
Calcium and Reactive Oxygen Species Signaling Interplays in Cardiac Physiology and Pathologies



Abstract
:1. Introduction
2. ROS
The Alpha and Omega of Mitochondrial ROS
3. Calcium
3.1. Influx and Efflux of Ca2+ in Mitochondria
3.2. Key Ca2+ Targets and Roles in the Regulation of Mitochondrial Bioenergetics
4. The Interplay between Ca2+ and ROS
5. Cardiac Muscle, ROS, and Ca2+ Signaling
5.1. Calcium and ROS in Heart Failure
5.2. Mitochondrial ROS vs. ER ROS
5.3. In Vivo Mouse Model of Postmyocardial Infarction
5.4. Drug Targeting of Mitochondrial Ca2+ and Homeostasis
5.5. Cardiomyopathy with Mitochondrial Dysfunction-Associated Genes
5.5.1. MTO1
5.5.2. AGK
5.5.3. SLC25A4
5.5.4. MT-TL1
5.5.5. MT-TK
5.5.6. TAFAZZIN
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bravo-Sagua, R.; Parra, V.; López-Crisosto, C.; Díaz, P.; Quest, A.F.G.; Lavandero, S. Calcium Transport and Signaling in Mitochondria. In Comprehensive Physiology; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2017; pp. 623–634. ISBN 978-0-470-65071-4. [Google Scholar]
- Kohlhaas, M.; Nickel, A.G.; Maack, C. Mitochondrial Energetics and Calcium Coupling in the Heart. J. Physiol. 2017, 595, 3753–3763. [Google Scholar] [CrossRef]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The Versatility and Universality of Calcium Signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A Mutual Interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef]
- D’Oria, R.; Schipani, R.; Leonardini, A.; Natalicchio, A.; Perrini, S.; Cignarelli, A.; Laviola, L.; Giorgino, F. The Role of Oxidative Stress in Cardiac Disease: From Physiological Response to Injury Factor. Oxid. Med. Cell. Longev. 2020, 2020, 5732956. [Google Scholar] [CrossRef]
- Santulli, G.; Marks, A.R. Essential Roles of Intracellular Calcium Release Channels in Muscle, Brain, Metabolism, and Aging. Curr. Mol. Pharmacol. 2015, 8, 206–222. [Google Scholar] [CrossRef]
- Santana, L.F.; Cheng, H.; Gómez, A.M.; Cannell, M.B.; Lederer, W.J. Relation between the Sarcolemmal Ca2+ Current and Ca2+ Sparks and Local Control Theories for Cardiac Excitation-Contraction Coupling. Circ. Res. 1996, 78, 166–171. [Google Scholar] [CrossRef]
- Modesti, L.; Danese, A.; Angela Maria Vitto, V.; Ramaccini, D.; Aguiari, G.; Gafà, R.; Lanza, G.; Giorgi, C.; Pinton, P. Mitochondrial Ca2+ Signaling in Health, Disease and Therapy. Cells 2021, 10, 1317. [Google Scholar] [CrossRef]
- Nickel, A.G.; von Hardenberg, A.; Hohl, M.; Löffler, J.R.; Kohlhaas, M.; Becker, J.; Reil, J.-C.; Kazakov, A.; Bonnekoh, J.; Stadelmaier, M.; et al. Reversal of Mitochondrial Transhydrogenase Causes Oxidative Stress in Heart Failure. Cell Metab. 2015, 22, 472–484. [Google Scholar] [CrossRef]
- Gauthier, L.D.; Greenstein, J.L.; Cortassa, S.; O’Rourke, B.; Winslow, R.L. A Computational Model of Reactive Oxygen Species and Redox Balance in Cardiac Mitochondria. Biophys. J. 2013, 105, 1045–1056. [Google Scholar] [CrossRef] [Green Version]
- Köhler, A.C.; Sag, C.M.; Maier, L.S. Reactive Oxygen Species and Excitation–Contraction Coupling in the Context of Cardiac Pathology. J. Mol. Cell. Cardiol. 2014, 73, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Dietl, A.; Maack, C. Targeting Mitochondrial Calcium Handling and Reactive Oxygen Species in Heart Failure. Curr. Heart Fail. Rep. 2017, 14, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C. Defining Roles of Specific Reactive Oxygen Species (ROS) in Cell Biology and Physiology. Nat. Rev. Mol. Cell Biol. 2022, 23, 499–515. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Abramov, A.Y. Role of Mitochondrial ROS in the Brain: From Physiology to Neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Hempel, N.; Trebak, M. Crosstalk between Calcium and Reactive Oxygen Species Signaling in Cancer. Cell Calcium 2017, 63, 70–96. [Google Scholar] [CrossRef]
- Juan, C.A.; Pérez de la Lastra, J.M.; Plou, F.J.; Pérez-Lebeña, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642. [Google Scholar] [CrossRef]
- Vermot, A.; Petit-Härtlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxid. Basel Switz. 2021, 10, 890. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Margreiter, R.; Ausserlechner, M.J.; Hagenbuchner, J. The Complex Interplay between Mitochondria, ROS and Entire Cellular Metabolism. Antioxidants 2022, 11, 1995. [Google Scholar] [CrossRef]
- Cheung, E.C.; Vousden, K.H. The Role of ROS in Tumour Development and Progression. Nat. Rev. Cancer 2022, 22, 280–297. [Google Scholar] [CrossRef]
- Sies, H.; Chance, B. The Steady State Level of Catalase Compound I in Isolated Hemoglobin-Free Perfused Rat Liver. FEBS Lett. 1970, 11, 172–176. [Google Scholar] [CrossRef] [Green Version]
- Pratt, D.A.; Tallman, K.A.; Porter, N.A. Free Radical Oxidation of Polyunsaturated Lipids: New Mechanistic Insights and the Development of Peroxyl Radical Clocks. Acc. Chem. Res. 2011, 44, 458–467. [Google Scholar] [CrossRef]
- Bagur, R.; Hajnóczky, G. Intracellular Ca2+ Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef]
- Jones, D.P.; Sies, H. The Redox Code. Antioxid. Redox Signal. 2015, 23, 734–746. [Google Scholar] [CrossRef]
- Paulsen, C.E.; Carroll, K.S. Cysteine-Mediated Redox Signaling: Chemistry, Biology, and Tools for Discovery. Chem. Rev. 2013, 113, 4633–4679. [Google Scholar] [CrossRef] [PubMed]
- Bak, D.W.; Weerapana, E. Cysteine-Mediated Redox Signalling in the Mitochondria. Mol. Biosyst. 2015, 11, 678–697. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Fu, L.; Liu, K.; Tian, C.; Wu, Z.; Jung, Y.; Ferreira, R.B.; Carroll, K.S.; Blackwell, T.K.; Yang, J. Global Profiling of Distinct Cysteine Redox Forms Reveals Wide-Ranging Redox Regulation in C. Elegans. Nat. Commun. 2021, 12, 1415. [Google Scholar] [CrossRef]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative Stress and Autophagy: The Clash between Damage and Metabolic Needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of Reactive Oxygen Species Generation by Mitochondria Oxidizing Different Substrates. Redox Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef]
- Fato, R.; Bergamini, C.; Leoni, S.; Lenaz, G. Mitochondrial Production of Reactive Oxygen Species: Role of Complex I and Quinone Analogues. BioFactors 2008, 32, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Schultz, B.E.; Chan, S.I. Structures and Proton-Pumping Strategies of Mitochondrial Respiratory Enzymes. Annu. Rev. Biophys. Biomol. Struct. 2001, 30, 23–65. [Google Scholar] [CrossRef] [PubMed]
- Grgic, L.; Zwicker, K.; Kashani-Poor, N.; Kerscher, S.; Brandt, U. Functional Significance of Conserved Histidines and Arginines in the 49-KDa Subunit of Mitochondrial Complex I. J. Biol. Chem. 2004, 279, 21193–21199. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D. Mitochondrial Generation of Superoxide and Hydrogen Peroxide as the Source of Mitochondrial Redox Signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive Oxygen Species (ROS) as Pleiotropic Physiological Signalling Agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Kudin, A.P.; Bimpong-Buta, N.Y.-B.; Vielhaber, S.; Elger, C.E.; Kunz, W.S. Characterization of Superoxide-Producing Sites in Isolated Brain Mitochondria. J. Biol. Chem. 2004, 279, 4127–4135. [Google Scholar] [CrossRef]
- Cox, A.G.; Winterbourn, C.C.; Hampton, M.B. Mitochondrial Peroxiredoxin Involvement in Antioxidant Defence and Redox Signalling. Biochem. J. 2009, 425, 313–325. [Google Scholar] [CrossRef]
- Molavian, H.; Madani Tonekaboni, A.; Kohandel, M.; Sivaloganathan, S. The Synergetic Coupling among the Cellular Antioxidants Glutathione Peroxidase/Peroxiredoxin and Other Antioxidants and Its Effect on the Concentration of H2O2. Sci. Rep. 2015, 5, 13620. [Google Scholar] [CrossRef]
- Angelova, P.R.; Abramov, A.Y. Functional Role of Mitochondrial Reactive Oxygen Species in Physiology. Free Radic. Biol. Med. 2016, 100, 81–85. [Google Scholar] [CrossRef]
- Starkov, A.A.; Fiskum, G.; Chinopoulos, C.; Lorenzo, B.J.; Browne, S.E.; Patel, M.S.; Beal, M.F. Mitochondrial α-Ketoglutarate Dehydrogenase Complex Generates Reactive Oxygen Species. J. Neurosci. 2004, 24, 7779–7788. [Google Scholar] [CrossRef]
- Finkel, T. Signal Transduction by Reactive Oxygen Species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Suski, J.M.; Lebiedzinska, M.; Bonora, M.; Pinton, P.; Duszynski, J.; Wieckowski, M.R. Relation between Mitochondrial Membrane Potential and ROS Formation. Methods Mol. Biol. 2012, 810, 183–205. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial Formation of Reactive Oxygen Species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High Protonic Potential Actuates a Mechanism of Production of Reactive Oxygen Species in Mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Brandt, U. Energy Converting NADH:Quinone Oxidoreductase (Complex I). Annu. Rev. Biochem. 2006, 75, 69–92. [Google Scholar] [CrossRef]
- Kussmaul, L.; Hirst, J. The Mechanism of Superoxide Production by NADH:Ubiquinone Oxidoreductase (Complex I) from Bovine Heart Mitochondria. Proc. Natl. Acad. Sci. USA 2006, 103, 7607–7612. [Google Scholar] [CrossRef]
- Fato, R.; Bergamini, C.; Bortolus, M.; Maniero, A.L.; Leoni, S.; Ohnishi, T.; Lenaz, G. Differential Effects of Mitochondrial Complex I Inhibitors on Production of Reactive Oxygen Species. Biochim. Biophys. Acta 2009, 1787, 384–392. [Google Scholar] [CrossRef]
- Diquigiovanni, C.; Bergamini, C.; Diaz, R.; Liparulo, I.; Bianco, F.; Masin, L.; Baldassarro, V.A.; Rizzardi, N.; Tranchina, A.; Buscherini, F.; et al. A Novel Mutation in SPART Gene Causes a Severe Neurodevelopmental Delay Due to Mitochondrial Dysfunction with Complex I Impairments and Altered Pyruvate Metabolism. FASEB J. 2019, 33, 11284–11302. [Google Scholar] [CrossRef]
- Walker, F.O. Huntington’s Disease. Semin. Neurol. 2007, 27, 143–150. [Google Scholar] [CrossRef]
- Paillusson, S.; Gomez-Suaga, P.; Stoica, R.; Little, D.; Gissen, P.; Devine, M.J.; Noble, W.; Hanger, D.P.; Miller, C.C.J. α-Synuclein Binds to the ER–Mitochondria Tethering Protein VAPB to Disrupt Ca2+ Homeostasis and Mitochondrial ATP Production. Acta Neuropathol. 2017, 134, 129–149. [Google Scholar] [CrossRef]
- Raffaello, A.; Mammucari, C.; Gherardi, G.; Rizzuto, R. Calcium at the Center of Cell Signaling: Interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes. Trends Biochem. Sci. 2016, 41, 1035–1049. [Google Scholar] [CrossRef] [Green Version]
- Patergnani, S.; Suski, J.M.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Giorgi, C.; Marchi, S.; Missiroli, S.; Poletti, F.; et al. Calcium Signaling around Mitochondria Associated Membranes (MAMs). Cell Commun. Signal. 2011, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close Contacts with the Endoplasmic Reticulum as Determinants of Mitochondrial Ca2+ Responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Gang, X.; He, G.; Liu, Y.; Wang, Y.; Zhao, X.; Wang, G. The Molecular Mechanisms Underlying Mitochondria-Associated Endoplasmic Reticulum Membrane-Induced Insulin Resistance. Front. Endocrinol. 2020, 11, 592129. [Google Scholar] [CrossRef]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative Genomics Identifies MCU as an Essential Component of the Mitochondrial Calcium Uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef]
- De Stefani, D.; Rizzuto, R.; Pozzan, T. Enjoy the Trip: Calcium in Mitochondria Back and Forth. Annu. Rev. Biochem. 2016, 85, 161–192. [Google Scholar] [CrossRef]
- Yoo, J. Structural Basis of Ca2+ Uptake by Mitochondrial Calcium Uniporter in Mitochondria: A Brief Review. BMB Rep. 2022, 55, 528–534. [Google Scholar] [CrossRef]
- Zhuo, W.; Zhou, H.; Guo, R.; Yi, J.; Zhang, L.; Yu, L.; Sui, Y.; Zeng, W.; Wang, P.; Yang, M. Structure of Intact Human MCU Supercomplex with the Auxiliary MICU Subunits. Protein Cell 2021, 12, 220–229. [Google Scholar] [CrossRef]
- Marchi, S.; Pinton, P. The Mitochondrial Calcium Uniporter Complex: Molecular Components, Structure and Physiopathological Implications. J. Physiol. 2014, 592, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C.B.; Tsai, C.-W.; Tsai, M.-F. The Conserved Aspartate Ring of MCU Mediates MICU1 Binding and Regulation in the Mitochondrial Calcium Uniporter Complex. eLife 2019, 8, e41112. [Google Scholar] [CrossRef]
- Fan, M.; Zhang, J.; Tsai, C.-W.; Orlando, B.J.; Rodriguez, M.; Xu, Y.; Liao, M.; Tsai, M.-F.; Feng, L. Structure and Mechanism of the Mitochondrial Ca2+ Uniporter Holocomplex. Nature 2020, 582, 129–133. [Google Scholar] [CrossRef]
- Mammucari, C.; Raffaello, A.; Vecellio Reane, D.; Gherardi, G.; De Mario, A.; Rizzuto, R. Mitochondrial Calcium Uptake in Organ Physiology: From Molecular Mechanism to Animal Models. Pflüg. Arch.—Eur. J. Physiol. 2018, 470, 1165–1179. [Google Scholar] [CrossRef] [PubMed]
- Lasorsa, F.M.; Pinton, P.; Palmieri, L.; Fiermonte, G.; Rizzuto, R.; Palmieri, F. Recombinant Expression of the Ca(2+)-Sensitive Aspartate/Glutamate Carrier Increases Mitochondrial ATP Production in Agonist-Stimulated Chinese Hamster Ovary Cells. J. Biol. Chem. 2003, 278, 38686–38692. [Google Scholar] [CrossRef] [PubMed]
- Nesci, S. Mitochondrial Permeability Transition, F1 FO-ATPase and Calcium: An Enigmatic Triangle. EMBO Rep. 2017, 18, 1265–1267. [Google Scholar] [CrossRef]
- McCormack, J.G.; Halestrap, A.P.; Denton, R.M. Role of Calcium Ions in Regulation of Mammalian Intramitochondrial Metabolism. Physiol. Rev. 1990, 70, 391–425. [Google Scholar] [CrossRef] [PubMed]
- Denton, R.M. Regulation of Mitochondrial Dehydrogenases by Calcium Ions. Biochim. Biophys. Acta 2009, 1787, 1309–1316. [Google Scholar] [CrossRef]
- Denton, R.M.; Randle, P.J.; Martin, B.R. Stimulation by Calcium Ions of Pyruvate Dehydrogenase Phosphate Phosphatase. Biochem. J. 1972, 128, 161–163. [Google Scholar] [CrossRef]
- Rutter, G.A.; Denton, R.M. Regulation of NAD+-Linked Isocitrate Dehydrogenase and 2-Oxoglutarate Dehydrogenase by Ca2+ Ions within Toluene-Permeabilized Rat Heart Mitochondria. Interactions with Regulation by Adenine Nucleotides and NADH/NAD+ Ratios. Biochem. J. 1988, 252, 181–189. [Google Scholar] [CrossRef]
- Foskett, J.K.; Mak, D.-O.D. Regulation of IP3R Channel Gating by Ca2+ and Ca2+ Binding Proteins. Curr. Top. Membr. 2010, 66, 235–272. [Google Scholar] [CrossRef]
- Cárdenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgó, J.; Müller, M.; Vais, H.; Cheung, K.-H.; Yang, J.; Parker, I.; et al. Essential Regulation of Cell Bioenergetics by Constitutive InsP3 Receptor Ca2+ Transfer to Mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef]
- Filadi, R.; Leal, N.S.; Schreiner, B.; Rossi, A.; Dentoni, G.; Pinho, C.M.; Wiehager, B.; Cieri, D.; Calì, T.; Pizzo, P.; et al. TOM70 Sustains Cell Bioenergetics by Promoting IP3R3-Mediated ER to Mitochondria Ca2+ Transfer. Curr. Biol. CB 2018, 28, 369–382.e6. [Google Scholar] [CrossRef] [Green Version]
- Rossi, A.; Pizzo, P.; Filadi, R. Calcium, Mitochondria and Cell Metabolism: A Functional Triangle in Bioenergetics. Biochim. Biophys. Acta BBA—Mol. Cell Res. 2019, 1866, 1068–1078. [Google Scholar] [CrossRef] [PubMed]
- Mallilankaraman, K.; Cárdenas, C.; Doonan, P.J.; Chandramoorthy, H.C.; Irrinki, K.M.; Golenár, T.; Csordás, G.; Madireddi, P.; Yang, J.; Müller, M.; et al. MCUR1 Is an Essential Component of Mitochondrial Ca2+ Uptake That Regulates Cellular Metabolism. Nat. Cell Biol. 2012, 14, 1336–1343. [Google Scholar] [CrossRef] [PubMed]
- Di Lisa, F.; Bernardi, P. A CaPful of Mechanisms Regulating the Mitochondrial Permeability Transition. J. Mol. Cell. Cardiol. 2009, 46, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Madesh, M.; Hajnóczky, G. VDAC-Dependent Permeabilization of the Outer Mitochondrial Membrane by Superoxide Induces Rapid and Massive Cytochrome c Release. J. Cell Biol. 2001, 155, 1003–1016. [Google Scholar] [CrossRef] [PubMed]
- Petronilli, V.; Penzo, D.; Scorrano, L.; Bernardi, P.; Lisa, F.D. The Mitochondrial Permeability Transition, Release of Cytochrome c and Cell Death: Correlation with the duration of pore openings in situ. J. Biol. Chem. 2001, 276, 12030–12034. [Google Scholar] [CrossRef] [PubMed]
- Di Lisa, F.; Bernardi, P. Mitochondrial Function and Myocardial Aging. A Critical Analysis of the Role of Permeability Transition. Cardiovasc. Res. 2005, 66, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Di Venosa, N.; Federici, A.; Ruggiero, F.M. Decrease in Mitochondrial Complex I Activity in Ischemic/Reperfused Rat Heart: Involvement of Reactive Oxygen Species and Cardiolipin. Circ. Res. 2004, 94, 53–59. [Google Scholar] [CrossRef]
- Nomura, K.; Imai, H.; Koumura, T.; Kobayashi, T.; Nakagawa, Y. Mitochondrial Phospholipid Hydroperoxide Glutathione Peroxidase Inhibits the Release of Cytochrome c from Mitochondria by Suppressing the Peroxidation of Cardiolipin in Hypoglycaemia-Induced Apoptosis. Biochem. J. 2000, 351, 183–193. [Google Scholar] [CrossRef]
- Shigenaga, M.K.; Hagen, T.M.; Ames, B.N. Oxidative Damage and Mitochondrial Decay in Aging. Proc. Natl. Acad. Sci. USA 1994, 91, 10771–10778. [Google Scholar] [CrossRef] [Green Version]
- Ben-Kasus Nissim, T.; Zhang, X.; Elazar, A.; Roy, S.; Stolwijk, J.A.; Zhou, Y.; Motiani, R.K.; Gueguinou, M.; Hempel, N.; Hershfinkel, M.; et al. Mitochondria Control Store-Operated Ca2+ Entry through Na+ and Redox Signals. EMBO J. 2017, 36, 797–815. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Antunes, F.; Canali, R.; Rettori, D.; Cadenas, E. Voltage-Dependent Anion Channels Control the Release of the Superoxide Anion from Mitochondria to Cytosol. J. Biol. Chem. 2003, 278, 5557–5563. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Weaver, D.; Eisner, V.; Várnai, P.; Hunyady, L.; Ma, J.; Csordás, G.; Hajnóczky, G. Switch from ER-Mitochondrial to SR-Mitochondrial Calcium Coupling during Muscle Differentiation. Cell Calcium 2012, 52, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Demydenko, K.; Ekhteraei-Tousi, S.; Roderick, H.L. Inositol 1,4,5-Trisphosphate Receptors in Cardiomyocyte Physiology and Disease. Philos. Trans. R. Soc. B Biol. Sci. 2022, 377, 20210319. [Google Scholar] [CrossRef]
- Nakayama, H.; Bodi, I.; Maillet, M.; DeSantiago, J.; Domeier, T.L.; Mikoshiba, K.; Lorenz, J.N.; Blatter, L.A.; Bers, D.M.; Molkentin, J.D. The IP3 Receptor Regulates Cardiac Hypertrophy in Response to Select Stimuli. Circ. Res. 2010, 107, 659–666. [Google Scholar] [CrossRef]
- Paillard, M.; Tubbs, E.; Thiebaut, P.-A.; Gomez, L.; Fauconnier, J.; Da Silva, C.C.; Teixeira, G.; Mewton, N.; Belaidi, E.; Durand, A.; et al. Depressing Mitochondria-Reticulum Interactions Protects Cardiomyocytes from Lethal Hypoxia-Reoxygenation Injury. Circulation 2013, 128, 1555–1565. [Google Scholar] [CrossRef]
- Nickel, A.; Löffler, J.; Maack, C. Myocardial Energetics in Heart Failure. Basic Res. Cardiol. 2013, 108, 358. [Google Scholar] [CrossRef]
- Huss, J.M.; Kelly, D.P. Mitochondrial Energy Metabolism in Heart Failure: A Question of Balance. J. Clin. Investig. 2005, 115, 547–555. [Google Scholar] [CrossRef]
- O-Uchi, J.; Jhun, B.S.; Xu, S.; Hurst, S.; Raffaello, A.; Liu, X.; Yi, B.; Zhang, H.; Gross, P.; Mishra, J.; et al. Adrenergic Signaling Regulates Mitochondrial Ca2+ Uptake Through Pyk2-Dependent Tyrosine Phosphorylation of the Mitochondrial Ca2+ Uniporter. Antioxid. Redox Signal. 2014, 21, 863–879. [Google Scholar] [CrossRef]
- Holmström, K.M.; Pan, X.; Liu, J.C.; Menazza, S.; Liu, J.; Nguyen, T.T.; Pan, H.; Parks, R.J.; Anderson, S.; Noguchi, A.; et al. Assessment of Cardiac Function in Mice Lacking the Mitochondrial Calcium Uniporter. J. Mol. Cell. Cardiol. 2015, 85, 178–182. [Google Scholar] [CrossRef] [Green Version]
- Elrod, J.W.; Wong, R.; Mishra, S.; Vagnozzi, R.J.; Sakthievel, B.; Goonasekera, S.A.; Karch, J.; Gabel, S.; Farber, J.; Force, T.; et al. Cyclophilin D Controls Mitochondrial Pore-Dependent Ca(2+) Exchange, Metabolic Flexibility, and Propensity for Heart Failure in Mice. J. Clin. Investig. 2010, 120, 3680–3687. [Google Scholar] [CrossRef]
- Odagiri, K.; Katoh, H.; Kawashima, H.; Tanaka, T.; Ohtani, H.; Saotome, M.; Urushida, T.; Satoh, H.; Hayashi, H. Local Control of Mitochondrial Membrane Potential, Permeability Transition Pore and Reactive Oxygen Species by Calcium and Calmodulin in Rat Ventricular Myocytes. J. Mol. Cell. Cardiol. 2009, 46, 989–997. [Google Scholar] [CrossRef]
- Tocchetti, C.G.; Stanley, B.A.; Murray, C.I.; Sivakumaran, V.; Donzelli, S.; Mancardi, D.; Pagliaro, P.; Gao, W.D.; van Eyk, J.; Kass, D.A.; et al. Playing with Cardiac “Redox Switches”: The “HNO Way” to Modulate Cardiac Function. Antioxid. Redox Signal. 2011, 14, 1687–1698. [Google Scholar] [CrossRef]
- Griffiths, E.J.; Balaska, D.; Cheng, W.H.Y. The Ups and Downs of Mitochondrial Calcium Signalling in the Heart. Biochim. Biophys. Acta 2010, 1797, 856–864. [Google Scholar] [CrossRef]
- Zhang, M.; Prosser, B.L.; Bamboye, M.A.; Gondim, A.N.S.; Santos, C.X.; Martin, D.; Ghigo, A.; Perino, A.; Brewer, A.C.; Ward, C.W.; et al. Contractile Function During Angiotensin-II Activation: Increased Nox2 Activity Modulates Cardiac Calcium Handling via Phospholamban Phosphorylation. J. Am. Coll. Cardiol. 2015, 66, 261–272. [Google Scholar] [CrossRef]
- Kim, T.Y.; Terentyeva, R.; Roder, K.H.F.; Li, W.; Liu, M.; Greener, I.; Hamilton, S.; Polina, I.; Murphy, K.R.; Clements, R.T.; et al. SK Channel Enhancers Attenuate Ca2+-Dependent Arrhythmia in Hypertrophic Hearts by Regulating Mito-ROS-Dependent Oxidation and Activity of RyR. Cardiovasc. Res. 2017, 113, 343–353. [Google Scholar] [CrossRef]
- Wagner, S.; Dantz, C.; Flebbe, H.; Azizian, A.; Sag, C.M.; Engels, S.; Möllencamp, J.; Dybkova, N.; Islam, T.; Shah, A.M.; et al. NADPH Oxidase 2 Mediates Angiotensin II-Dependent Cellular Arrhythmias via PKA and CaMKII. J. Mol. Cell. Cardiol. 2014, 75, 206–215. [Google Scholar] [CrossRef]
- Ago, T.; Liu, T.; Zhai, P.; Chen, W.; Li, H.; Molkentin, J.D.; Vatner, S.F.; Sadoshima, J. A Redox-Dependent Pathway for Regulating Class II HDACs and Cardiac Hypertrophy. Cell 2008, 133, 978–993. [Google Scholar] [CrossRef]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of Cyclophilin D Reveals a Critical Role for Mitochondrial Permeability Transition in Cell Death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef]
- Nakagawa, T.; Shimizu, S.; Watanabe, T.; Yamaguchi, O.; Otsu, K.; Yamagata, H.; Inohara, H.; Kubo, T.; Tsujimoto, Y. Cyclophilin D-Dependent Mitochondrial Permeability Transition Regulates Some Necrotic but Not Apoptotic Cell Death. Nature 2005, 434, 652–658. [Google Scholar] [CrossRef]
- Maack, C.; Kartes, T.; Kilter, H.; Schäfers, H.-J.; Nickenig, G.; Böhm, M.; Laufs, U. Oxygen Free Radical Release in Human Failing Myocardium Is Associated with Increased Activity of Rac1-GTPase and Represents a Target for Statin Treatment. Circulation 2003, 108, 1567–1574. [Google Scholar] [CrossRef]
- Dey, S.; Sidor, A.; O’Rourke, B. Compartment-Specific Control of Reactive Oxygen Species Scavenging by Antioxidant Pathway Enzymes. J. Biol. Chem. 2016, 291, 11185–11197. [Google Scholar] [CrossRef]
- Belch, J.J.; Bridges, A.B.; Scott, N.; Chopra, M. Oxygen Free Radicals and Congestive Heart Failure. Br. Heart J. 1991, 65, 245–248. [Google Scholar] [CrossRef]
- Michels, G.; Khan, I.F.; Endres-Becker, J.; Rottlaender, D.; Herzig, S.; Ruhparwar, A.; Wahlers, T.; Hoppe, U.C. Regulation of the Human Cardiac Mitochondrial Ca2+ Uptake by 2 Different Voltage-Gated Ca2+ Channels. Circulation 2009, 119, 2435–2443. [Google Scholar] [CrossRef]
- Weber, C.R.; Piacentino, V.; Houser, S.R.; Bers, D.M. Dynamic Regulation of Sodium/Calcium Exchange Function in Human Heart Failure. Circulation 2003, 108, 2224–2229. [Google Scholar] [CrossRef]
- Palty, R.; Sekler, I. The Mitochondrial Na(+)/Ca(2+) Exchanger. Cell Calcium 2012, 52, 9–15. [Google Scholar] [CrossRef]
- De Marchi, U.; Santo-Domingo, J.; Castelbou, C.; Sekler, I.; Wiederkehr, A.; Demaurex, N. NCLX Protein, but Not LETM1, Mediates Mitochondrial Ca2+ Extrusion, Thereby Limiting Ca2+-Induced NAD(P)H Production and Modulating Matrix Redox State. J. Biol. Chem. 2014, 289, 20377–20385. [Google Scholar] [CrossRef]
- Bers, D.M. Altered Cardiac Myocyte Ca Regulation in Heart Failure. Physiol. Bethesda Md 2006, 21, 380–387. [Google Scholar] [CrossRef]
- Kohlhaas, M.; Liu, T.; Knopp, A.; Zeller, T.; Ong, M.F.; Böhm, M.; O’Rourke, B.; Maack, C. Elevated Cytosolic Na+ Increases Mitochondrial Formation of Reactive Oxygen Species in Failing Cardiac Myocytes. Circulation 2010, 121, 1606–1613. [Google Scholar] [CrossRef] [PubMed]
- Rydström, J. Mitochondrial NADPH, Transhydrogenase and Disease. Biochim. Biophys. Acta 2006, 1757, 721–726. [Google Scholar] [CrossRef] [Green Version]
- Münzel, T.; Camici, G.G.; Maack, C.; Bonetti, N.R.; Fuster, V.; Kovacic, J.C. Impact of Oxidative Stress on the Heart and Vasculature: Part 2 of a 3-Part Series. J. Am. Coll. Cardiol. 2017, 70, 212–229. [Google Scholar] [CrossRef]
- Xi, Q.; Cheranov, S.Y.; Jaggar, J.H. Mitochondria-Derived Reactive Oxygen Species Dilate Cerebral Arteries by Activating Ca2+ Sparks. Circ. Res. 2005, 97, 354–362. [Google Scholar] [CrossRef]
- Li, J.; An, C.; Zheng, H.; Lei, T.; Zhang, N.; Zheng, Y.; Yang, M. Leukocyte Telomere Length and Risk of Papillary Thyroid Carcinoma. J. Clin. Endocrinol. Metab. 2019, 104, 2712–2718. [Google Scholar] [CrossRef]
- Zhou, L.; Aon, M.A.; Liu, T.; O’Rourke, B. Dynamic Modulation of Ca2+ Sparks by Mitochondrial Oscillations in Isolated Guinea Pig Cardiomyocytes under Oxidative Stress. J. Mol. Cell. Cardiol. 2011, 51, 632–639. [Google Scholar] [CrossRef]
- Cheranov, S.Y.; Jaggar, J.H. Mitochondrial Modulation of Ca2+ Sparks and Transient KCa Currents in Smooth Muscle Cells of Rat Cerebral Arteries. J. Physiol. 2004, 556, 755. [Google Scholar] [CrossRef]
- Yan, Y.; Liu, J.; Wei, C.; Li, K.; Xie, W.; Wang, Y.; Cheng, H. Bidirectional Regulation of Ca2+ Sparks by Mitochondria-Derived Reactive Oxygen Species in Cardiac Myocytes. Cardiovasc. Res. 2008, 77, 432–441. [Google Scholar] [CrossRef]
- Plant, D.R.; Lynch, G.S.; Williams, D.A. Hydrogen Peroxide Increases Depolarization-Induced Contraction of Mechanically Skinned Slow Twitch Fibres from Rat Skeletal Muscles. J. Physiol. 2002, 539, 883–891. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Temsah, R.M.; Netticadan, T. Role of Oxidative Stress in Cardiovascular Diseases. J. Hypertens. 2000, 18, 655–673. [Google Scholar] [CrossRef]
- Eisner, V.; Csordás, G.; Hajnóczky, G. Interactions between Sarco-Endoplasmic Reticulum and Mitochondria in Cardiac and Skeletal Muscle—Pivotal Roles in Ca2+ and Reactive Oxygen Species Signaling. J. Cell Sci. 2013, 126, 2965–2978. [Google Scholar] [CrossRef]
- Groenendyk, J.; Sreenivasaiah, P.K.; Kim, D.H.; Agellon, L.B.; Michalak, M. Biology of Endoplasmic Reticulum Stress in the Heart. Circ. Res. 2010, 107, 1185–1197. [Google Scholar] [CrossRef] [Green Version]
- Sevier, C.S.; Kaiser, C.A. Ero1 and Redox Homeostasis in the Endoplasmic Reticulum. Biochim. Biophys. Acta BBA—Mol. Cell Res. 2008, 1783, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Kania, E.; Pajak, B.; Orzechowski, A. Calcium Homeostasis and ER Stress in Control of Autophagy in Cancer Cells. Biomed. Res. Int. 2015, 2015, 352794. [Google Scholar] [CrossRef]
- Wagner, S.; Maier, L.S.; Bers, D.M. Role of Sodium and Calcium Dysregulation in Tachyarrhythmias in Sudden Cardiac Death. Circ. Res. 2015, 116, 1956–1970. [Google Scholar] [CrossRef]
- Gorski, P.A.; Ceholski, D.K.; Hajjar, R.J. Altered Myocardial Calcium Cycling and Energetics in Heart Failure—A Rational Approach for Disease Treatment. Cell Metab. 2015, 21, 183–194. [Google Scholar] [CrossRef]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.-S. Calcium, ATP, and ROS: A Mitochondrial Love-Hate Triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Marks, A.R. Calcium Cycling Proteins and Heart Failure: Mechanisms and Therapeutics. J. Clin. Investig. 2013, 123, 46–52. [Google Scholar] [CrossRef]
- Moschella, M.C.; Marks, A.R. Inositol 1,4,5-Trisphosphate Receptor Expression in Cardiac Myocytes. J. Cell Biol. 1993, 120, 1137–1146. [Google Scholar] [CrossRef]
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial Calcium Overload Is a Key Determinant in Heart Failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394. [Google Scholar] [CrossRef]
- Rizzuto, R.; Duchen, M.R.; Pozzan, T. Flirting in Little Space: The ER/Mitochondria Ca2+ Liaison. Sci. STKE Signal Transduct. Knowl. Environ. 2004, 2004, re1. [Google Scholar] [CrossRef]
- Min, C.K.; Yeom, D.R.; Lee, K.-E.; Kwon, H.-K.; Kang, M.; Kim, Y.-S.; Park, Z.Y.; Jeon, H.; Kim, D.H. Coupling of Ryanodine Receptor 2 and Voltage-Dependent Anion Channel 2 Is Essential for Ca2+ Transfer from the Sarcoplasmic Reticulum to the Mitochondria in the Heart. Biochem. J. 2012, 447, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Pieske, B.; Houser, S.R. [Na+]i Handling in the Failing Human Heart. Cardiovasc. Res. 2003, 57, 874–886. [Google Scholar] [CrossRef]
- Viatchenko-Karpinski, S.; Kornyeyev, D.; El-Bizri, N.; Budas, G.; Fan, P.; Jiang, Z.; Yang, J.; Anderson, M.E.; Shryock, J.C.; Chang, C.-P.; et al. Intracellular Na+ Overload Causes Oxidation of CaMKII and Leads to Ca2+ Mishandling in Isolated Ventricular Myocytes. J. Mol. Cell. Cardiol. 2014, 76, 247–256. [Google Scholar] [CrossRef]
- Peiling Yang, S.; Ngeow, J. Familial Non-Medullary Thyroid Cancer: Unraveling the Genetic Maze. Endocr. Relat. Cancer 2016, 23, R577–R595. [Google Scholar] [CrossRef]
- Wagner, S.; Dybkova, N.; Rasenack, E.C.L.; Jacobshagen, C.; Fabritz, L.; Kirchhof, P.; Maier, S.K.G.; Zhang, T.; Hasenfuss, G.; Brown, J.H.; et al. Ca2+/Calmodulin-Dependent Protein Kinase II Regulates Cardiac Na+ Channels. J. Clin. Investig. 2006, 116, 3127–3138. [Google Scholar] [CrossRef]
- Scirica, B.M.; Morrow, D.A.; Hod, H.; Murphy, S.A.; Belardinelli, L.; Hedgepeth, C.M.; Molhoek, P.; Verheugt, F.W.A.; Gersh, B.J.; McCabe, C.H.; et al. Effect of Ranolazine, an Antianginal Agent with Novel Electrophysiological Properties, on the Incidence of Arrhythmias in Patients with Non ST-Segment Elevation Acute Coronary Syndrome: Results from the Metabolic Efficiency With Ranolazine for Less Ischemia in Non ST-Elevation Acute Coronary Syndrome Thrombolysis in Myocardial Infarction 36 (MERLIN-TIMI 36) Randomized Controlled Trial. Circulation 2007, 116, 1647–1652. [Google Scholar] [CrossRef]
- Banerjee, K.; Ghosh, R.K.; Kamatam, S.; Banerjee, A.; Gupta, A. Role of Ranolazine in Cardiovascular Disease and Diabetes: Exploring beyond Angina. Int. J. Cardiol. 2017, 227, 556–564. [Google Scholar] [CrossRef]
- Morrow, D.A.; Scirica, B.M.; Sabatine, M.S.; de Lemos, J.A.; Murphy, S.A.; Jarolim, P.; Theroux, P.; Bode, C.; Braunwald, E. B-Type Natriuretic Peptide and the Effect of Ranolazine in Patients with Non-ST-Segment Elevation Acute Coronary Syndromes: Observations from the MERLIN-TIMI 36 (Metabolic Efficiency with Ranolazine for Less Ischemia in Non-ST Elevation Acute Coronary-Thrombolysis in Myocardial Infarction 36) Trial. J. Am. Coll. Cardiol. 2010, 55, 1189–1196. [Google Scholar] [CrossRef]
- Aldakkak, M.; Camara, A.K.S.; Heisner, J.S.; Yang, M.; Stowe, D.F. Ranolazine Reduces Ca2+ Overload and Oxidative Stress and Improves Mitochondrial Integrity to Protect against Ischemia Reperfusion Injury in Isolated Hearts. Pharmacol. Res. 2011, 64, 381–392. [Google Scholar] [CrossRef]
- Grempler, R.; Thomas, L.; Eckhardt, M.; Himmelsbach, F.; Sauer, A.; Sharp, D.E.; Bakker, R.A.; Mark, M.; Klein, T.; Eickelmann, P. Empagliflozin, a Novel Selective Sodium Glucose Cotransporter-2 (SGLT-2) Inhibitor: Characterisation and Comparison with Other SGLT-2 Inhibitors. Diabetes Obes. Metab. 2012, 14, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Baartscheer, A.; Schumacher, C.A.; van Borren, M.M.G.J.; Belterman, C.N.W.; Coronel, R.; Fiolet, J.W.T. Increased Na+/H+-Exchange Activity Is the Cause of Increased [Na+]i and Underlies Disturbed Calcium Handling in the Rabbit Pressure and Volume Overload Heart Failure Model. Cardiovasc. Res. 2003, 57, 1015–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhalla, A.K.; Hill, M.F.; Singal, P.K. Role of Oxidative Stress in Transition of Hypertrophy to Heart Failure. J. Am. Coll. Cardiol. 1996, 28, 506–514. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Bauersachs, J.; Galuppo, P.; Fraccarollo, D.; Christ, M.; Ertl, G. Improvement of Left Ventricular Remodeling and Function by Hydroxymethylglutaryl Coenzyme a Reductase Inhibition with Cerivastatin in Rats with Heart Failure after Myocardial Infarction. Circulation 2001, 104, 982–985. [Google Scholar] [CrossRef] [PubMed]
- Hayashidani, S.; Tsutsui, H.; Shiomi, T.; Suematsu, N.; Kinugawa, S.; Ide, T.; Wen, J.; Takeshita, A. Fluvastatin, a 3-Hydroxy-3-Methylglutaryl Coenzyme a Reductase Inhibitor, Attenuates Left Ventricular Remodeling and Failure after Experimental Myocardial Infarction. Circulation 2002, 105, 868–873. [Google Scholar] [CrossRef]
- Kjekshus, J.; Apetrei, E.; Barrios, V.; Böhm, M.; Cleland, J.G.F.; Cornel, J.H.; Dunselman, P.; Fonseca, C.; Goudev, A.; Grande, P.; et al. Rosuvastatin in Older Patients with Systolic Heart Failure. N. Engl. J. Med. 2007, 357, 2248–2261. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, P.W.; Pieske, B.; Anstrom, K.J.; Ezekowitz, J.; Hernandez, A.F.; Butler, J.; Lam, C.S.P.; Ponikowski, P.; Voors, A.A.; Jia, G.; et al. Vericiguat in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2020, 382, 1883–1893. [Google Scholar] [CrossRef]
- Chiles, R.; Al-Horani, R.A. Vericiguat: A New Hope for Heart Failure Patients. Cardiovasc. Ther. 2022, 2022, 1554875. [Google Scholar] [CrossRef]
- Xia, J.; Hui, N.; Tian, L.; Liang, C.; Zhang, J.; Liu, J.; Wang, J.; Ren, X.; Xie, X.; Wang, K. Development of Vericiguat: The First Soluble Guanylate Cyclase (SGC) Stimulator Launched for Heart Failure with Reduced Ejection Fraction (HFrEF). Biomed. Pharmacother. 2022, 149, 112894. [Google Scholar] [CrossRef]
- Shaikh, T.G.; Jawed, S.; Rahmat, Z.S.; Ahmed, S.H.; Waseem, S.; Ullah, I.; Irfan, M.; Asghar, M.S. Efficacy and Safety of Vericiguat for Treatment of Heart Failure: A Systematic Review. Curr. Probl. Cardiol. 2023, 101586, ahead-of-print. [Google Scholar] [CrossRef]
- Ghezzi, D.; Baruffini, E.; Haack, T.B.; Invernizzi, F.; Melchionda, L.; Dallabona, C.; Strom, T.M.; Parini, R.; Burlina, A.B.; Meitinger, T.; et al. Mutations of the Mitochondrial-TRNA Modifier MTO1 Cause Hypertrophic Cardiomyopathy and Lactic Acidosis. Am. J. Hum. Genet. 2012, 90, 1079–1087. [Google Scholar] [CrossRef] [Green Version]
- Sengers, R.C.A.; Trijbels, J.M.F.; Willems, J.L.; Daniels, O.; Stadhouders, A.M. Congenital Cataract and Mitochondrial Myopathy of Skeletal and Heart Muscle Associated with Lactic Acidosis after Exercise. J. Pediatr. 1975, 86, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Bakker, T.C.M. Positive Genetic Correlation between Female Preference and Preferred Male Ornament in Sticklebacks. Nature 1993, 363, 255–257. [Google Scholar] [CrossRef]
- Wortmann, S.B.; Champion, M.P.; van den Heuvel, L.; Barth, H.; Trutnau, B.; Craig, K.; Lammens, M.; Schreuder, M.F.; Taylor, R.W.; Smeitink, J.A.M.; et al. Mitochondrial DNA m.3242G > A Mutation, an under Diagnosed Cause of Hypertrophic Cardiomyopathy and Renal Tubular Dysfunction? Eur. J. Med. Genet. 2012, 55, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Viscomi, C.; Zeviani, M. MtDNA-Maintenance Defects: Syndromes and Genes. J. Inherit. Metab. Dis. 2017, 40, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Barth, P.G.; Scholte, H.R.; Berden, J.A.; Van Der Klei-Van Moorsel, J.M.; Luyt-Houwen, I.E.M.; Van’T Veer-Korthof, E.T.; Van Der Harten, J.J.; Sobotka-Plojhar, M.A. An X-Linked Mitochondrial Disease Affecting Cardiac Muscle, Skeletal Muscle and Neutrophil Leucocytes. J. Neurol. Sci. 1983, 62, 327–355. [Google Scholar] [CrossRef]
- Mayr, J.A.; Haack, T.B.; Graf, E.; Zimmermann, F.A.; Wieland, T.; Haberberger, B.; Superti-Furga, A.; Kirschner, J.; Steinmann, B.; Baumgartner, M.R.; et al. Lack of the Mitochondrial Protein Acylglycerol Kinase Causes Sengers Syndrome. Am. J. Hum. Genet. 2012, 90, 314–320. [Google Scholar] [CrossRef]
- Khademhosseini, A.; Langer, R. A Decade of Progress in Tissue Engineering. Nat. Protoc. 2016, 11, 1775–1781. [Google Scholar] [CrossRef]
- Wang, X.; Hu, G. Human Embryos in a Dish—Modeling Early Embryonic Development with Pluripotent Stem Cells. Cell Regen. 2022, 11, 4. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Branco, M.A.; Dias, T.P.; Cabral, J.M.S.; Pinto-do-Ó, P.; Diogo, M.M. Human Multilineage Pro-Epicardium/Foregut Organoids Support the Development of an Epicardium/Myocardium Organoid. Nat. Commun. 2022, 13, 6981. [Google Scholar] [CrossRef] [PubMed]
- Tenreiro, M.F.; Louro, A.F.; Alves, P.M.; Serra, M. Next Generation of Heart Regenerative Therapies: Progress and Promise of Cardiac Tissue Engineering. NPJ Regen. Med. 2021, 6, 30. [Google Scholar] [CrossRef] [PubMed]
- Final Report Summary—INDIVUHEART (Individualized Early Risk Assessment for Heart Diseases)|FP7|CORDIS|European Commission. Available online: https://cordis.europa.eu/project/id/340248/reporting (accessed on 24 January 2023).
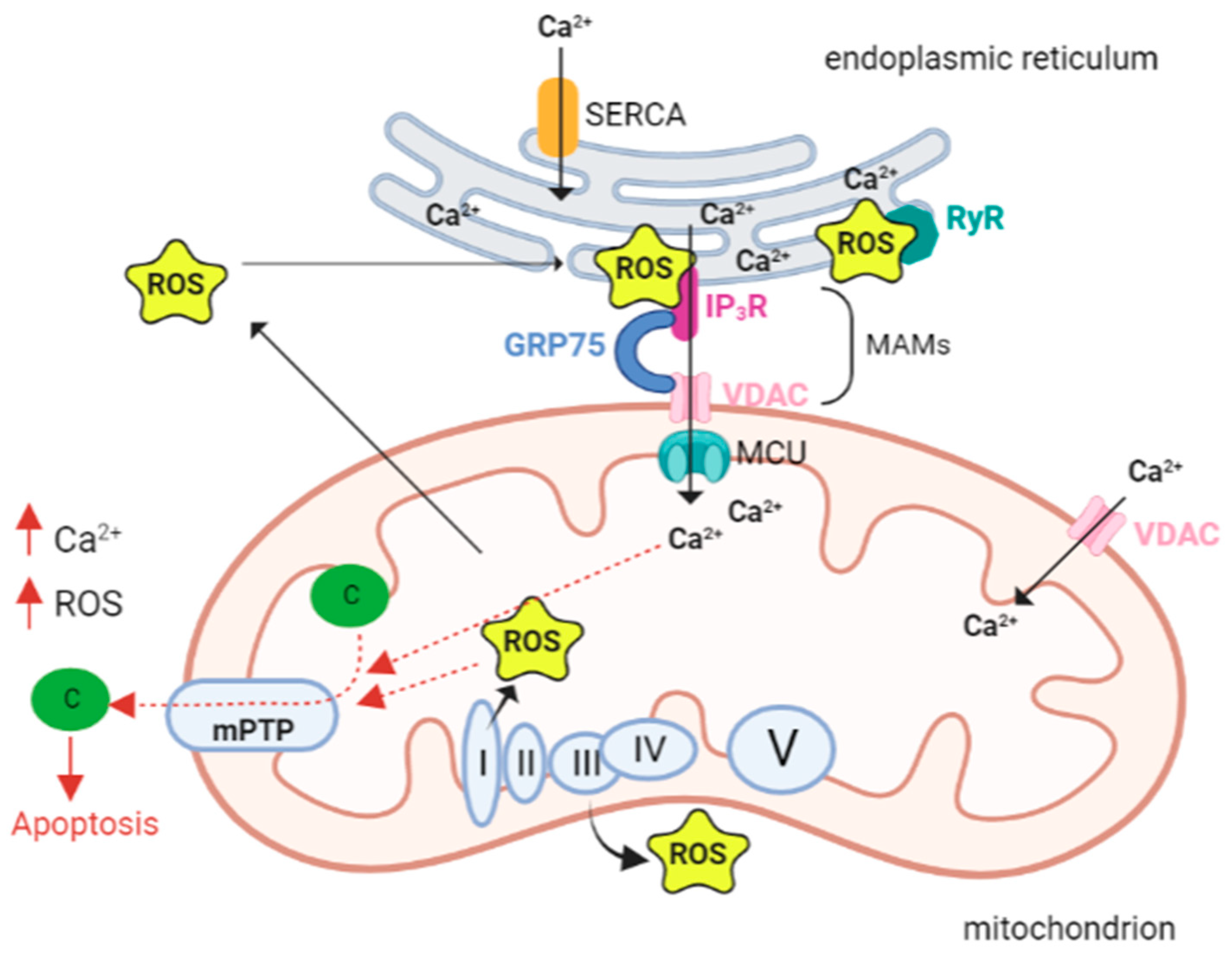

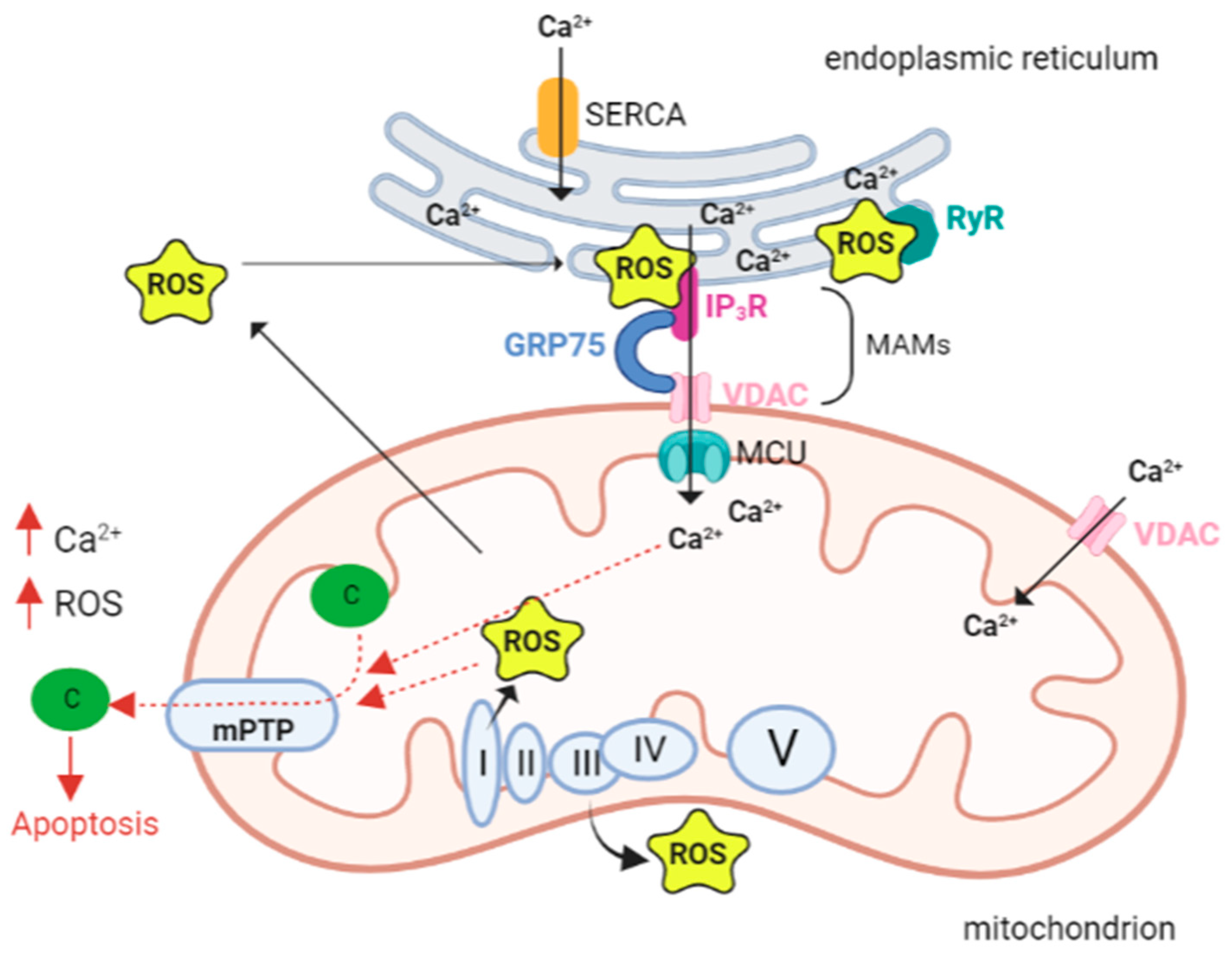{kind=link}
| Chromosome Location | Phenotype | Phenotype MIM Number | Inheritance | Gene/Locus | Gene/Locus MIM Number | References |
|---|---|---|---|---|---|---|
| 6q13 | Combined oxidative phosphorylation deficiency 10 | 614702 | AR | MTO1 | 614667 | [151] |
| 7q34 | Sengers syndrome | 212350 | AR | AGK | 610345 | [152] |
| 4q35.1 | Mitochondrial DNA depletion syndrome 12B (cardiomyopathic type) | 615418 | AR | SLC25A4 | 103220 | [153] |
| Mt | Hypertrophic cardiomyopathy with kidney anomalies due to mtDNA mutations | Mitochondrial inheritance | MT-TL1 | [154] | ||
| mt | Mitochondrial DNA- related cardiomyopathy and hearing loss | Mitochondrial inheritance | MT-TK | [155] | ||
| Xq28 | Barth syndrome | 302060 | XLR | TAFAZZIN | 300394 | [156] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Nicolo, B.; Cataldi-Stagetti, E.; Diquigiovanni, C.; Bonora, E. Calcium and Reactive Oxygen Species Signaling Interplays in Cardiac Physiology and Pathologies. Antioxidants 2023, 12, 353. https://doi.org/10.3390/antiox12020353
De Nicolo B, Cataldi-Stagetti E, Diquigiovanni C, Bonora E. Calcium and Reactive Oxygen Species Signaling Interplays in Cardiac Physiology and Pathologies. Antioxidants. 2023; 12(2):353. https://doi.org/10.3390/antiox12020353
Chicago/Turabian StyleDe Nicolo, Bianca, Erica Cataldi-Stagetti, Chiara Diquigiovanni, and Elena Bonora. 2023. "Calcium and Reactive Oxygen Species Signaling Interplays in Cardiac Physiology and Pathologies" Antioxidants 12, no. 2: 353. https://doi.org/10.3390/antiox12020353
APA StyleDe Nicolo, B., Cataldi-Stagetti, E., Diquigiovanni, C., & Bonora, E. (2023). Calcium and Reactive Oxygen Species Signaling Interplays in Cardiac Physiology and Pathologies. Antioxidants, 12(2), 353. https://doi.org/10.3390/antiox12020353