
Phosphorylation of the Human DNA Glycosylase NEIL2 Is Affected by Oxidative Stress and Modulates Its Activity

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Stably Transfected Cell Lines
2.2. Metabolic Labeling with [32P]Orthophosphate
2.3. Immunoblotting Analysis
2.4. Bioinformatics
2.5. Cloning and Purification of NEIL2
2.6. In Vitro Kinase Assays
2.7. Radiolabeling and Annealing of Oligonucleotides
2.8. In Vitro DNA Repair Activity Assays
2.9. Proximity Ligation Assay
2.10. Statistics
3. Results
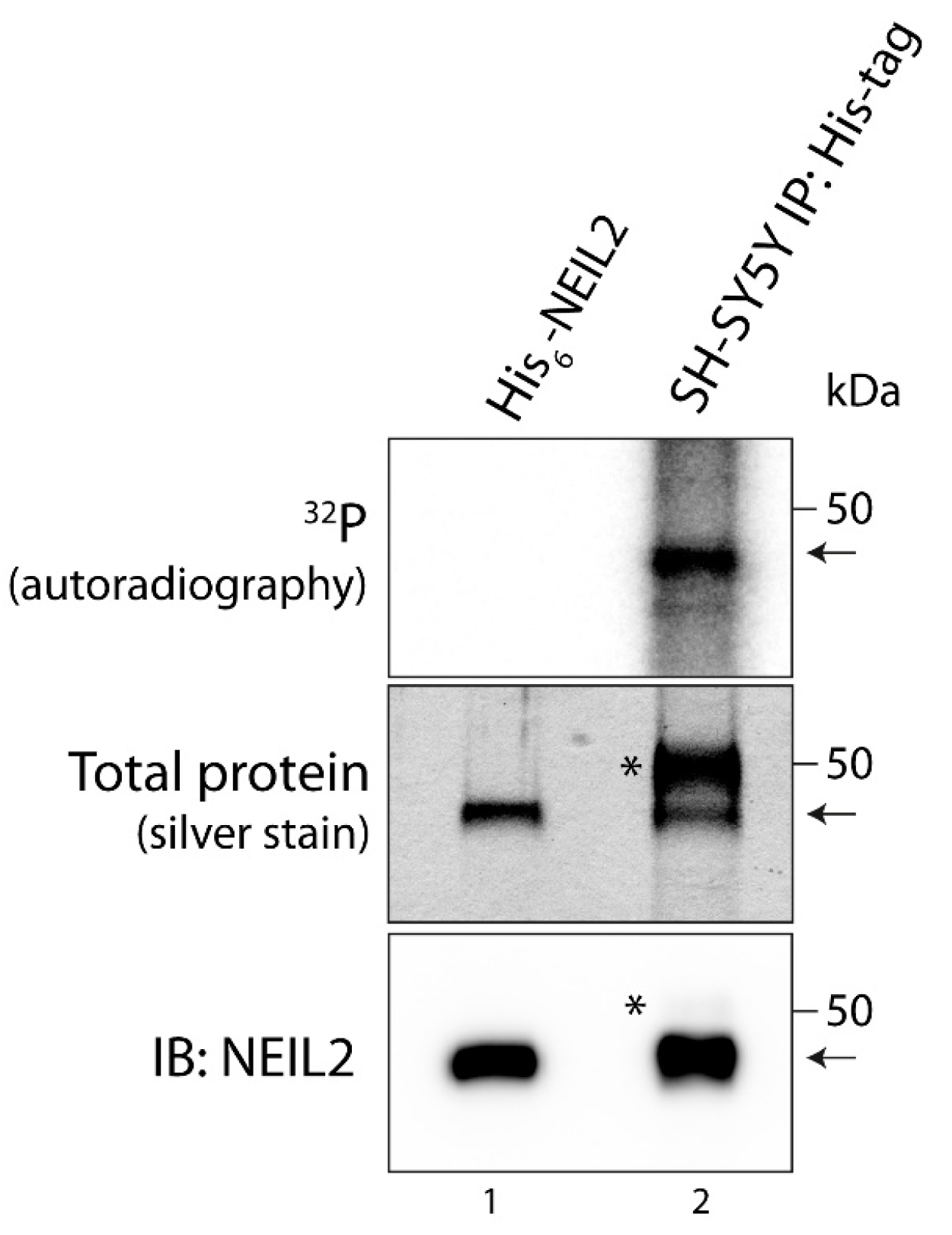
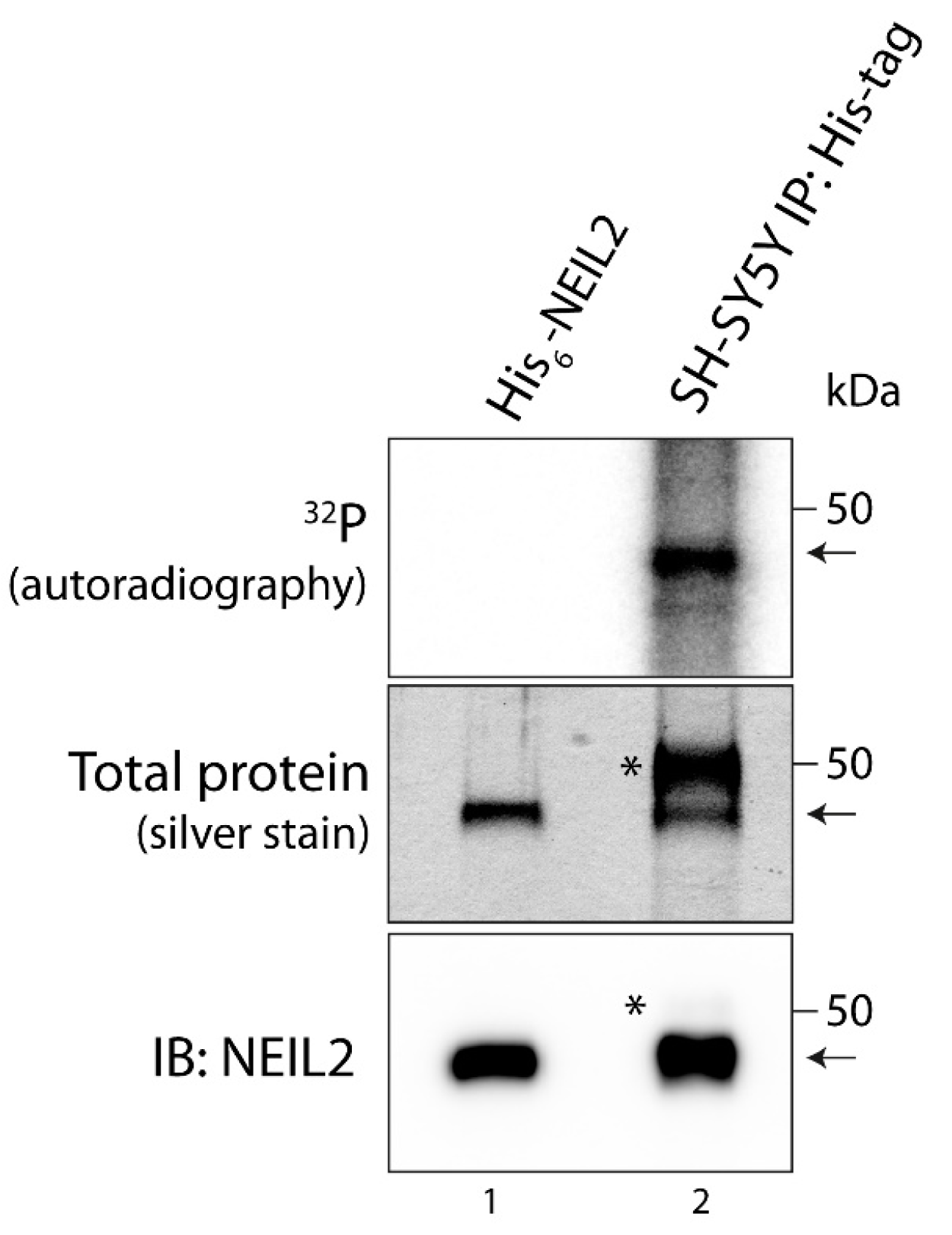
3.1. NEIL2 Is Phosphorylated in SH-SY5Y Cells
3.2. In Silico Prediction of Responsible Kinases
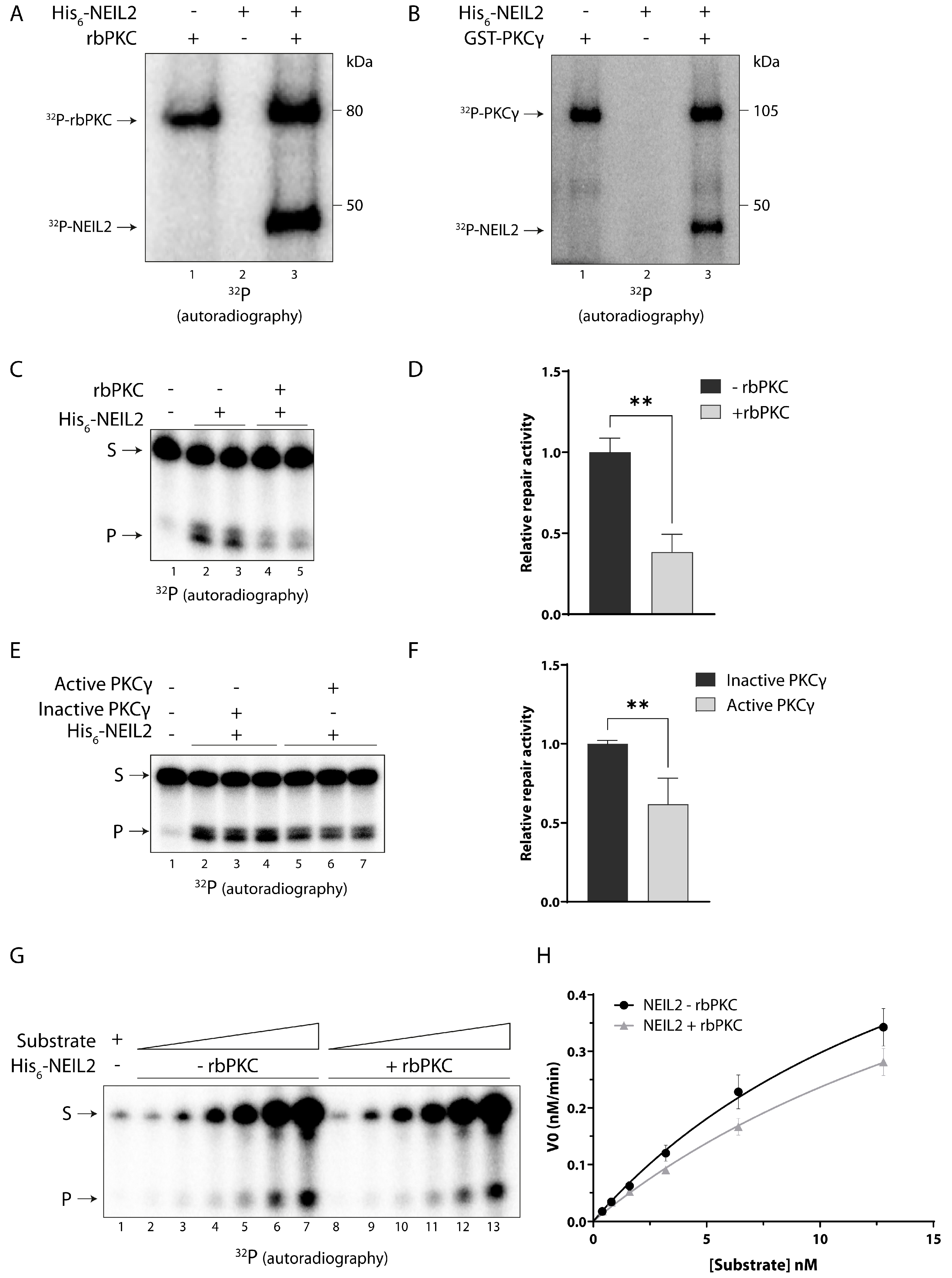
3.3. NEIL2 Is Phosphorylated by PKC and Phosphorylation Decreases the Repair Activity of NEIL2 In Vitro
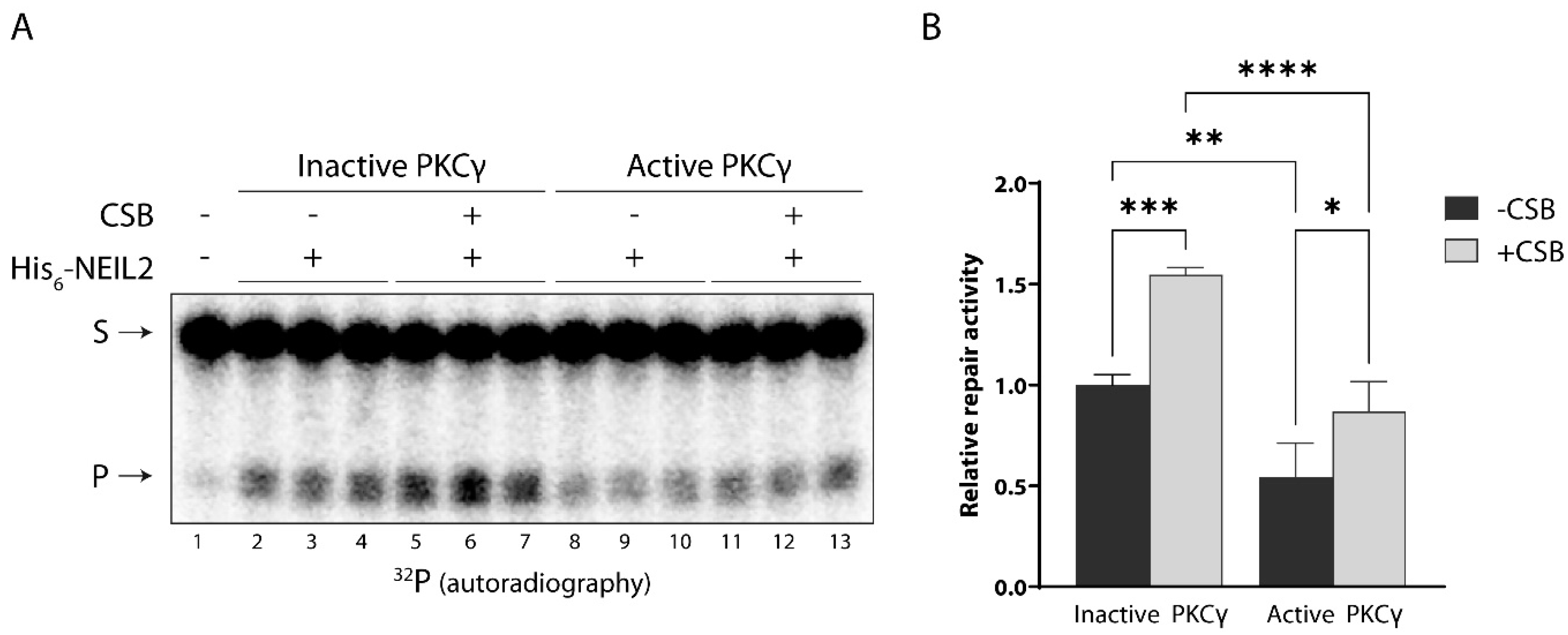
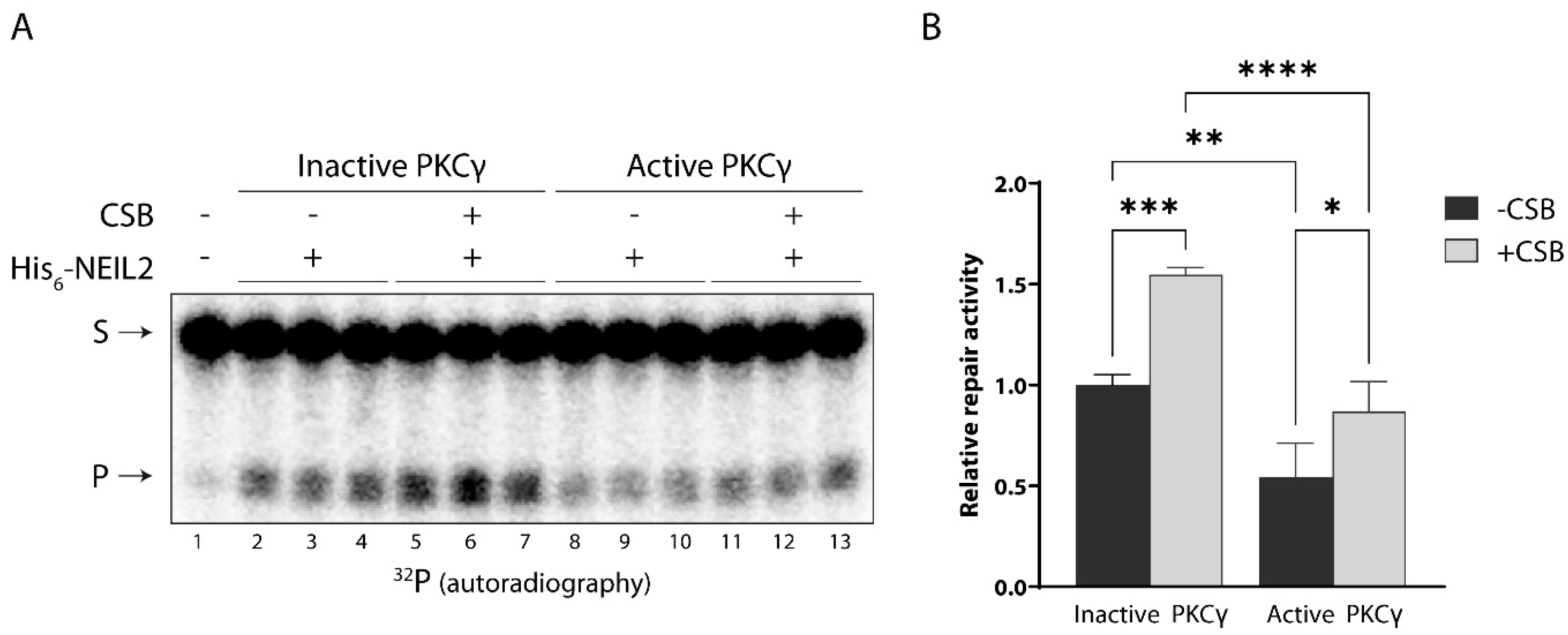
3.4. CSB Stimulates NEIL2 Activity Similarly in the Presence and Absence of PKC Phosphorylation In Vitro
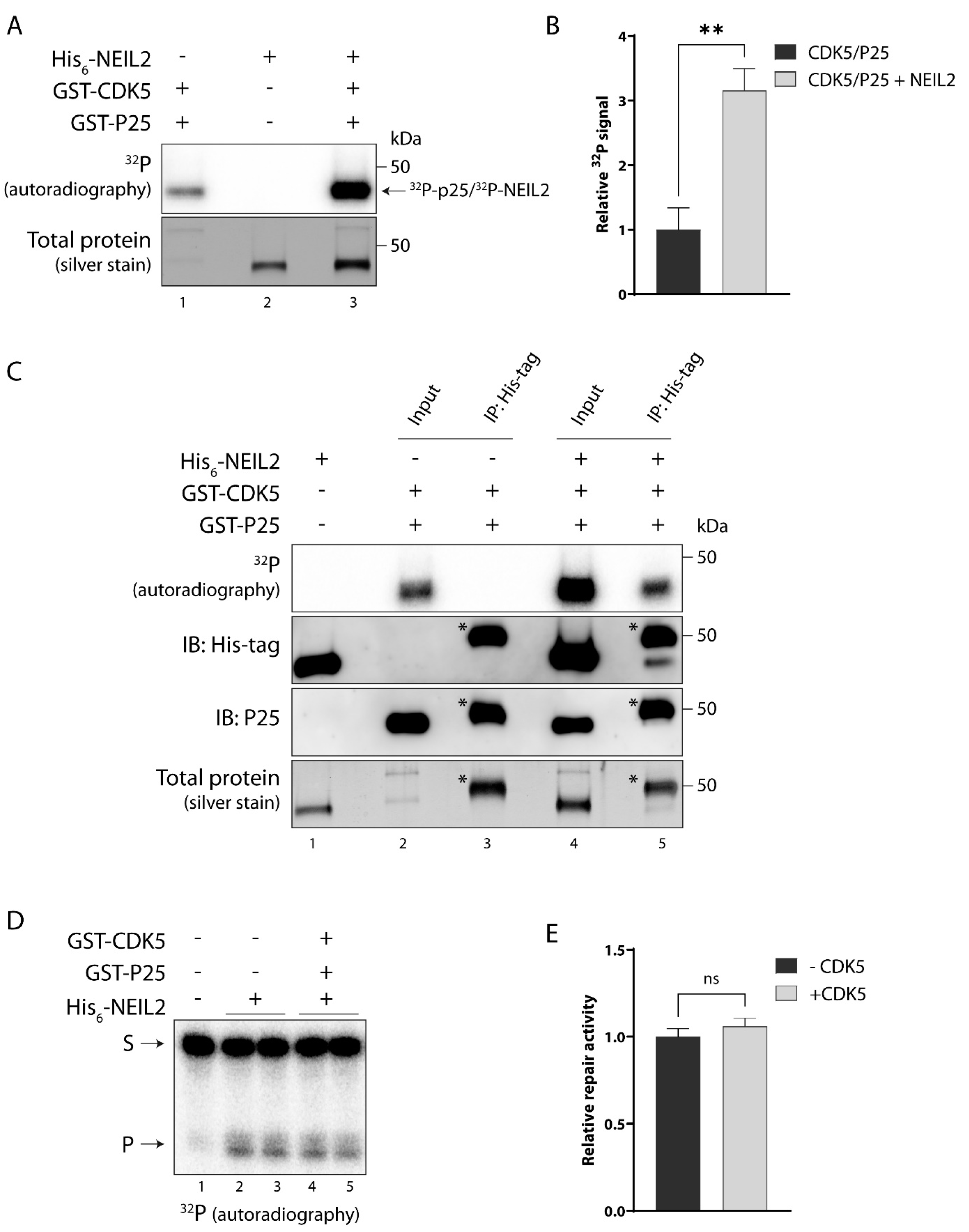
3.5. NEIL2 Is Phosphorylated by CDK5 In Vitro, but It Does Not Affect NEIL2 Activity
3.6. NEIL2 Interacts with and Is Phosphorylated by PKC and CDK5 in SH-SY5Y Cells
3.7. Oxidative Stress Induces Dephosphorylation of NEIL2 in SH-SY5Y Cells
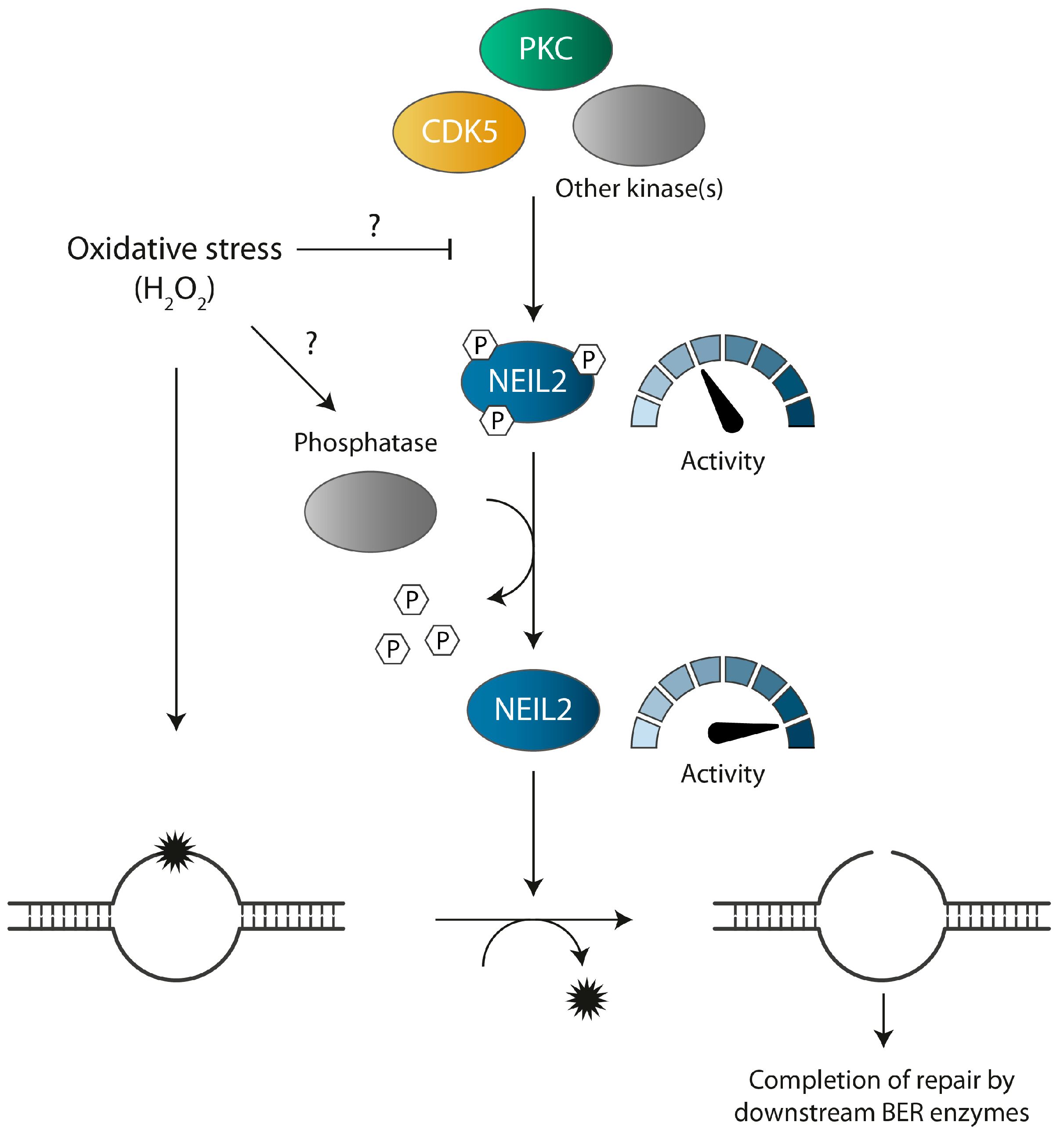
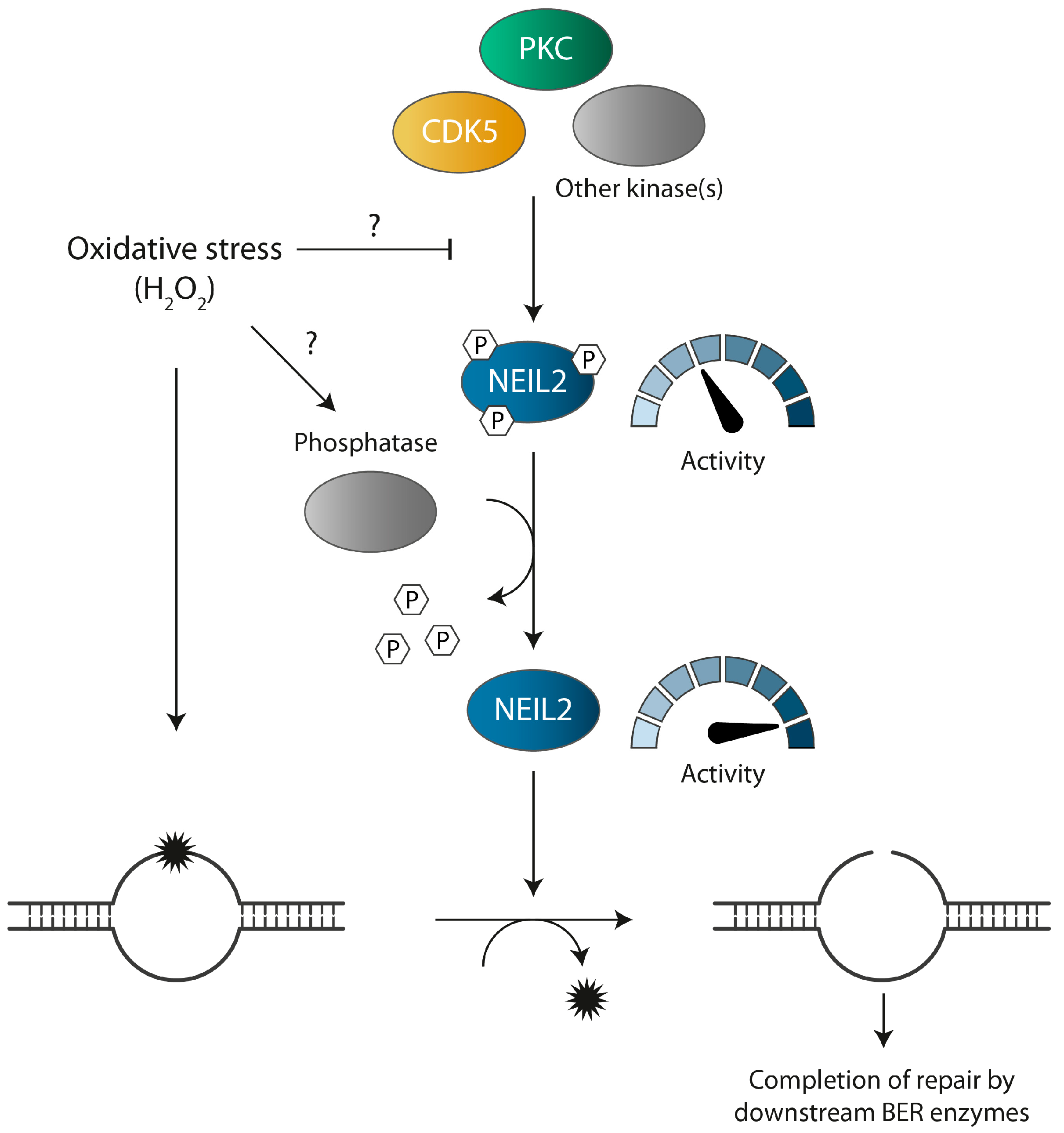
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Englander, E.W.; Ma, H. Differential modulation of base excision repair activities during brain ontogeny: Implications for repair of transcribed DNA. Mech. Ageing Dev. 2006, 127, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Rolseth, V.; Rundén-Pran, E.; Luna, L.; McMurray, C.; Bjørås, M.; Ottersen, O.P. Widespread distribution of DNA glycosylases removing oxidative DNA lesions in human and rodent brains. DNA Repair 2008, 7, 1578–1588. [Google Scholar] [CrossRef] [PubMed]
- Sykora, P.; Wilson, D.M., 3rd; Bohr, V.A. Base excision repair in the mammalian brain: Implication for age related neurodegeneration. Mech. Ageing Dev. 2013, 134, 440–448. [Google Scholar] [CrossRef]
- Wang, H.; Dharmalingam, P.; Vasquez, V.; Mitra, J.; Boldogh, I.; Rao, K.S.; Kent, T.A.; Mitra, S.; Hegde, M.L. Chronic oxidative damage together with genome repair deficiency in the neurons is a double whammy for neurodegeneration: Is damage response signaling a potential therapeutic target? Mech. Ageing Dev. 2017, 161 Pt A, 163–176. [Google Scholar] [CrossRef]
- Grin, I.R.; Zharkov, D.O. Eukaryotic endonuclease VIII-like proteins: New components of the base excision DNA repair system. Biochem. (Mosc.) 2011, 76, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Bandaru, V.; Bond, J.P.; Jaruga, P.; Zhao, X.; Christov, P.P.; Burrows, C.J.; Rizzo, C.J.; Dizdaroglu, M.; Wallace, S.S. The mouse ortholog of NEIL3 is a functional DNA glycosylase in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 4925–4930. [Google Scholar] [CrossRef] [PubMed]
- Dou, H.; Mitra, S.; Hazra, T.K. Repair of Oxidized Bases in DNA Bubble Structures by Human DNA Glycosylases NEIL1 and NEIL2. J. Biol. Chem. 2003, 278, 49679–49684. [Google Scholar] [CrossRef] [PubMed]
- Hazra, T.K.; Kow, Y.W.; Hatahet, Z.; Imhoff, B.; Boldogh, I.; Mokkapati, S.K.; Mitra, S.; Izumi, T. Identification and Characterization of a Novel Human DNA Glycosylase for Repair of Cytosine-derived Lesions. J. Biol. Chem. 2002, 277, 30417–30420. [Google Scholar] [CrossRef]
- Hailer, M.K.; Slade, P.G.; Martin, B.D.; Rosenquist, T.A.; Sugden, K.D. Recognition of the oxidized lesions spiroiminodihydantoin and guanidinohydantoin in DNA by the mammalian base excision repair glycosylases NEIL1 and NEIL2. DNA Repair 2005, 4, 41–50. [Google Scholar] [CrossRef]
- Makasheva, K.; Endutkin, A.V.; Zharkov, D.O. Requirements for DNA bubble structure for efficient cleavage by helix–two-turn–helix DNA glycosylases. Mutagenesis 2019, 35, 119–128. [Google Scholar] [CrossRef]
- Hazra, T.K.; Izumi, T.; Boldogh, I.; Imhoff, B.; Kow, Y.W.; Jaruga, P.; Dizdaroglu, M.; Mitra, S. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc. Natl. Acad. Sci. USA 2002, 99, 3523–3528. [Google Scholar] [CrossRef] [PubMed]
- Bacolla, A.; Sengupta, S.; Ye, Z.; Yang, C.; Mitra, J.; De-Paula, R.B.; Hegde, M.L.; Ahmed, Z.; Mort, M.; Cooper, D.N.; et al. Heritable pattern of oxidized DNA base repair coincides with pre-targeting of repair complexes to open chromatin. Nucleic Acids Res. 2020, 49, 221–243. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.L.; Hegde, P.M.; Bellot, L.J.; Mandal, S.M.; Hazra, T.K.; Li, G.-M.; Boldogh, I.; Tomkinson, A.E.; Mitra, S. Prereplicative repair of oxidized bases in the human genome is mediated by NEIL1 DNA glycosylase together with replication proteins. Proc. Natl. Acad. Sci. USA 2013, 110, E3090–E3099. [Google Scholar] [CrossRef]
- Dou, H.; Theriot, C.A.; Das, A.; Hegde, M.L.; Matsumoto, Y.; Boldogh, I.; Hazra, T.K.; Bhakat, K.K.; Mitra, S. Interaction of the Human DNA Glycosylase NEIL1 with Proliferating Cell Nuclear Antigen. J. Biol. Chem. 2008, 283, 3130–3140. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Wakamiya, M.; Venkova-Canova, T.; Pandita, R.K.; Aguilera-Aguirre, L.; Sarker, A.H.; Singh, D.K.; Hosoki, K.; Wood, T.G. Neil2-null Mice Accumulate Oxidized DNA Bases in the Transcriptionally Active Sequences of the Genome and Are Susceptible to Innate Inflammation. J. Biol. Chem. 2015, 290, 24636–24648. [Google Scholar] [CrossRef]
- Banerjee, D.; Mandal, S.M.; Das, A.; Hegde, M.L.; Das, S.; Bhakat, K.K.; Boldogh, I.; Sarkar, P.S.; Mitra, S.; Hazra, T.K. Preferential Repair of Oxidized Base Damage in the Transcribed Genes of Mammalian Cells. J. Biol. Chem. 2011, 286, 6006–6016. [Google Scholar] [CrossRef]
- Eckenroth, B.E.; Cao, V.B.; Averill, A.M.; Dragon, J.A.; Doublié, S. Unique Structural Features of Mammalian NEIL2 DNA Glycosylase Prime Its Activity for Diverse DNA Substrates and Environments. Structure 2020, 29, 29–42.e4. [Google Scholar] [CrossRef]
- Hegde, M.L.; Izumi, T.; Mitra, S. Oxidized base damage and single-strand break repair in mammalian genomes: Role of disordered regions and posttranslational modifications in early enzymes. Prog. Mol. Biol. Transl. Sci. 2012, 110, 123–153. [Google Scholar]
- Das, A.; Wiederhold, L.; Leppard, J.B.; Kedar, P.; Prasad, R.; Wang, H.; Boldogh, I.; Karimi-Busheri, F.; Weinfeld, M.; Tomkinson, A.E.; et al. NEIL2-initiated, APE-independent repair of oxidized bases in DNA: Evidence for a repair complex in human cells. DNA Repair 2006, 5, 1439–1448. [Google Scholar] [CrossRef]
- Aamann, M.D.; Hvitby, C.; Popuri, V.; Muftuoglu, M.; Lemminger, L.; Skeby, C.K.; Keijzers, G.; Ahn, B.; Bjørås, M.; Bohr, V.A.; et al. Cockayne Syndrome group B protein stimulates NEIL2 DNA glycosylase activity. Mech. Ageing Dev. 2014, 135, 1–14. [Google Scholar] [CrossRef]
- Mandal, S.M.; Hegde, M.L.; Chatterjee, A.; Hegde, P.M.; Szczesny, B.; Banerjee, D.; Boldogh, I.; Gao, R.; Falkenberg, M.; Gustafsson, C.M.; et al. Role of Human DNA Glycosylase Nei-like 2 (NEIL2) and Single Strand Break Repair Protein Polynucleotide Kinase 3′-Phosphatase in Maintenance of Mitochondrial Genome. J. Biol. Chem. 2012, 287, 2819–2829. [Google Scholar] [CrossRef] [Green Version]
- Schomacher, L.; Han, D.; Musheev, M.U.; Arab, K.; Kienhöfer, S.; von Seggern, A.; Niehrs, C. Neil DNA glycosylases promote substrate turnover by Tdg during DNA demethylation. Nat. Struct. Mol. Biol. 2016, 23, 116–124. [Google Scholar] [CrossRef]
- Müller, U.; Bauer, C.; Siegl, M.; Rottach, A.; Leonhardt, H. TET-mediated oxidation of methylcytosine causes TDG or NEIL glycosylase dependent gene reactivation. Nucleic Acids Res. 2014, 42, 8592–8604. [Google Scholar] [CrossRef]
- Tapryal, N.; Shahabi, S.; Chakraborty, A.; Hosoki, K.; Wakamiya, M.; Sarkar, G.; Sharma, G.; Cardenas, V.J.; Boldogh, I.; Sur, S.; et al. Intrapulmonary administration of purified NEIL2 abrogates NF-κB-mediated inflammation. J. Biol. Chem. 2021, 296, 100723. [Google Scholar] [CrossRef] [PubMed]
- Scheffler, K.; Jalland, C.M.; Benestad, S.L.; Moldal, T.; Ersdal, C.; Gunnes, G.; Suganthan, R.; Bjørås, M.; Tranulis, M.A. DNA glycosylase Neil2 contributes to genomic responses in the spleen during clinical prion disease. Free Radic. Biol. Med. 2020, 152, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Sayed, I.; Chakraborty, A.; El-Hafeez, A.A.; Sharma, A.; Sahan, A.; Huang, W.; Sahoo, D.; Ghosh, P.; Hazra, T.; Das, S. The DNA Glycosylase NEIL2 Suppresses Fusobacterium-Infection-Induced Inflammation and DNA Damage in Colonic Epithelial Cells. Cells 2020, 9, 1980. [Google Scholar] [CrossRef]
- Carter, R.J.; Parsons, J.L. Base Excision Repair, a Pathway Regulated by Posttranslational Modifications. Mol. Cell Biol. 2016, 36, 1426–1437. [Google Scholar] [CrossRef]
- Limpose, K.L.; Corbett, A.H.; Doetsch, P.W. BERing the burden of damage: Pathway crosstalk and posttranslational modification of base excision repair proteins regulate DNA damage management. DNA Repair 2017, 56, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.; Cao, V.B.; Doublié, S. Phosphorylation Sites Identified in the NEIL1 DNA Glycosylase Are Potential Targets for the JNK1 Kinase. PLoS ONE 2016, 11, e0157860. [Google Scholar] [CrossRef]
- Sengupta, S.; Yang, C.; Hegde, M.L.; Hegde, P.M.; Mitra, J.; Pandey, A.; Dutta, A.; Datarwala, A.T.; Bhakat, K.K.; Mitra, S. Acetylation of oxidized base repair-initiating NEIL1 DNA glycosylase required for chromatin-bound repair complex formation in the human genome increases cellular resistance to oxidative stress. DNA Repair 2018, 66–67, 1–10. [Google Scholar] [CrossRef]
- Edmonds, M.J.; Carter, R.J.; Nickson, C.M.; Williams, S.C.; Parsons, J.L. Ubiquitylation-dependent regulation of NEIL1 by Mule and TRIM26 is required for the cellular DNA damage response. Nucleic Acids Res. 2016, 45, 726–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhakat, K.K.; Hazra, T.K.; Mitra, S. Acetylation of the human DNA glycosylase NEIL2 and inhibition of its activity. Nucleic Acids Res. 2004, 32, 3033–3039. [Google Scholar] [CrossRef] [PubMed]
- Blom, N.S.; Gammeltoft, S.; Brunak, S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 1999, 294, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Blom, N.; Sicheritz-Pontén, T.; Gupta, R.; Gammeltoft, S.; Brunak, S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 2004, 4, 1633–1649. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2014, 43, D512–D520. [Google Scholar] [CrossRef]
- Christiansen, M.; Stevnsner, T.; Modin, C.; Martensen, P.M.; Brosh, R.M., Jr.; Bohr, V.A. Functional consequences of mutations in the conserved SF2 motifs and post-translational phosphorylation of the CSB protein. Nucleic Acids Res. 2003, 31, 963–973. [Google Scholar] [CrossRef]
- Xie, H.R.; Hu, L.; Li, G. SH-SY5Y human neuroblastoma cell line: In vitro cell model of dopaminergic neurons in Parkinson’s disease. Chin. Med. J. (Engl.) 2010, 123, 1086–1092. [Google Scholar] [PubMed]
- Kovalevich, J.; Langford, D. Considerations for the use of SH-SY5Y neuroblastoma cells in neurobiology. Methods Mol. Biol. 2013, 1078, 9–21. [Google Scholar] [CrossRef]
- Hegde, M.L.; Hegde, P.M.; Holthauzen, L.M.; Hazra, T.K.; Rao, K.J.; Mitra, S. Specific Inhibition of NEIL-initiated repair of oxidized base damage in human genome by copper and iron: Potential etiological linkage to neurodegenerative diseases. J. Biol. Chem. 2010, 285, 28812–28825. [Google Scholar] [CrossRef]
- Jämsä, A.; Agerman, K.; Radesäter, A.-C.; Ottervald, J.; Malmström, J.; Hiller, G.; Liu, G.; Vasänge, M. Identification of non-muscle myosin heavy chain as a substrate for Cdk5 and tool for drug screening. J. Biomed. Sci. 2009, 16, 55. [Google Scholar] [CrossRef]
- Leli, U.; Shea, T.B.; Cataldo, A.; Hauser, G.; Grynspan, F.; Beermann, M.L.; Liepkalns, V.A.; Nixon, R.A.; Parker, P.J. Differential expression and subcellular localization of protein kinase C alpha, beta, gamma, delta, and epsilon isoforms in SH-SY5Y neuroblastoma cells: Modifications during differentiation. J. Neurochem. 1993, 60, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Attoff, K.; Johansson, Y.; Cediel-Ulloa, A.; Lundqvist, J.; Gupta, R.; Caiment, F.; Gliga, A.; Forsby, A. Acrylamide alters CREB and retinoic acid signalling pathways during differentiation of the human neuroblastoma SH-SY5Y cell line. Sci. Rep. 2020, 10, 16714. [Google Scholar] [CrossRef]
- Steinberg, S.F. Structural Basis of Protein Kinase C Isoform Function. Physiol. Rev. 2008, 88, 1341–1378. [Google Scholar] [CrossRef]
- Freschi, L.; Osseni, M.; Landry, C.R. Functional Divergence and Evolutionary Turnover in Mammalian Phosphoproteomes. PLOS Genet. 2014, 10, e10040622014. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.; Kusalik, A.; Napper, S. Computational Analysis of the Predicted Evolutionary Conservation of Human Phosphorylation Sites. PLoS ONE 2016, 11, e0152809. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Schomacher, L.; Schüle, K.M.; Mallick, M.; Musheev, M.U.; Karaulanov, E.; Krebs, L.; von Seggern, A.; Niehrs, C. NEIL1 and NEIL2 DNA glycosylases protect neural crest development against mitochondrial oxidative stress. eLife 2019, 8, e49044. [Google Scholar] [CrossRef] [PubMed]
- Dhavan, R.; Tsai, L. A decade of CDK5. Nat. Rev. Mol. Cell Biol. 2001, 2, 749–759. [Google Scholar] [CrossRef]
- Stuart, S.A.; Houel, S.; Lee, T.; Wang, N.; Old, W.M.; Ahn, N.G. A Phosphoproteomic Comparison of B-RAFV600E and MKK1/2 Inhibitors in Melanoma Cells. Mol. Cell Proteom. 2015, 14, 1599–1615. [Google Scholar] [CrossRef]
- Bian, Y.; Song, C.; Cheng, K.; Dong, M.; Wang, F.; Huang, J.; Sun, D.; Wang, L.; Ye, M.; Zou, H. An enzyme assisted RP-RPLC approach for in-depth analysis of human liver phosphoproteome. J. Proteom. 2014, 96, 253–262. [Google Scholar] [CrossRef]
- Zhou, H.; Di Palma, S.; Preisinger, C.; Peng, M.; Polat, A.N.; Heck, A.J.R.; Mohammed, S. Toward a Comprehensive Characterization of a Human Cancer Cell Phosphoproteome. J. Proteome Res. 2013, 12, 260–271. [Google Scholar] [CrossRef]
- Mertins, P.; Cptac, N.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beli, P.; Lukashchuk, N.; Wagner, S.A.; Weinert, B.T.; Olsen, J.V.; Baskcomb, L.; Mann, M.; Jackson, S.P.; Choudhary, C. Proteomic Investigations Reveal a Role for RNA Processing Factor THRAP3 in the DNA Damage Response. Mol. Cell 2012, 46, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Chilumuri, A.; Markiv, A.; Milton, N.G. Immunocytochemical staining of endogenous nuclear proteins with the HIS-1 anti-poly-histidine monoclonal antibody: A potential source of error in His-tagged protein detection. Acta Histochem. 2014, 116, 1022–1028. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, N.; Xie, J. An endogenous ‘non-specific’ protein detected by a His-tag antibody is human transcription regulator YY1. Data Brief 2015, 2, 52–55. [Google Scholar] [CrossRef] [PubMed]
- Meijer, L.; Borgne, A.; Mulner, O.; Chong, J.P.J.; Blow, J.J.; Inagaki, N.; Inagaki, M.; Delcros, J.-G.; Moulinoux, J.-P. Biochemical and Cellular Effects of Roscovitine, a Potent and Selective Inhibitor of the Cyclin-Dependent Kinases cdc2, cdk2 and cdk5. Eur. J. Biochem. 1997, 243, 527–536. [Google Scholar] [CrossRef]
- Bach, S.; Knockaert, M.; Reinhardt, J.; Lozach, O.; Schmitt, S.; Baratte, B.; Koken, M.; Coburn, S.P.; Tang, L.; Jiang, T.; et al. Roscovitine Targets, Protein Kinases and Pyridoxal Kinase. J. Biol. Chem. 2005, 280, 31208–31219. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.A.; Lee, H.C.; Lee, J.-J.; Hong, M.-N.; Park, M.-J.; Lee, Y.-S.; Choi, H.-D.; Kim, N.; Ko, Y.-G. Effects of combined radiofrequency radiation exposure on levels of reactive oxygen species in neuronal cells. J. Radiat. Res. 2013, 55, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Sykora, P.; Yang, J.-L.; Ferrarelli, L.K.; Tian, J.; Tadokoro, T.; Kulkarni, A.; Weissman, L.; Keijzers, G.; Wilson, D.M.; Mattson, M.P.; et al. Modulation of DNA base excision repair during neuronal differentiation. Neurobiol. Aging 2013, 34, 1717–1727. [Google Scholar] [CrossRef]
- Pregi, N.; Belluscio, L.M.; Berardino, B.G.; Castillo, D.S.; Cánepa, E.T. Oxidative stress-induced CREB upregulation promotes DNA damage repair prior to neuronal cell death protection. Mol. Cell. Biochem. 2016, 425, 9–24. [Google Scholar] [CrossRef] [PubMed]
- Yousefzadeh, M.; Henpita, C.; Vyas, R.; Soto-Palma, C.; Robbins, P.; Niedernhofer, L. DNA damage-how and why we age? Elife 2021, 10, e62852. [Google Scholar] [CrossRef]
- Almeida, K.H.; Sobol, R.W. A unified view of base excision repair: Lesion-dependent protein complexes regulated by post-translational modification. DNA Repair 2007, 6, 695–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Imam, S.Z.; Hashiguchi, K.; Souza-Pinto, N.; Bohr, V.A. Phosphorylation of human oxoguanine DNA glycosylase (-OGG1) modulates its function. Nucleic Acids Res. 2005, 33, 3271–3282. [Google Scholar] [CrossRef] [PubMed]
- Yacoub, A.; Kelley, M.R.; Deutsch, W.A. The DNA repair activity of human redox/repair protein APE/Ref-1 is inactivated by phosphorylation. Cancer Res. 1997, 57, 5457–5459. [Google Scholar] [PubMed]
- Huang, E.; Qu, D.; Zhang, Y.; Venderova, K.; Haque, M.E.; Rousseaux, M.W.; Slack, R.S.; Woulfe, J.M.; Park, D.S. The role of Cdk5-mediated apurinic/apyrimidinic endonuclease 1 phosphorylation in neuronal death. Nature 2010, 12, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, F.; Luna, L.; Bjørås, M.; Seeberg, E. Human OGG1 undergoes serine phosphorylation and associates with the nuclear matrix and mitotic chromatin in vivo. Nucleic Acids Res. 2002, 30, 2349–2357. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Chattopadhyay, R.; Bhakat, K.K.; Boldogh, I.; Kohno, K.; Prasad, R.; Wilson, S.H.; Hazra, T.K. Stimulation of NEIL2-mediated Oxidized Base Excision Repair via YB-1 Interaction during Oxidative Stress. J. Biol. Chem. 2007, 282, 28474–28484. [Google Scholar] [CrossRef]
- Parsons, J.; Khoronenkova, S.; Dianova, I.I.; Ternette, N.; Kessler, B.; Datta, P.K.; Dianov, G.L. Phosphorylation of PNKP by ATM prevents its proteasomal degradation and enhances resistance to oxidative stress. Nucleic Acids Res. 2012, 40, 11404–11415. [Google Scholar] [CrossRef]
- Dong, Z.; Tomkinson, A. ATM mediates oxidative stress-induced dephosphorylation of DNA ligase IIIα. Nucleic Acids Res. 2006, 34, 5721. [Google Scholar] [CrossRef]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., III; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef]
- Cicchillitti, L.; Fasanaro, P.; Biglioli, P.; Capogrossi, M.C.; Martelli, F. Oxidative Stress Induces Protein Phosphatase 2A-dependent Dephosphorylation of the Pocket Proteins pRb, p107, and p130. J. Biol. Chem. 2003, 278, 19509–19517. [Google Scholar] [CrossRef]
- Yamazaki, T.; Liu, L.; Manley, J.L. Oxidative stress induces Ser 2 dephosphorylation of the RNA polymerase II CTD and premature transcription termination. Transcription 2021, 12, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.-H.; de Pablo, Y.; Vincent, F.; Shah, K. Deregulated Cdk5 promotes oxidative stress and mitochondrial dysfunction. J. Neurochem. 2008, 107, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Strocchi, P.; Pession, A.; Dozza, B. Up-regulation of cDK5/p35 by oxidative stress in human neuroblastoma IMR-32 cells. J. Cell Biochem. 2003, 88, 758–765. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishna, R.; Jaken, S. Protein kinase C signaling and oxidative stress. Free Radic. Biol. Med. 2000, 28, 1349–1361. [Google Scholar] [CrossRef] [PubMed]
- Agarwal-Mawal, A.; Paudel, H. Neuronal Cdc2-like protein kinase (Cdk5/p25) is associated with protein phosphatase 1 and phosphorylates inhibitor-2. J. Biol. Chem. 2001, 276, 23712–23718. [Google Scholar] [CrossRef] [PubMed]
- Zambrano, C.A.; Egaña, J.T.; Núñez, M.T.; Maccioni, R.B.; González-Billault, C. Oxidative stress promotes τ dephosphorylation in neuronal cells: The roles of cdk5 and PP1. Free Radic. Biol. Med. 2004, 36, 1393–1402. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kinase | Score Range | No. of Predicted Sites | Predicted Sites |
|---|---|---|---|
| PKC | 0.56–0.87 | 8 | S28, S29, T25, S198, S101, T312, S250, S134 |
| PKA | 0.51–0.69 | 5 | S134, S39, S162, S36, S29 |
| CDK5 | 0.50–0.61 | 5 | T191, S68, T70, S162, S187 |
| Kinase | −rbPKC | +rbPKC |
|---|---|---|
| KM (nM) | 17.98 ± 4.1 | 25.05 ± 5.6 |
| kcat (×103 nM/min) | 33.27 ± 5.1 | 33.16 ± 5.4 |
| kcat/KM (min−1 nM−1) | 1.85 ± 1.2 | 1.32 ± 1.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Myrup Holst, C.; Brøndum Andersen, N.; Thinggaard, V.; Tilken, M.; Lautrup, S.; Tesauro, C.; Stevnsner, T. Phosphorylation of the Human DNA Glycosylase NEIL2 Is Affected by Oxidative Stress and Modulates Its Activity. Antioxidants 2023, 12, 355. https://doi.org/10.3390/antiox12020355
Myrup Holst C, Brøndum Andersen N, Thinggaard V, Tilken M, Lautrup S, Tesauro C, Stevnsner T. Phosphorylation of the Human DNA Glycosylase NEIL2 Is Affected by Oxidative Stress and Modulates Its Activity. Antioxidants. 2023; 12(2):355. https://doi.org/10.3390/antiox12020355
Chicago/Turabian StyleMyrup Holst, Camilla, Nanna Brøndum Andersen, Vibeke Thinggaard, Mine Tilken, Sofie Lautrup, Cinzia Tesauro, and Tinna Stevnsner. 2023. "Phosphorylation of the Human DNA Glycosylase NEIL2 Is Affected by Oxidative Stress and Modulates Its Activity" Antioxidants 12, no. 2: 355. https://doi.org/10.3390/antiox12020355
APA StyleMyrup Holst, C., Brøndum Andersen, N., Thinggaard, V., Tilken, M., Lautrup, S., Tesauro, C., & Stevnsner, T. (2023). Phosphorylation of the Human DNA Glycosylase NEIL2 Is Affected by Oxidative Stress and Modulates Its Activity. Antioxidants, 12(2), 355. https://doi.org/10.3390/antiox12020355