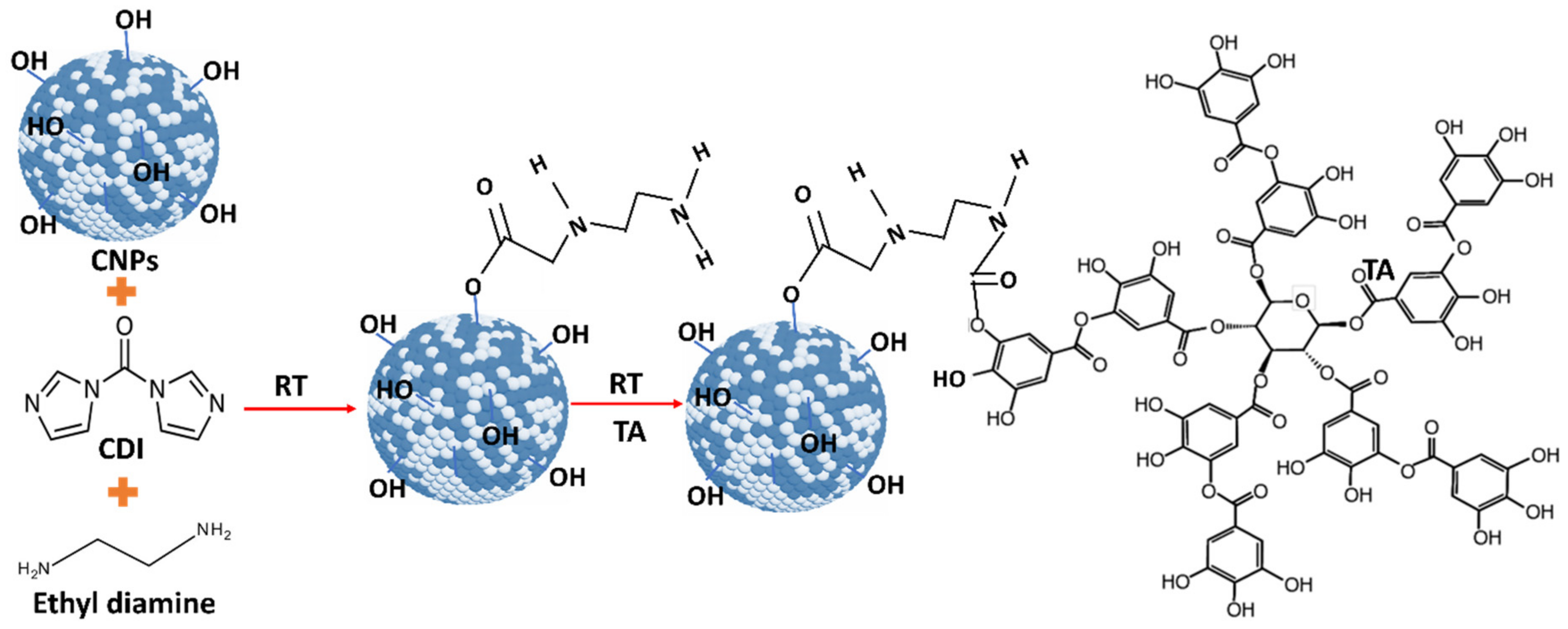
2.2. Tannic Acid Conjugation with CNP
The cerium oxide nanoparticle was synthesized by a wet chemical method based on work published elsewhere [
19]. Briefly, cerium nitrate hexahydrate (Ce(NO
3)
3·6H
2O) and hydrogen peroxide (3
v/
v %) were used as reactants for nanoparticle synthesis. Initial preparation involved dissolving cerium nitrate hexahydrate in deionized water, followed by adding hydrogen peroxide to the cerium nitrate solution and mixing at room temperature. In combination with hydrogen peroxide, cerium ions are oxidized, resulting in a yellow solution, which becomes clear as particles undergo condensation and solution dispersion. The oxidation process occurs at room temperature and fully aged nanoparticles are ready for use after 6 to 8 weeks.
As-synthesized water-dispersed-cerium oxide nanoparticles (CNPs) were used for conjugation. Initially, 100 μL of synthesized 5 mM CNPs was diluted into 900 μL of DMSO solvent. 100 μL of CDI (500 mM) solution was added to CNPs to activate the OH group on CNPs. This solution was continuously shaken for 1 h using shake-plate equipment, at room temperature. After activation of the OH group, 250 μL of ethyl diamine was added to activated CNPs, and it was continuously shaken for another 2 h. Then, 225 µL of 0.5 mg/mL tannic acid solution was added to the CNPs solution and it was continuously stirred overnight to conjugate the tannic acid with CNPs. In ethyl diamine, one end of the amine group is attached to the CDI-CNPs [
20], and another end is attached to the OH group in the TA molecule (
Figure 1). Any un-conjugated nanoparticles and drug molecules were removed by the dialysis process performed for 24 h against RNAase-free water. The tannic acid-conjugated CNPs (CNP-TA) samples were stored at −4 °C up to further use.
2.3. CNP-TA Physicochemical Characterization
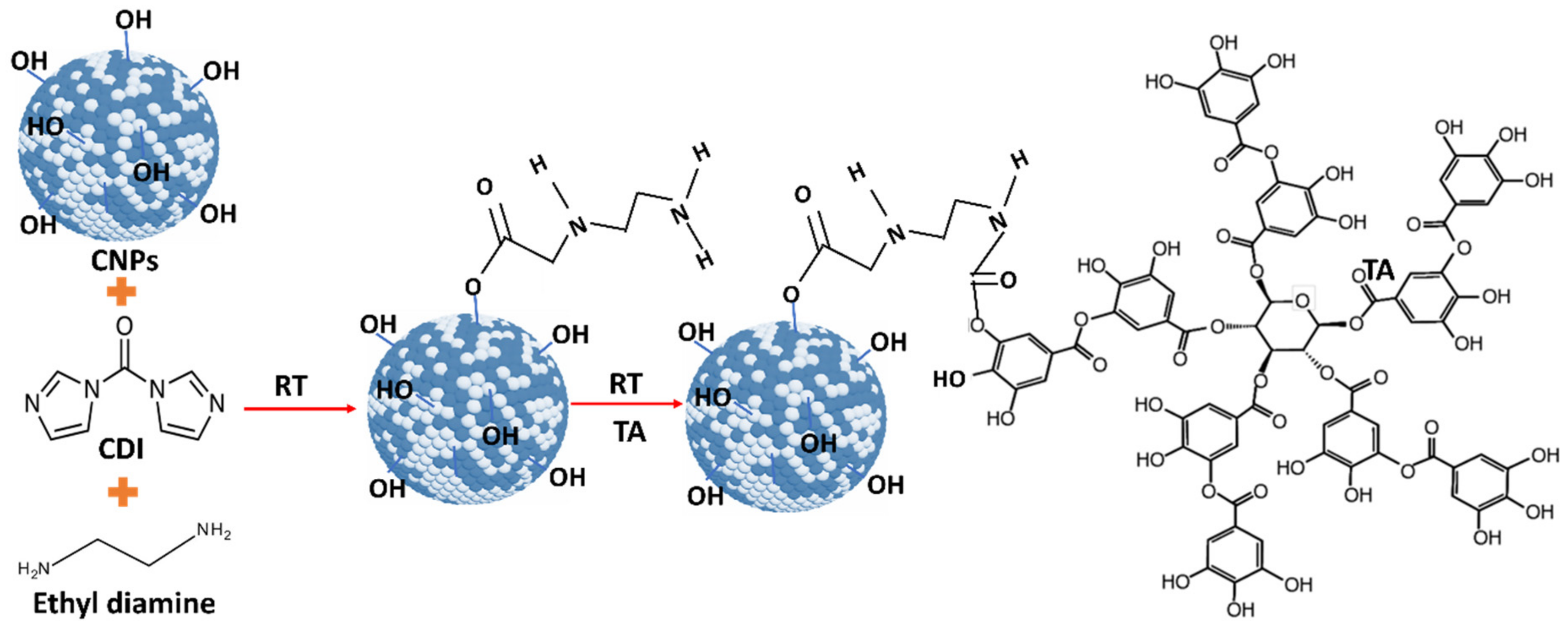
Before and after TA conjugation with CNPs, samples were subjected to various characterizations. UV–Vis spectra of samples were analyzed. The surface properties of the oxidation state of pure and TA-conjugated CNPs were examined by 5400 PHI ESCA X-ray photoelectron spectroscopy (XPS) (RBD Instruments Inc., Bend, OR, USA) with Mg-Kα X-radiation (1253.6 eV) at 350 W and pressure 10−9 Torr maintained during the measurement. C-1S peak at 284.6 eV was used as a reference to compensate for any peak shift due to charging. The size and morphology of the drug-conjugated samples were analyzed by 300 keV high-resolution transmission electron microscopy (HR-TEM) with an FEI Tecnai F30 EDX analyzer (Jeol Inc., Peabody, MA, USA). Diluted pure and drug-conjugated samples drop cast on a carbon-coated 400 mesh copper grid and dried under the IR lamp. After, SOD activity was analyzed for the pure and drug-conjugated CNPs samples. The SOD activity was tested on pure, drug-conjugated CNPs and the positive kit control using the Dojindo SOD assay kit (Rockville, MD, USA). The different concentrations of pure and drug-conjugated CNPs were prepared using deionized water and examined for SOD activity. The absorbance at 450 nm was measured while xanthine oxide and samples interacted in the presence of WST-1 dye. Linear fitting was applied to these data, and the slope value was determined. The percentage of SOD activity for conjugated samples was calculated from the estimated slope value and compared against SOD control data. The experiment was performed in triplicate in each condition of the samples. Further, the zeta potential of TA-conjugated CNPs was analyzed using a Nano-ZS zeta sizer (Malvern Instruments, Swavesey, Cambridge, UK) instrument. Additionally, the conjugated samples were subjected to inductively coupled plasma mass spectrometry (ICP-MS) analysis to determine the concentration of cerium. The fixed volume of drug-conjugated samples was digested in 35% nitric acid solution and incubated for 48 h at 80 °C in a conventional hot air oven. The digested sample was diluted further with deionized water to maintain a 5% nitric acid concentration in the final solution. To quantify the cerium concentration in the synthesized nanoparticles, the Agilent 7700X (Agilent Technologies Inc., Santa Clara, CA, USA) inductively coupled plasma-mass spectrometry facility was used at Florida International University. Bond character and chemistry for TA, CNP and CNP-TA samples were analyzed using Fourier-Transformation Infrared Spectroscopy (FTIR). IR spectra were recorded in the range of 4000–500 cm−1, with a Perkin-Elmer Spectrum One FTIR (Waltham, MA, USA) by dispersing in KBr.
2.4. Cell Culture and UVB Irradiation
Photoprotection assessment of free TA, CNPs, and CNP-TA was evaluated in L929 fibroblasts (ATCC CCL1™, Manassas, VA, USA). Cells were cultured in DMEM supplemented with 10% (v/v) fetal bovine serum, penicillin (100 U/mL), and streptomycin (100 µg/mL), maintained at 37 °C under 5% CO2 atmosphere. For the irradiation process, the culture medium was replaced by HBSS buffer. Then, the cells were exposed to UVB (UVB lamp: TL40 W/12 RS; Philips; 290–315 nm; peak intensity 312 nm) at an intensity of 500 mJ/cm2, monitored using a radiometer (VLX-3W, Vilber Lourmat, Eberhardzell, Baden-Wurttemberg, Germany).
2.5. Cell Viability
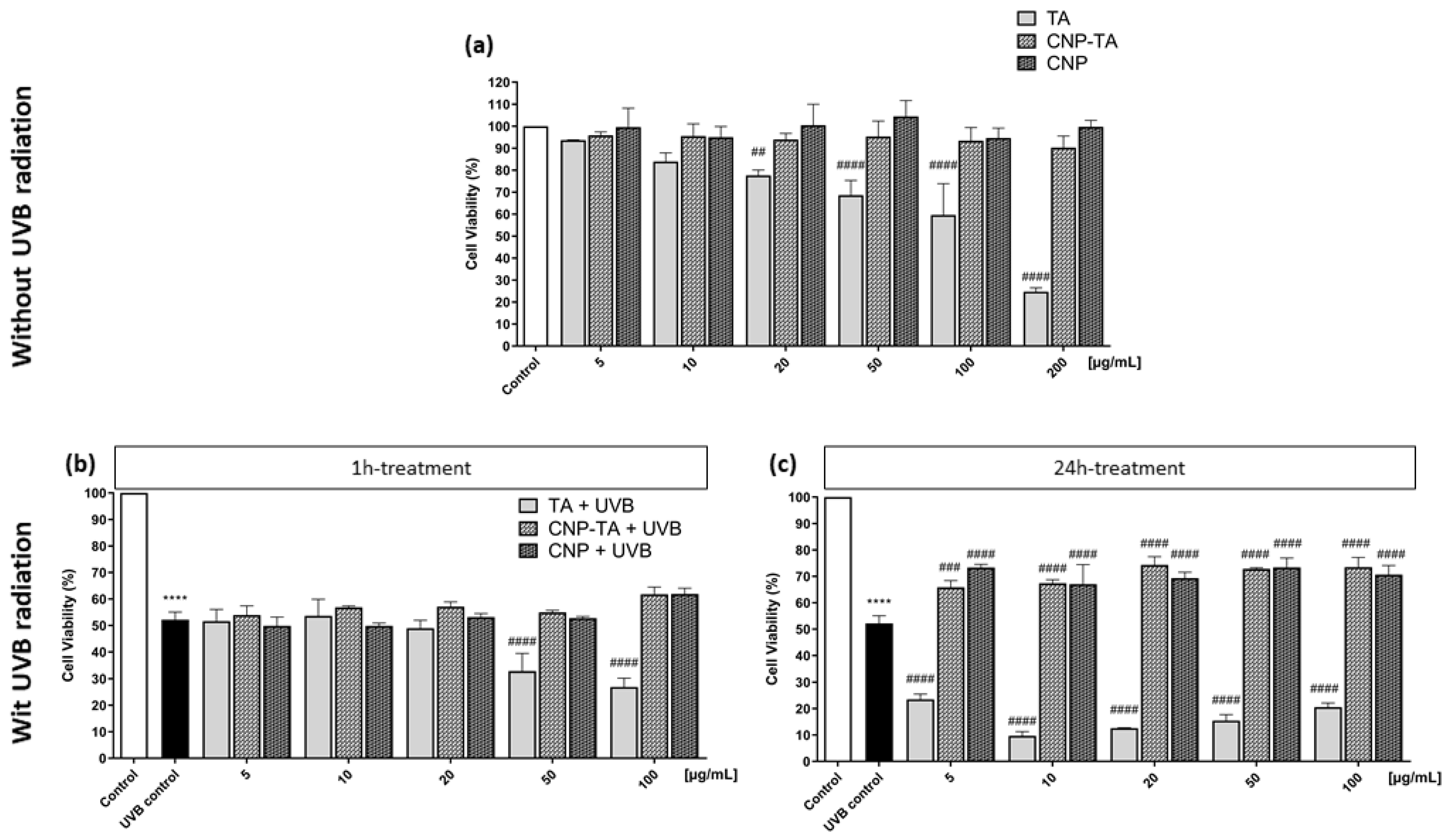
To evaluate the effects of free TA, CNPs, and CNP-TA on the viability of L929 fibroblasts, cells grown in 96-well plates (2.5 × 10
5 cells/mL) were treated with different concentrations of the samples (5, 10, 20, 50, 100 and 200 µg/mL) for 24 h. For the evaluation of cell viability in fibroblasts treated and irradiated, cells cultivated in 24-well plates (2.5 × 10
5 cells/mL) were treated for 1 h or 24 h with different concentrations of the samples (5, 10, 20, 50, and 100 µg/mL), exposed to UVB and incubated for 24 h. After the incubation period of the respective experiments, the neutral red assay was performed to assess cell viability [
21]. The cells were incubated for 3 h with neutral red (40 µg/mL), fixed with formaldehyde (2%) and CaCl
2 (1%) for 5 min, and the neutral red retained in the cells was solubilized with 50% ethanol/1% acetic acid. Measurements were taken at 540 nm (Bio-Tek
®, Power Wave XS, Hampton, VA, USA).
2.6. Reactive Oxygen Species Detection
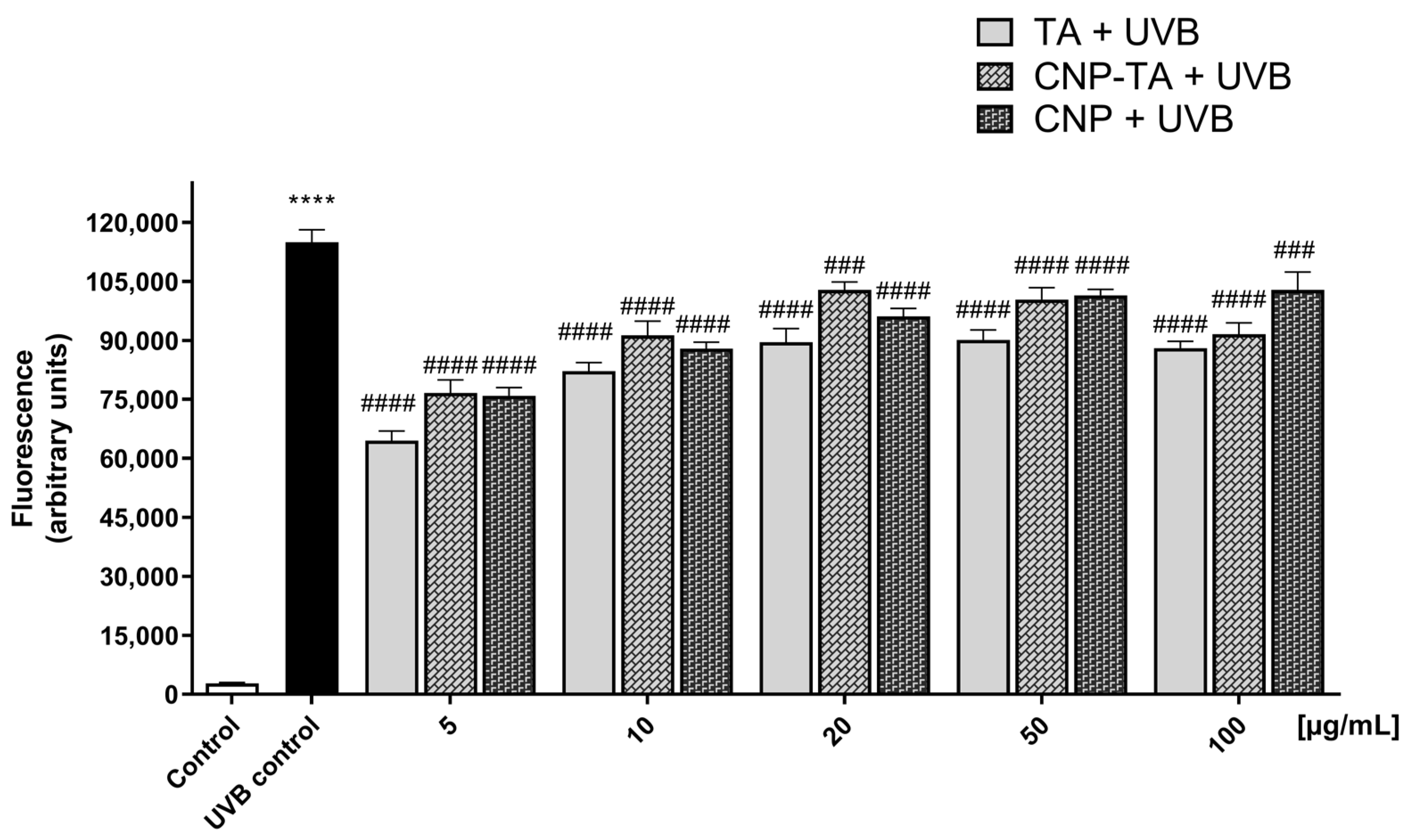
To evaluate ROS generation L929 fibroblasts cultured in 96-well plates (2.5 × 105 cells/mL) were treated for 24 h with free TA, CNPs, and CNP-TA (5, 10, 20, 50, and 100 µg/mL), then incubated with H2DCF-DA (10 µM) for 45 min at 37 °C. Then, the cells were exposed to UVB, and measurements were immediately taken. Fluorescence was detected at 488/525 nm excitation/emission (Victor® X3, Perkin Elmer, Waltham, MA, USA).
2.7. Endogenous Antioxidant Defense System Evaluation
The endogenous antioxidant defense system was analyzed by evaluating the enzymatic activity of CAT and the levels of glutathione (GSH). Briefly, L929 fibroblasts were cultured in 6-well plates (4 × 10
5 cells/mL), treated for 24 h with free TA, CNPs, and CNP-TA (10 and 20 µg/mL), exposed to UVB and incubated for 24 h. Afterward, cell lysates were prepared by scraping the cells in lysis buffer [10 mM Tris-HCl (pH 7.4) and 1% triton X-100], followed by sonication for 60 s and centrifugation at 10,000×
g/10 min/4 °C. The supernatants were collected, and the amount of protein was estimated [
22]. For CAT activity [
23], potassium phosphate buffer (50 mM, pH = 7.0), cell lysate (equivalent to 50 μg of protein), and H
2O
2 (30 mM) were added to a quartz cuvette. The reaction was monitored at 240 nm for 5 min at 25 °C (Shimadzu, UV-1700, Columbus, GA, USA), and the results were expressed according to the calibration curve of the bovine liver CAT standard (E.C. 1.11.1.6) (0.391–50 U/mL). For the GSH assay [
24], in a 96-well black plate were added 180 μL of sodium phosphate buffer (100 mM, 5 mM EDTA, pH 8.0), 10 μL of cell lysate and 10 μL of ortho-phthalaldehyde. Fluorescence was detected after 15 min of incubation at room temperature at 350/420 nm excitation/emission (Victor
® X3, Perkin Elmer, Waltham, MA, USA), and the results were expressed according to the GSH standard calibration curve (1.953–1000 μg/mL).
2.8. Cellular Photodamage Evaluation
To assess lipid peroxidation, L929 fibroblasts cultured in 96-well plates (2.5 × 10
5 cells/mL) were treated for 24 h with free TA, CNPs and CNP-TA (10 µg/mL), exposed to UVB and incubated for more 24 h. Then, the cells were incubated with DPPP (20 μM) for 30 min at 37 °C. The fluorescence was determined immediately after at 351/460 nm excitation/emission (Victor
® X3, PerkinElmer, Waltham, MA, USA) and the amount of protein was estimated [
22].
To assess DNA damage, DNA condensation and fragmentation were evaluated by acridine orange staining and agarose gel electrophoresis, respectively. For DNA condensation analysis, L929 fibroblasts cultured in 24-well plates under glass coverslips (2.5 × 10
5 cells/mL) were treated for 24 h with free TA, CNPs and CNP-TA (10 µg/mL), exposed to UVB and incubated for another 24 h. Then, the cells were stained with acridine orange (1 μg/mL), incubated at room temperature for 10 min, and the images were recorded under a fluorescence microscope (Olympus
®, BX51, Olympus America Inc., Melville, NY, USA). The images were also analyzed quantitatively by the ImageJ software version 1.51 (National Institutes of Health, Bethesda, MD, USA). For the analysis of DNA fragmentation, L929 fibroblasts cultured in 6-well plates (4 × 10
5 cells/mL) were treated for 24 h with free TA, CNPs and CNP-TA (10 µg/mL), exposed to UVB and incubated for another 24 h. DNA isolation and electrophoresis process were carried out as described by [
12], using a modified methodology proposed by [
25]. The DNA isolates were subjected to electrophoresis in a 0.7% agarose gel containing SYBR
® Safe DNA dye for 1 h at 80 V. The images of the obtained gels were recorded using the GelDoc
® XR+ Imaging System (Bio-Rad Laboratories Inc., Hercules, CA, USA).
2.9. Wound Healing Scratch Assay
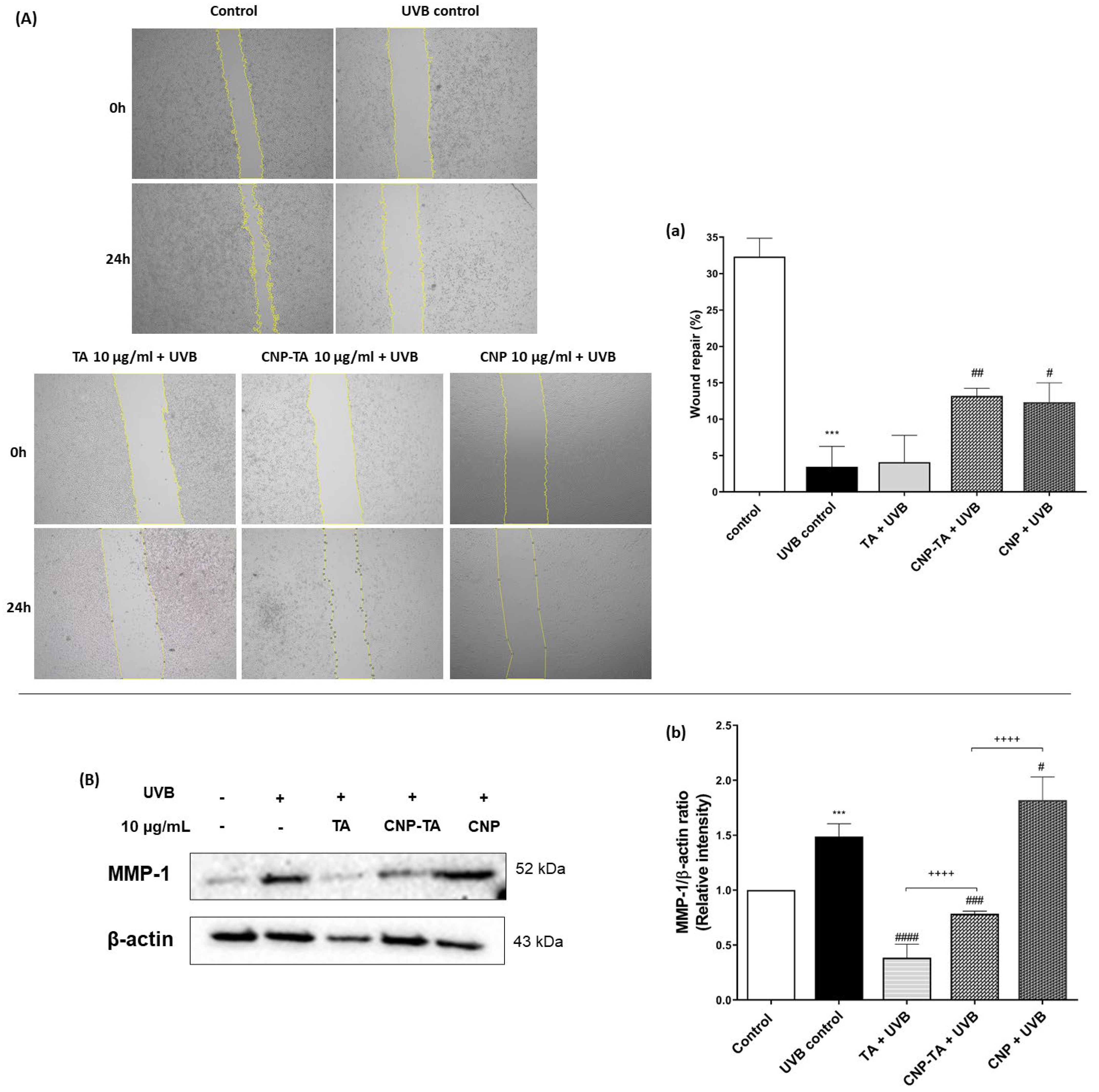
To assess cell proliferation after UVB exposure, a sterile 200-µL tip was used to make a straight scratch on the monolayer of L929 fibroblasts plated in 24-well plates (2.5 × 105 cells/mL). Then, the cells were treated for 24 h with free TA, CNPs and CNP-TA (10 µg/mL) and exposed to UVB. Then, DMEM containing 0.5% fetal bovine serum was added to the wells, and pictures were taken immediately after irradiation (0 h) and 24 h after irradiation. Wound repopulation was visualized under an optical microscope (Olympus®, CKX31, Olympus America Inc., Melville, NY, USA) and images were captured at 5× magnification. The wound repopulation area was measured using ImageJ software version 1.51 with a wound healing plugin.
2.10. TGF-β Levels Detection
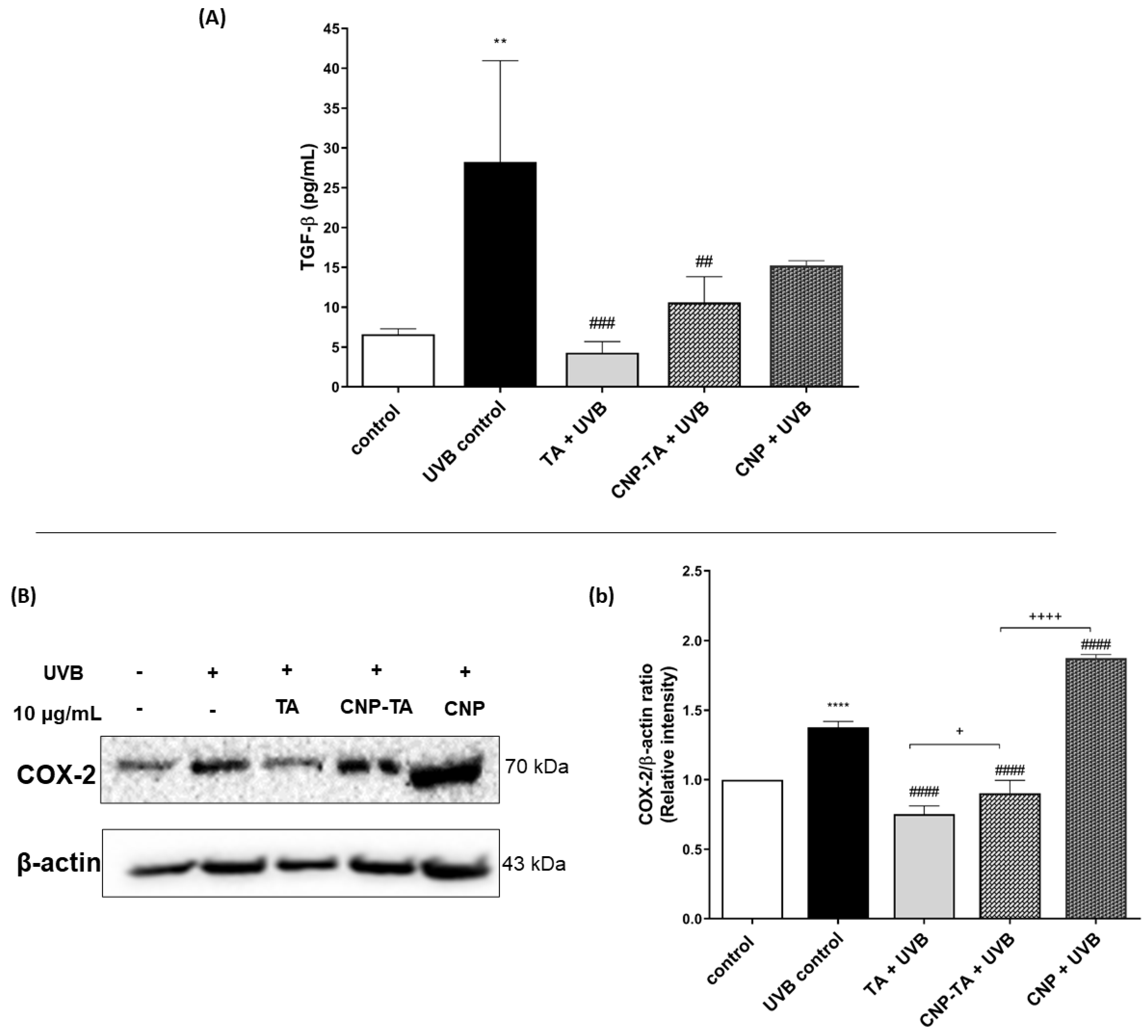
To assess TGF-β levels, L929 fibroblasts cultured in 6-well plates (4 × 105 cells/mL) were treated for 24 h with free TA, CNPs and CNP-TA (10 µg/mL), exposed to UVB and incubated for another 24 h. Then, the supernatants were used for TGF-β1 quantification using an ELISA kit (enzyme-linked immunosorbent assay), according to the manufacturer’s instructions (88–8350, ThermoFisher, Vienna, Austria). Recombinant human/mouse TGF-β was used as standard for the calibration curve.
2.11. Western Blot
L929 fibroblasts cultured in 6-well plates (4 × 10
5 cells/mL) were treated for 24 h with free TA, CNPs and CNP-TA (10 µg/mL), exposed to UVB and incubated for another 24 h. Afterward, cell lysates were prepared with lysis buffer [150 mM sodium chloride, 5 mM EDTA, 50 mM tris-HCl (pH 8.0), 1% triton X-100, 5% dodecyl sulfate sodium and 1% protease inhibitor cocktail]. Cell lysates were further sonicated for 2 min (60% amplitude), centrifuged at 10,000×
g/20 min/4 °C and the amount of protein was estimated [
22]. Equal amounts of cell lysates were diluted in sample buffer [5% mercaptoethanol, 5% bromophenol blue, 75 mM tris-HCl (pH 6.8), 2% sodium dodecyl sulfate and 10% glycerol], heated to 100 °C for 5 min and subjected to 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Then, the proteins were transferred to a nitrocellulose membrane (GE-Healthcare, Germany) in transfer buffer (25 mM tris, 192 mM glycine, 20% methanol and 0.01% sodium dodecyl sulfate). Membranes were blocked with 5% albumin in TBS-T buffer [25 mM Tris-HCl (pH 7.4), 150 mM NaCl and 0.1% tween 20] for 1 h and incubated overnight at 4 °C with the anti-MMP-1, anti-COX-2 and anti-β-actin primary mouse antibodies. Then, the membranes were incubated for 1 h with HRP-conjugated goat anti-rabbit IgG secondary antibody. The ECL reagent was used to detect the antigen-antibody complex formed. Images were recorded using the ChemiDoc
® XRS+ Imaging System (Bio-Rad Laboratories Inc., Hercules, CA, USA).
2.12. Statistical Analysis
Results were expressed as the mean ± standard deviation of triplicates from three independent experiments. Data were analyzed on GraphPad Prism® 5 using one-way ANOVA for different groups followed by Tukey’s test. Values of p < 0.05 were accepted as statistically significant.
4. Discussion
Skin photodamage originates from photon-induced changes in the skin, affecting either epidermis and dermis, attributed to continuous exposure to UV radiation, including UVA (400–315 nm) and UVB (315–280 nm) wavelengths [
28]. The UVB exposure causes an imbalance between the removal of excessive ROS UVB-generated and/or an impaired ability to detoxify the reactive intermediates by the endogenous antioxidant defense system, inducing oxidative stress in skin cells [
29]. Oxidative stress is the first step for photoaging and photocarcinogenesis generation. Photoaging or extrinsic aging is one of the results of photodamage, which is a consequence of exposure to outdoor agents mainly UV irradiation, overlaying the effects of natural aging (intrinsic aging) which is a constant process composed of gradual and irreversible tissue degeneration. Generally characterized by loss of elasticity, disorganized and fragmented collagen fibers, and dermal connective tissue injury [
30].
The oxidative stress theory of aging is built on the structural damage-based hypothesis that the accumulation of oxidative damage to macromolecules (carbohydrates, lipids, proteins, and DNA) is associated with functional loss. This occurs probably because of senescent cells that acquire a senescence-associated secretory phenotype, including the secretion of interleukins, chemokines, and growth factors, which is involved in inflammatory events, and the secretion of degradative enzymes such as matrix metalloproteases (MMPs) [
31]. Moreover, photocarcinogenesis is another consequence of photodamage, involving biochemical events that ultimately may cause skin cancer. These events are initiated by UV radiation, which is a complete carcinogen, as it initiates cancer through DNA damage via the formation of DNA photoproducts and activates oncogenes and/or silencing tumor suppressor genes, but also promotes cancer growth through inflammatory processes inherent in cumulative UV exposure [
2,
32].
The administration of exogenous antioxidants is a strategy used to scavenge excessive ROS and, consequently, prevent an overload of the endogenous antioxidant defense system and minimize oxidative damage in cellular components. A well-studied group of antioxidants is polyphenols, secondary metabolites of plants found largely in fruits, vegetables, cereals, and beverages. Among them, TA, a hydrolyzable tannin, has been reported to possess a variety of biological properties, including antioxidant and anti-inflammatory effects [
33,
34,
35]. Another class of antioxidant that recently gained attention is nanoparticles that demonstrate enzyme-like activities, mimicking natural enzymes or nanozymes [
36]. CNPs exhibit this property because it exists in a mixed valence state (Ce
3+ and Ce
4+), and due to the low redox potential of these states, allows CNPs to have self-regenerative redox cycling properties, which is evidenced as CAT and SOD-mimetic activities. Enabling them to combat excessive ROS production, as demonstrated by several studies [
37,
38,
39].
In addition, it has been shown in many studies that the density of Ce
3+ redox states in a given cerium oxide nanomaterial determines, in significant part, the catalytic behavior of the material towards unique ROS species. Therefore, building from these basis studies, particle formulations with controlled Ce
3+ state densities, and thereby surface sensitivity towards ROS scavenging, may be produced by controlling the particle synthesis method. In earlier work from our group, a unique approach to synthesizing spherical particles with substantial colloidal stability and a significant presence of Ce
3+ states in its crystal [
19] was reported. In the present study, a similar synthesis was performed and subjected to a conjugation process (room temperature bioconjugation chemistry using CDI compound) with TA, aiming the investigation of the efficacy of CNP-TA in preventing photodamage in skin cells. For this, the in vitro model of L929 fibroblasts exposed to UVB-irradiation was selected, given that fibroblasts are the majority cell type comprising the dermal layer of the skin.
For CNP-TA synthesis, the activated OH group on CNPs reacted with CDI to form an active imidazole carbamate intermediate compound. Similarly, the activated OH group on TA molecules is attached to another end-amine group in the EDI molecules to form a carbamate bond. According to UV–Vis spectra results, CNP-TA nanoparticles showed peaks of TA molecule and its conjugation with CNPs. The breadth of the observed peak may arise from the secondary boning of TA molecules and their irregular aggregate structuring on the particle surface. Indicating the successful conjugation of TA on the CNP surface. From XPS results, CNP-TA decreased the ratio of Ce3+/Ce4+, compared to bare CNPs. This change is expected from the conjugation procedure, wherein the particles are put in a more alkaline condition during the conjugation and surface sites are utilized for the conjugation itself. Moreover, TA conjugation increased its size by 30% (approximately 10 nm), probably due to the association of TA molecules on the CNP surface.
For photoprotective activity, several experiments were performed to analyze the potential of CNP-TA to decrease the oxidative stress induced by UVB radiation. The 24 h treatments of either bare CNPs or CNP-TA were able to prevent the decrease in cell viability; this reflected the ability of CNP-TA to prevent the UVB-induced oxidative stress in L929 irradiated cells. In general, the induction of ROS can occur by endogenous or exogenous sources. UV radiation is the main exogenous source of ROS increase, which causes oxidative modification of skin cellular macromolecules [
29]. Either TA alone, bare CNPs or CNP-TA were able to decrease ROS, this result is in line with previous findings of TA [
12] and bare CNPs [
13] to block ROS production in UVB-irradiated L929 fibroblasts. We found that the combination of CNPs with TA maintained the effect of diminishing ROS in the cellular environment.
To analyze the ability of the cell to cope with ROS production, it is important to analyze the endogenous antioxidant defense levels. This mechanism protects cells from free radical toxicity. The SOD and CAT are important antioxidant enzymes, SOD catalyzes the dismutation of superoxide anion (O
2−) into hydrogen peroxide (H
2O
2), and CAT decomposes H
2O
2 into water and oxygen, preventing hydroxyl (OH
.) radicals production [
40]. Whereas GSH, an endogenous antioxidant agent, acts as a cofactor of several detoxifying enzymes (e.g., glutathione peroxidase), scavenges OH
. radicals and singlet oxygen (
1O
2), and regenerates the antioxidant vitamins C and E back to their active forms [
41]. Here, we showed that although UVB depletes cellular levels of CAT and GSH, TA and CNP-TA treatments were able to prevent the decrease in these antioxidant defense components in UVB-irradiated fibroblasts. This result might be explained due to the ability of CNPs to act as SOD and CAT-like enzymes, preventing the depletion of endogenous antioxidant defense in cells exposed to UVB.
Additionally, the increase in ROS is related to the increase in oxidative damages to macromolecules, which leads to photodamage. Lipid peroxidation alters the cytoplasmatic membrane fluidity, leading to cell death, whereas DNA damage is among the initial steps in photocarcinogenesis and photoaging [
41,
42]. Here, we found that CNP-TA treatment was able to prevent UVB-induced oxidative damage in lipids and DNA molecules. The DNA fragmentation experiment using agarose gel electrophoresis can generate two different types of DNA drag patterns. If the cells are in the process of necrosis, with a non-specific DNA cleavage, the DNA pattern on the gel is a shear. However, if the cell is in the apoptosis process, with the activation of endonucleases that cleave the DNA into fragments of approximately 200-base pairs intervals, the DNA on the gel will resemble a ladder pattern [
43]. In our experiment, in the UVB-irradiated group, the DNA on the gel was a continuous smear rather than a ladder, suggesting that the cells are in necrosis process. However, in the treatment groups, mainly for CNPs and CNP-TA, there was a decrease in the smear pattern, suggesting a protection against the DNA oxidative damage.
The alterations in the dermis mainly are due to collagen degradation and remodeling of the dermal extracellular matrix by MMPs, enzymes produced by keratinocytes, fibroblasts and neutrophils in response to UV radiation and inflammation [
28,
44]. MMP-1 is a type of metalloproteinase that directly cleaves collagen fibers, which will later be cleaved into smaller fragments by MMP-2, MMP-3 and MMP-9 [
45], producing disorganization of collagen fibers in the dermis [
1]. Indeed, Fisher et al. [
46] reported that MMP-1 expression was increased in human UV-irradiated skin along with collagen degradation. These biological processes translate to clinical signs including wrinkle formation and reduced wound healing [
30]. Here, we showed that CNP-TA treatment was able to decrease MMP-1 expression in UVB-irradiated cells. The reduction in MMP-1 was probably correlated to the improved cell proliferation at wound healing assay in L929-UVB irradiated cells treated with CNP-TA.
As inflammation is intrinsically associated with photoaging and photocarcinogenesis evolution [
47], it is important to analyze inflammatory mediators that are increased in UV exposure. COX-2 is involved in arachidonic acid metabolism and mediates various inflammatory responses. COX-2 is upregulated in the skin after UV irradiation, with peak expression after 24 h [
48]. There is also an age-dependent increase in UV-induced COX-2 expression that can result in chronic low-grade inflammation over time [
49]. The increase in COX-2 upregulates the secretion of prostaglandin 2 (PGE2), which is a major pro-inflammatory cytokine responsible for immune cell infiltration in the skin [
50]. Moreover, TGF-β is an important mediator in the acute and chronic inflammatory effects of solar radiation [
51]. Chainiaux et al. [
52] reported that repeated exposure to UVB induced a TGF-β-driven premature senescence in human dermal fibroblasts. We found that CNP-TA treatment was able to decrease COX-2 expression and TGF-β secretion in UVB-irradiated fibroblasts, indicating there may be a decrease in the inflammatory response.
In conclusion, this work demonstrated the synthesis of a novel delivery system for TA conjugated with CNPs and its photoprotective potential. The CNP-TA exhibited less cytotoxicity, compared to free TA, in which free TA demonstrates a cytotoxic effect in a dose-dependent manner, which was not observed with CNP-TA. In addition, the CNP-TA system showed better photoprotective activity when compared to bare CNPs, exhibiting better ROS scavenging activity, better prevention against depletion of endogenous antioxidant defense (CAT and GSH), and better prevention against oxidative damage to lipids and DNA. Furthermore, CNP-TA, improved cell proliferation, decreased MMP-1 expression, the main enzyme involved in collagen degradation, and decreased the secretion of TGF-β and COX-2 expression, suggesting the decrease in UVB-mediated inflammatory process. These results suggested that CNP-TA is a promising nanoparticulate system for preventing photodamage, minimizing the progression of photoaging and UVB-induced inflammation.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}