

NAD+ Homeostasis and NAD+-Consuming Enzymes: Implications for Vascular Health

Abstract
:1. Introduction
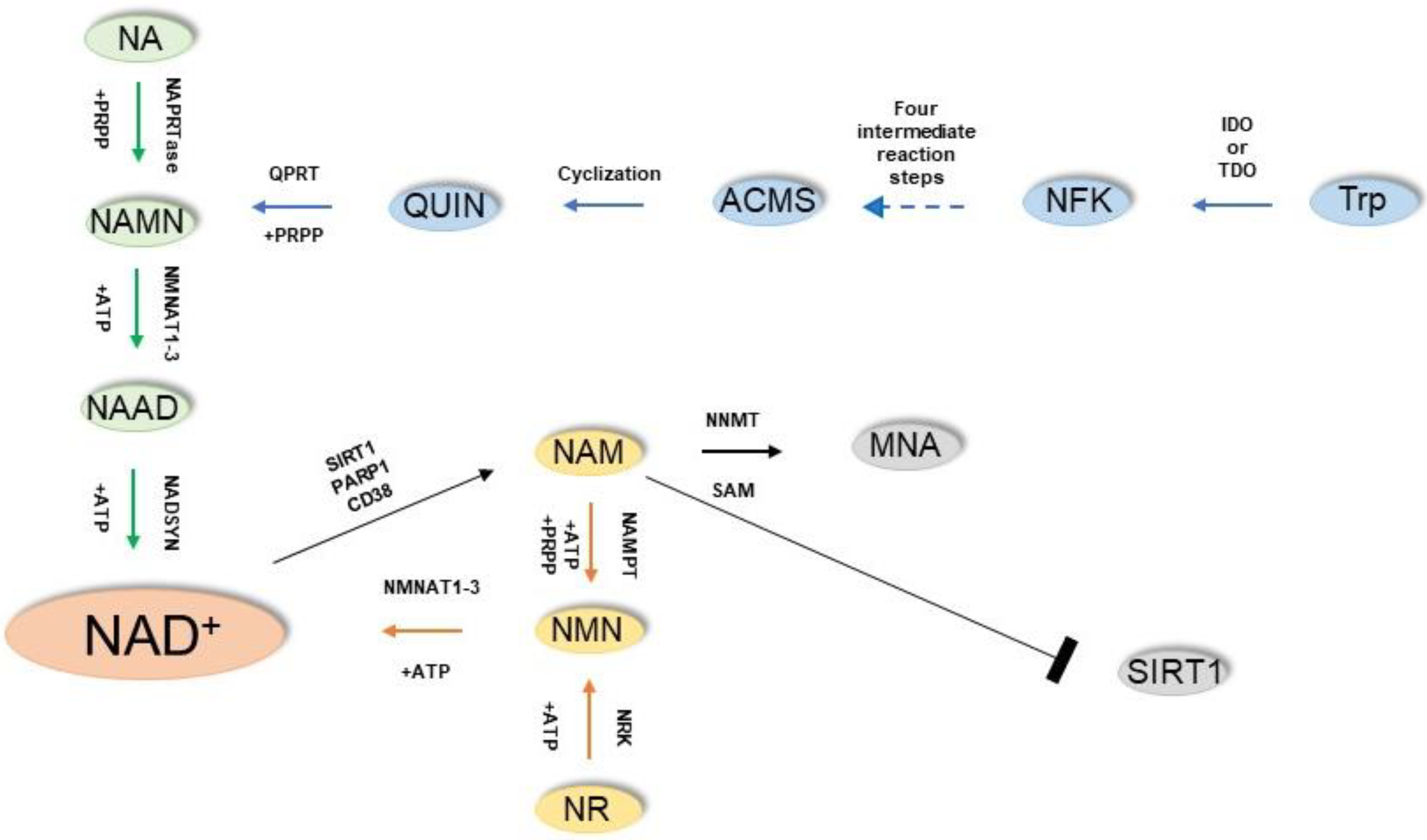
2. NAD+ Biosynthesis
3. NAD+ Use: Redox Reactions and NAD+-Dependent Enzymes
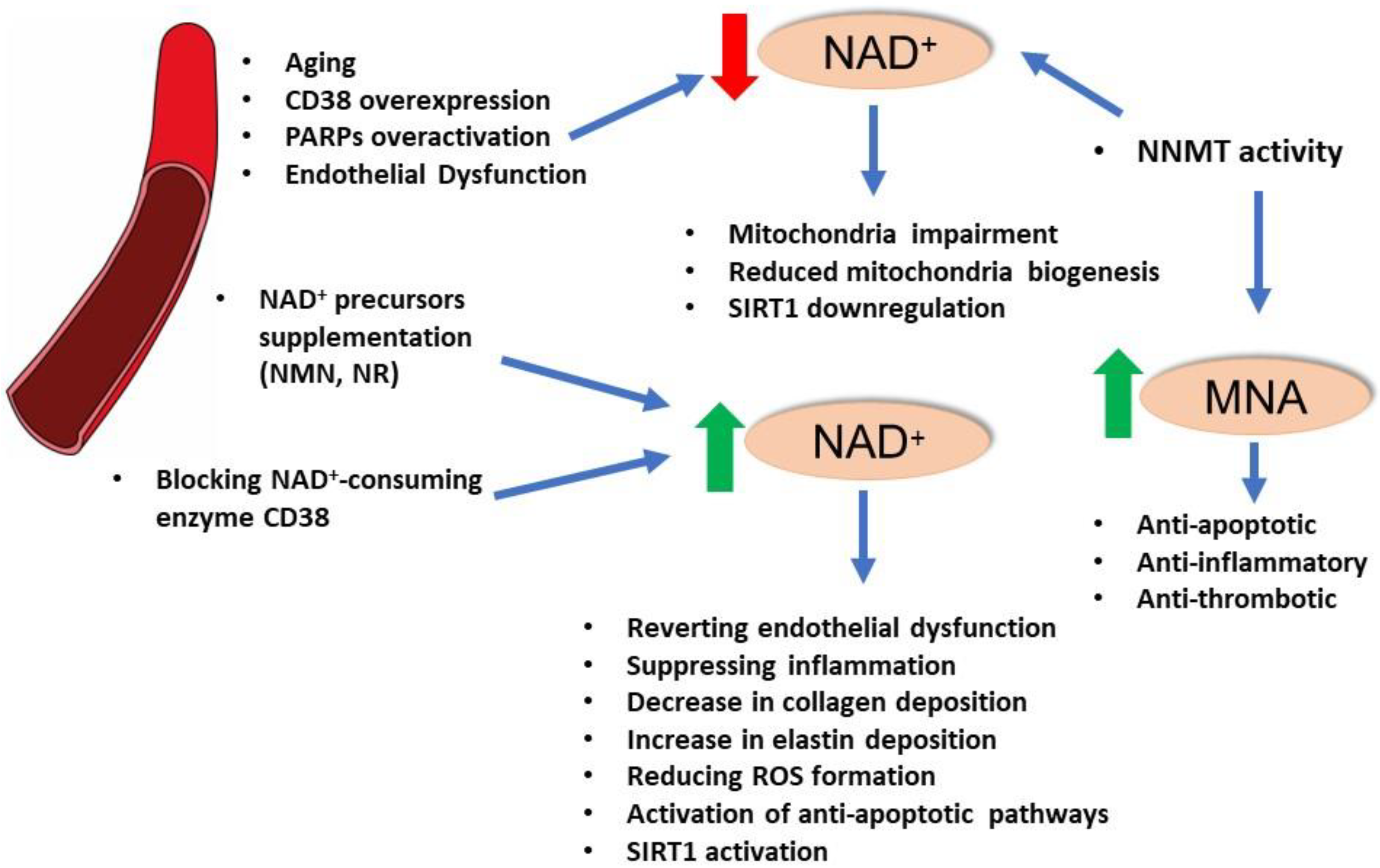
4. NAD+ and Vascular Function
5. NAD+-Dependent Enzymes and Vascular Function: SIRT1
6. NAD+-Dependent Enzymes and Vascular Function: PARPs

{kind=link}
{kind=link}
7. Enzymes of NAD+ Homeostasis: Nicotinamide N-methyltransferase (NNMT)
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence. Cell Metab. 2018, 27, 529–547. [Google Scholar] [CrossRef]
- Zapata-Perez, R.; Wanders, R.J.A.; van Karnebeek, C.D.M.; Houtkooper, R.H. NAD+ homeostasis in human health and disease. EMBO Mol. Med. 2021, 13, e13943. [Google Scholar] [CrossRef]
- D’Amours, D.; Desnoyers, S.; D’Silva, I.; Poirier, G.G. Poly(ADP-ribosyl) ation reactions in the regulation of nuclear functions. Biochem. J. 1999, 342 Pt 2, 249–268. [Google Scholar] [CrossRef]
- Bogan, K.L.; Brenner, C. Nicotinic acid, nicotinamide, and nicotinamide riboside: A molecular evaluation of NAD+ pre-cursor vitamins in human nutrition. Annu. Rev. Nutr. 2008, 28, 115–130. [Google Scholar] [CrossRef]
- Chu, X.; Raju, R.P. Regulation of NAD+ metabolism in aging and disease. Metabolism 2021, 126, 154923. [Google Scholar] [CrossRef]
- Fang, E.F.; Lautrup, S.; Hou, Y.; Demarest, T.G.; Croteau, D.L.; Mattson, M.P.; Bohr, V.A. NAD+ in Aging: Molecular Mechanisms and Translational Implications. Trends Mol. Med. 2017, 23, 899–916. [Google Scholar] [CrossRef]
- Radenkovic, D.; Reason; Verdin, E. Clinical Evidence for Targeting NAD Therapeutically. Pharmaceuticals 2020, 13, 247. [Google Scholar] [CrossRef]
- Zhang, M.; Ying, W. NAD+ Deficiency Is a Common Central Pathological Factor of a Number of Diseases and Aging: Mechanisms and Therapeutic Implications. Antioxid. Redox Signal. 2019, 30, 890–905. [Google Scholar] [CrossRef]
- Abdellatif, M.; Bugger, H.; Kroemer, G.; Sedej, S. NAD+ and Vascular Dysfunction: From Mechanisms to Therapeutic Opportunities. J. Lipid Atheroscler. 2022, 11, 111–132. [Google Scholar] [CrossRef]
- Griffiths, H.B.S.; Williams, C.; King, S.J.; Allison, S.J. Nicotinamide adenine dinucleotide (NAD+): Essential redox metabolite, co-substrate and an anti-cancer and anti-ageing therapeutic target. Biochem. Soc. Trans. 2020, 48, 733–744. [Google Scholar] [CrossRef]
- Pacinella, G.; Ciaccio, A.M.; Tuttolomondo, A. Endothelial Dysfunction and Chronic Inflammation: The Cornerstones of Vascular Alterations in Age-Related Diseases. Int. J. Mol. Sci. 2022, 23, 15722. [Google Scholar] [CrossRef] [PubMed]
- Begum, M.K.; Konja, D.; Singh, S.; Chlopicki, S.; Wang, Y. Endothelial SIRT1 as a Target for the Prevention of Arterial Aging: Promises and Challenges. J. Cardiovasc. Pharmacol. 2021, 78, S63–S77. [Google Scholar] [CrossRef] [PubMed]
- Palmer, R.D.; Elnashar, M.M.; Vaccarezza, M. Precursor comparisons for the upregulation of nicotinamide adenine dinucleotide. Novel approaches for better aging. Aging Med. 2021, 4, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Hegyi, J.; Schwartz, R.A.; Hegyi, V. Pellagra: Dermatitis, dementia, and diarrhea. Int. J. Dermatol. 2004, 43, 1–5. [Google Scholar] [CrossRef]
- Ummarino, S.; Mozzon, M.; Zamporlini, F.; Amici, A.; Mazzola, F.; Orsomando, G.; Ruggieri, S.; Raffaelli, N. Simultaneous quantitation of nicotinamide riboside, nicotinamide mononucleotide and nicotinamide adenine dinucleotide in milk by a novel enzyme-coupled assay. Food Chem. 2017, 221, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.F.; Yoshida, S.; Stein, L.R.; Grozio, A.; Kubota, S.; Sasaki, Y.; Redpath, P.; Migaud, M.E.; Apte, R.S.; Uchida, K.; et al. Long-Term Administration of Nicotinamide Mononucleotide Mitigates Age-Associated Physiological Decline in Mice. Cell Metab. 2016, 24, 795–806. [Google Scholar] [CrossRef]
- Uddin, G.M.; Youngson, N.A.; Chowdhury, S.S.; Hagan, C.; Sinclair, D.A.; Morris, M.J. Administration of Nicotinamide Mononucleotide (NMN) Reduces Metabolic Impairment in Male Mouse Offspring from Obese Mothers. Cells 2020, 9, 791. [Google Scholar] [CrossRef]
- Ramanathan, C.; Lackie, T.; Williams, D.H.; Simone, P.S.; Zhang, Y.; Bloomer, R.J. Oral Administration of Nicotinamide Mononucleotide Increases Nicotinamide Adenine Dinucleotide Level in an Animal Brain. Nutrients 2022, 14, 300. [Google Scholar] [CrossRef]
- Bieganowski, P.; Brenner, C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Pre-iss-Handler independent route to NAD+ in fungi and humans. Cell 2004, 117, 495–502. [Google Scholar] [CrossRef]
- Dietrich, L.S.; Muniz, O.; Powanda, M. NAD synthesis in animal tissues. J. Vitaminol. 1968, 14, 123–129. [Google Scholar] [CrossRef] [Green Version]
- Yoshino, J.; Baur, J.A.; Imai, S.-I. NAD+ Intermediates: The Biology and Therapeutic Potential of NMN and NR. Cell Metab. 2018, 27, 513–528. [Google Scholar] [CrossRef] [PubMed]
- Sharma, C.; Donu, D.; Cen, Y. Emerging Role of Nicotinamide Riboside in Health and Diseases. Nutrients 2022, 14, 3889. [Google Scholar] [CrossRef] [PubMed]
- Horwitt, M.K.; Harper, A.E.; Henderson, L.M. Niacin-tryptophan relationships for evaluating niacin equivalents. Am. J. Clin. Nutr. 1981, 34, 423–427. [Google Scholar] [CrossRef]
- Katsyuba, E.; Romani, M.; Hofer, D.; Auwerx, J. NAD+ homeostasis in health and disease. Nat. Metab. 2020, 2, 9–31. [Google Scholar] [CrossRef]
- Pirinen, E.; Cantó, C.; Jo, Y.S.; Morato, L.; Zhang, H.; Menzies, K.J.; Williams, E.G.; Mouchiroud, L.; Moullan, N.; Hagberg, C.; et al. Pharmacological Inhibition of Poly(ADP-Ribose) Polymerases Improves Fitness and Mitochondrial Function in Skeletal Muscle. Cell Metab. 2014, 19, 1034–1041. [Google Scholar] [CrossRef]
- Clement, J.; Wong, M.; Poljak, A.; Sachdev, P.; Braidy, N. The Plasma NAD+ Metabolome Is Dysregulated in “Normal” Aging. Rejuvenation Res 2019, 22, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.A.; Dominy, J.E.; Lee, Y.; Puigserver, P. The sirtuin family’s role in aging and age-associated pathologies. J. Clin. Investig. 2013, 123, 973–979. [Google Scholar] [CrossRef]
- Daiber, A.; Chlopicki, S. Revisiting pharmacology of oxidative stress and endothelial dysfunction in cardiovascular disease: Evidence for redox-based therapies. Free Radic. Biol. Med. 2020, 157, 15–37. [Google Scholar] [CrossRef]
- Szczesny-Malysiak, E.; Stojak, M.; Campagna, R.; Grosicki, M.; Jamrozik, M.; Kaczara, P.; Chlopicki, S. Bardoxolone Methyl Displays Detrimental Effects on Endothelial Bioenergetics, Suppresses Endothelial ET-1 Release, and Increases Endothelial Permeability in Human Microvascular Endothelium. Oxidative Med. Cell. Longev. 2020, 2020, 4678252. [Google Scholar] [CrossRef]
- Zapotoczny, B.; Braet, F.; Kus, E.; Ginda-Makela, K.; Klejevskaja, B.; Campagna, R.; Chlopicki, S.; Szymonski, M. Actin-spectrin scaffold supports open fenestrae in liver sinusoidal endothelial cells. Traffic 2019, 20, 932–942. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Y.; Sun, A.; Ge, J. The effects of nicotinamide adenine dinucleotide in cardiovascular diseases: Molecular mechanisms, roles and therapeutic potential. Genes Dis. 2021, 9, 959–972. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Martinez, M.; Cabail, M.Z. The PI3K/Akt Pathway in Meta-Inflammation. Int. J. Mol. Sci. 2022, 23, 15330. [Google Scholar] [CrossRef] [PubMed]
- Tossetta, G.; Fantone, S.; Gesuita, R.; Di Renzo, G.C.; Meyyazhagan, A.; Tersigni, C.; Scambia, G.; Di Simone, N.; Marzioni, D. HtrA1 in Gestational Diabetes Mellitus: A Possible Biomarker? Diagnostics 2022, 12, 2705. [Google Scholar] [CrossRef]
- Cecati, M.; Sartini, D.; Campagna, R.; Biagini, A.; Ciavattini, A.; Emanuelli, M.; Giannubilo, S.R. Molecular analysis of endometrial inflammation in preterm birth. Cell. Mol. Biol. 2017, 63, 51–57. [Google Scholar] [CrossRef]
- Rizvi, A.A.; Kathuria, A.; Al Mahmeed, W.; Al-Rasadi, K.; Al-Alawi, K.; Banach, M.; Banerjee, Y.; Ceriello, A.; Cesur, M.; Cosentino, F.; et al. Post-COVID syndrome, inflammation, and diabetes. J. Diabetes Complicat. 2022, 36, 108336. [Google Scholar] [CrossRef]
- Tossetta, G.; Fantone, S.; Delli Muti, N.; Balercia, G.; Ciavattini, A.; Giannubilo, S.R.; Marzioni, D. Preeclampsia and severe acute respiratory syndrome coronavirus 2 infection: A systematic review. J. Hypertens. 2022, 40, 1629–1638. [Google Scholar] [CrossRef]
- Fantone, S.; Tossetta, G.; Di Simone, N.; Tersigni, C.; Scambia, G.; Marcheggiani, F.; Giannubilo, S.R.; Marzioni, D. CD93 a potential player in cytotrophoblast and endothelial cell migration. Cell Tissue Res. 2021, 387, 123–130. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Kale, A.; Perrone, R.; Lopez-Dominguez, J.A.; Pisco, A.O.; Kasler, H.G.; Schmidt, M.S.; Heckenbach, I.; Kwok, R.; Wiley, C.D.; et al. Senescent cells promote tissue NAD+ decline during ageing via the activation of CD38+ macrophages. Nat. Metab. 2020, 2, 1265–1283. [Google Scholar] [CrossRef]
- Mateuszuk, L.; Campagna, R.; Kutryb-Zajac, B.; Kus, K.; Slominska, E.M.; Smolenski, R.T.; Chlopicki, S. Reversal of endothelial dysfunction by nicotinamide mononucleotide via extracellular conversion to nicotinamide riboside. Biochem. Pharmacol. 2020, 178, 114019. [Google Scholar] [CrossRef]
- Boslett, J.; Hemann, C.; Christofi, F.L.; Zweier, J.L. Characterization of CD38 in the major cell types of the heart: Endothelial cells highly express CD38 with activation by hypoxia-reoxygenation triggering NAD(P)H depletion. Am. J. Physiol. Physiol. 2018, 314, C297–C309. [Google Scholar] [CrossRef] [PubMed]
- Polzonetti, V.; Carpi, F.M.; Micozzi, D.; Pucciarelli, S.; Vincenzetti, S.; Napolioni, V. Population variability in CD38 activity: Correlation with age and significant effect of TNF-α -308G>A and CD38 184C>G SNPs. Mol. Genet. Metab. 2012, 105, 502–507. [Google Scholar] [CrossRef]
- Amici, S.A.; Young, N.A.; Narvaez-Miranda, J.; Jablonski, K.A.; Arcos, J.; Rosas, L.; Papenfuss, T.L.; Torrelles, J.B.; Jarjour, W.N.; Guerau-de-Arellano, M. CD38 Is Robustly Induced in Human Macrophages and Monocytes in Inflammatory Conditions. Front Immunol. 2018, 9, 1593. [Google Scholar] [CrossRef]
- Bitterman, K.J.; Anderson, R.M.; Cohen, H.Y.; Latorre-Esteves, M.; Sinclair, D.A. Inhibition of Silencing and Accelerated Aging by Nicotinamide, a Putative Negative Regulator of Yeast Sir2 and Human SIRT1. J. Biol. Chem. 2002, 277, 45099–45107. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Chai, H.; Chen, Y.; Cheng, Y.; Liu, X. Nicotinamide mononucleotide: An emerging nutraceutical against cardiac aging? Curr. Opin. Pharmacol. 2021, 60, 291–297. [Google Scholar] [CrossRef]
- Mitchell, S.J.; Bernier, M.; Aon, M.A.; Cortassa, S.; Kim, E.Y.; Fang, E.F.; Palacios, H.H.; Ali, A.; Navas-Enamorado, I.; Di Francesco, A.; et al. Nicotinamide Improves Aspects of Healthspan, but Not Lifespan, in Mice. Cell Metab. 2018, 27, 667–676.e4. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.; Schilling, E.; Grahnert, A.; Kölling, V.; Dorow, J.; Ceglarek, U.; Sack, U.; Hauschildt, S. Nicotinamide: A vitamin able to shift macrophage differentiation toward macrophages with restricted inflammatory features. Innate Immun. 2015, 21, 813–826. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Li, J.; Shen, Y.; Liu, G.; Zuo, S.; Tao, B.; Ji, Y.; Lu, A.; Lazarus, M.; Breyer, R.M.; et al. Niacin Promotes Cardiac Healing after Myocardial Infarction through Activation of the Myeloid Prostaglandin D2 Receptor Subtype 1. J. Pharmacol. Exp. Ther. 2017, 360, 435–444. [Google Scholar] [CrossRef]
- Huynh, P.K.; Wilder, J.; Hiller, S.; Hagaman, J.; Takahashi, N.; Maeda-Smithies, N.; Li, F. Beneficial effects of nicotinamide on hypertensive mice with impaired endothelial nitric oxide function. J. Exp. Nephrol. 2020, 1, 1. [Google Scholar]
- Mazzanti, L.; Vignini, A.; Emanuelli, M. A 25-Year Long Journey into the World of NO. In The First Outstanding 50 Years of “Universita Politecnica delle Marche”: Research Achievements in Life Sciences; Springer: Cham, Switzerland, 2020. [Google Scholar]
- Reiten, O.K.; Wilvang, M.A.; Mitchell, S.J.; Hu, Z.; Fang, E.F. Preclinical and clinical evidence of NAD+ precursors in health, disease, and ageing. Mech. Ageing Dev. 2021, 199, 111567. [Google Scholar] [CrossRef]
- de Picciotto, N.E.; Gano, L.B.; Johnson, L.C.; Martens, C.R.; Sindler, A.L.; Mills, K.F.; Imai, S.; Seals, D.R. Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell 2016, 15, 522–530. [Google Scholar] [CrossRef]
- Hong, G.; Zheng, D.; Zhang, L.; Ni, R.; Wang, G.; Fan, G.-C.; Lu, Z.; Peng, T. Administration of nicotinamide riboside prevents oxidative stress and organ injury in sepsis. Free. Radic. Biol. Med. 2018, 123, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Tarantini, S.; Valcarcel-Ares, M.N.; Toth, P.; Yabluchanskiy, A.; Tucsek, Z.; Kiss, T.; Hertelendy, P.; Kinter, M.; Ballabh, P.; Süle, Z.; et al. Nicotinamide mononucleotide (NMN) supplementation rescues cerebromicrovascular endothelial function and neurovascular coupling responses and improves cognitive function in aged mice. Redox Biol. 2019, 24, 101192. [Google Scholar] [CrossRef] [PubMed]
- Kiss, T.; Giles, C.B.; Tarantini, S.; Yabluchanskiy, A.; Balasubramanian, P.; Gautam, T.; Csipo, T.; Nyúl-Tóth, Á.; Lipecz, A.; Szabo, C.; et al. Nicotinamide mononucleotide (NMN) supplementation promotes anti-aging miRNA expression profile in the aorta of aged mice, predicting epigenetic rejuvenation and anti-atherogenic effects. GeroScience 2019, 41, 419–439. [Google Scholar] [CrossRef] [PubMed]
- Kiss, T.; Nyúl-Tóth, A.; Balasubramanian, P.; Tarantini, S.; Ahire, C.; Yabluchanskiy, A.; Csipo, T.; Farkas, E.; Wren, J.D.; Garman, L.; et al. Nicotinamide mononucleotide (NMN) supplementation promotes neurovascular rejuvenation in aged mice: Transcriptional footprint of SIRT1 activation, mitochondrial protection, anti-inflammatory, and anti-apoptotic effects. Geroscience 2020, 42, 527–546. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Huang, G.X.; Bonkowski, M.S.; Longchamp, A.; Li, C.; Schultz, M.B.; Kim, L.-J.; Osborne, B.; Joshi, S.; Lu, Y.; et al. Impairment of an Endothelial NAD+-H2S Signaling Network Is a Reversible Cause of Vascular Aging. Cell 2018, 173, 74–89.e20. [Google Scholar] [CrossRef]
- Lin, Q.; Zuo, W.; Liu, Y.; Wu, K.; Liu, Q. NAD+ and cardiovascular diseases. Clin. Chim. Acta 2021, 515, 104–110. [Google Scholar] [CrossRef]
- Schultz, M.B.; Sinclair, D.A. Why NAD+ Declines during Aging: It’s Destroyed. Cell Metab. 2016, 23, 965–966. [Google Scholar] [CrossRef]
- Clayton, Z.S.; Hutton, D.A.; Brunt, V.E.; VanDongen, N.S.; Ziemba, B.P.; Casso, A.G.; Greenberg, N.T.; Mercer, A.N.; Rossman, M.J.; Campisi, J.; et al. Apigenin restores endothelial function by ameliorating oxidative stress, reverses aortic stiffening, and mitigates vascular inflammation with aging. Am. J. Physiol. Heart Circ. Physiol. 2021, 321, H185–H196. [Google Scholar] [CrossRef]
- Zhang, H.-N.; Dai, Y.; Zhang, C.-H.; Omondi, A.M.; Ghosh, A.; Khanra, I.; Chakraborty, M.; Yu, X.-B.; Liang, J. Sirtuins family as a target in endothelial cell dysfunction: Implications for vascular ageing. Biogerontology 2020, 21, 495–516. [Google Scholar] [CrossRef]
- Hong, J.Y.; Lin, H. Sirtuin Modulators in Cellular and Animal Models of Human Diseases. Front. Pharmacol. 2021, 12, 735044. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD+ metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef] [PubMed]
- Donato, A.J.; Machin, D.R.; Lesniewski, L.A. Mechanisms of Dysfunction in the Aging Vasculature and Role in Age-Related Disease. Circ. Res. 2018, 123, 825–848. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Miguelez, P.; Looney, J.; Thomas, J.; Harshfield, G.; Pollock, J.S.; Harris, R.A. Sirt1 during childhood is associated with microvascular function later in life. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H1371–H1378. [Google Scholar] [CrossRef] [PubMed]
- Lipphardt, M.; Song, J.W.; Ratliff, B.B.; Dihazi, H.; Müller, G.A.; Goligorsky, M.S. Endothelial dysfunction is a superinducer of syndecan-4: Fibrogenic role of its ectodomain. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H484–H496. [Google Scholar] [CrossRef]
- Guo, Y.; Xu, C.; Man, A.W.C.; Bai, B.; Luo, C.; Huang, Y.; Xu, A.; Vanhoutte, P.M.; Wang, Y. Endothelial SIRT1 prevents age-induced impairment of vasodilator responses by enhancing the expression and activity of soluble guanylyl cyclase in smooth muscle cells. Cardiovasc. Res. 2019, 115, 678–690. [Google Scholar] [CrossRef]
- Mattagajasingh, I.; Kim, C.-S.; Naqvi, A.; Yamamori, T.; Hoffman, T.A.; Jung, S.-B.; DeRicco, J.; Kasuno, K.; Irani, K. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2007, 104, 14855–14860. [Google Scholar] [CrossRef] [PubMed]
- Habibian, J.; Ferguson, B.S. The Crosstalk between Acetylation and Phosphorylation: Emerging New Roles for HDAC Inhibitors in the Heart. Int. J. Mol. Sci. 2018, 20, 102. [Google Scholar] [CrossRef] [PubMed]
- Nisoli, E.; Tonello, C.; Cardile, A.; Cozzi, V.; Bracale, R.; Tedesco, L.; Falcone, S.; Valerio, A.; Cantoni, O.; Clementi, E.; et al. Calorie Restriction Promotes Mitochondrial Biogenesis by Inducing the Expression of eNOS. Science 2005, 310, 314–317. [Google Scholar] [CrossRef]
- Nagar, H.; Jung, S.-B.; Ryu, M.J.; Choi, S.-J.; Piao, S.; Song, H.-J.; Kang, S.K.; Shin, N.; Kim, D.W.; Jin, S.-A.; et al. CR6-Interacting Factor 1 Deficiency Impairs Vascular Function by Inhibiting the Sirt1-Endothelial Nitric Oxide Synthase Pathway. Antioxid. Redox Signal. 2017, 27, 234–249. [Google Scholar] [CrossRef]
- Testai, L.; Citi, V.; Martelli, A.; Brogi, S.; Calderone, V. Role of hydrogen sulfide in cardiovascular ageing. Pharmacol. Res. 2020, 160, 105125. [Google Scholar] [CrossRef] [PubMed]
- Franzoni, F.; Ghiadoni, L.; Galetta, F.; Plantinga, Y.; Lubrano, V.; Huang, Y.; Salvetti, G.; Regoli, F.; Taddei, S.; Santoro, G.; et al. Physical activity, plasma antioxidant capacity, and endothelium-dependent vasodilation in young and older men. Am. J. Hypertens. 2005, 18, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Donato, A.J.; Eskurza, I.; Silver, A.E.; Levy, A.S.; Pierce, G.L.; Gates, P.E.; Seals, D.R. Direct evidence of endothelial oxi-dative stress with aging in humans: Relation to impaired endothelium-dependent dilation and upregulation of nuclear factor-kappaB. Circ. Res. 2007, 100, 1659–1666. [Google Scholar] [CrossRef] [PubMed]
- Zu, Y.; Liu, L.; Lee, M.Y.; Xu, C.; Liang, Y.; Man, R.Y.; Vanhoutte, P.M.; Wang, Y. SIRT1 promotes proliferation and prevents senescence through targeting LKB1 in primary porcine aortic endothelial cells. Circ. Res. 2010, 106, 1384–1393. [Google Scholar] [CrossRef] [PubMed]
- Donato, A.J.; Walker, A.E.; Magerko, K.A.; Bramwell, R.C.; Black, A.D.; Henson, G.D.; Lawson, B.R.; Lesniewski, L.A.; Seals, D.R. Life-long caloric restriction reduces oxidative stress and preserves nitric oxide bioavailability and function in arteries of old mice. Aging Cell 2013, 12, 772–783. [Google Scholar] [CrossRef]
- Scisciola, L.; Sarno, F.; Carafa, V.; Cosconati, S.; Di Maro, S.; Ciuffreda, L.; De Angelis, A.; Stiuso, P.; Feoli, A.; Sbardella, G.; et al. Two novel SIRT1 activators, SCIC2 and SCIC2.1, enhance SIRT1-mediated effects in stress response and senescence. Epigenetics 2020, 15, 664–683. [Google Scholar] [CrossRef]
- Csiszar, A.; Tarantini, S.; Yabluchanskiy, A.; Balasubramanian, P.; Kiss, T.; Farkas, E.; Baur, J.A.; Ungvari, Z. Role of endothelial NAD+ deficiency in age-related vascular dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H1253–H1266. [Google Scholar] [CrossRef]
- Kiss, T.; Balasubramanian, P.; Valcarcel-Ares, M.N.; Tarantini, S.; Yabluchanskiy, A.; Csipo, T.; Lipecz, A.; Reglodi, D.; Zhang, X.A.; Bari, F.; et al. Nicotinamide mononucleotide (NMN) treatment attenuates oxidative stress and rescues angi-ogenic capacity in aged cerebromicrovascular endothelial cells: A potential mechanism for the prevention of vascular cog-nitive impairment. Geroscience 2019, 41, 619–630. [Google Scholar] [CrossRef]
- Frederick, D.W.; Davis, J.G.; Dávila, A., Jr.; Agarwal, B.; Michan, S.; Puchowicz, M.A.; Nakamaru-Ogiso, E.; Baur, J.A. Increasing NAD Synthesis in Muscle via Nicotinamide Phosphoribosyltransferase Is Not Sufficient to Promote Oxidative Metabolism. J. Biol. Chem. 2015, 290, 1546–1558. [Google Scholar] [CrossRef]
- Kraus, W.L.; Hottiger, M.O. PARP-1 and gene regulation: Progress and puzzles. Mol. Asp. Med. 2013, 34, 1109–1123. [Google Scholar] [CrossRef]
- Li, S.; Deng, J.; Sun, D.; Chen, S.; Yao, X.; Wang, N.; Zhang, J.; Gu, Q.; Zhang, S.; Wang, J.; et al. FBXW7 alleviates hyperglycemia-induced endothelial oxidative stress injury via ROS and PARP inhibition. Redox Biol. 2022, 58, 102530. [Google Scholar] [CrossRef]
- Marzioni, D.; Mazzucchelli, R.; Fantone, S.; Tossetta, G. NRF2 modulation in TRAMP mice: An in vivo model of prostate cancer. Mol. Biol. Rep. 2022, 50, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Li, W.-L.; Liu, J.-M.; Miao, C.-Y. NAMPT and NAMPT-controlled NAD Metabolism in Vascular Repair. J. Cardiovasc. Pharmacol. 2016, 67, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Tossetta, G.; Marzioni, D. Natural and synthetic compounds in Ovarian Cancer: A focus on NRF2/KEAP1 pathway. Pharmacol. Res. 2022, 183, 106365. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Bai, P.; Little, P.J.; Liu, P. Poly(ADP-ribose) Polymerase 1 (PARP1) in Atherosclerosis: From Molecular Mechanisms to Therapeutic Implications. Med. Res. Rev. 2014, 34, 644–675. [Google Scholar] [CrossRef]
- Zong, W.; Gong, Y.; Sun, W.; Li, T.; Wang, Z.-Q. PARP1: Liaison of Chromatin Remodeling and Transcription. Cancers 2022, 14, 4162. [Google Scholar] [CrossRef]
- Palazzo, L.; Suskiewicz, M.J.; Ahel, I. Serine ADP-ribosylation in DNA-damage response regulation. Curr. Opin. Genet. Dev. 2021, 71, 106–113. [Google Scholar] [CrossRef]
- Vida, A.; Márton, J.; Mikó, E.; Bai, P. Metabolic roles of poly(ADP-ribose) polymerases. Semin. Cell Dev. Biol. 2017, 63, 135–143. [Google Scholar] [CrossRef]
- Pacher, P.; Szabo, C. Role of the Peroxynitrite-Poly(ADP-Ribose) Polymerase Pathway in Human Disease. Am. J. Pathol. 2008, 173, 2–13. [Google Scholar] [CrossRef]
- Berger, N.A. Poly(ADP-Ribose) in the Cellular Response to DNA Damage. Radiat. Res. 1985, 101, 4–15. [Google Scholar] [CrossRef]
- Oliver, F.J.; Menissier-de Murcia, J.; de Murcia, G. Poly(ADP-Ribose) Polymerase in the Cellular Response to DNA Damage, Apoptosis, and Disease. Am. J. Hum. Genet. 1999, 64, 1282–1288. [Google Scholar] [CrossRef] [Green Version]
- Groelly, F.J.; Fawkes, M.; Dagg, R.A.; Blackford, A.N.; Tarsounas, M. Targeting DNA damage response pathways in cancer. Nat. Rev. Cancer 2022, 23, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Kim, J.H.; Frizzell, K.M.; Kraus, W.L.; Lin, H. Clickable NAD analogues for labeling substrate proteins of poly(ADP-ribose) polymerases. J. Am. Chem. Soc. 2010, 132, 9363–9372. [Google Scholar] [CrossRef] [PubMed]
- Bai, P.; Cantó, C. The Role of PARP-1 and PARP-2 Enzymes in Metabolic Regulation and Disease. Cell Metab. 2012, 16, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Virág, L.; Robaszkiewicz, A.; Rodriguez-Vargas, J.M.; Oliver, F.J. Poly(ADP-ribose) signaling in cell death. Mol. Asp. Med. 2013, 34, 1153–1167. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Szabó, C. Role of Poly(ADP-ribose) polymerase 1 (PARP-1) in Cardiovascular Diseases: The Therapeutic Potential of PARP Inhibitors. Cardiovasc. Drug Rev. 2007, 25, 235–260. [Google Scholar] [CrossRef]
- Fulco, M.; Cen, Y.; Zhao, P.; Hoffman, E.P.; McBurney, M.W.; Sauve, A.A.; Sartorelli, V. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev. Cell 2008, 14, 661–673. [Google Scholar] [CrossRef]
- Gerhart-Hines, Z.; Dominy, J.E., Jr.; Blattler, S.M.; Jedrychowski, M.P.; Banks, A.S.; Lim, J.H.; Chim, H.; Gygi, S.P.; Puigserver, P. The cAMP/PKA pathway rapidly activates SIRT1 to promote fatty acid oxidation independently of changes in NAD(+). Mol Cell 2011, 44, 851–863. [Google Scholar] [CrossRef]
- von Lukowicz, T.; Hassa, P.O.; Lohmann, C.; Borén, J.; Braunersreuther, V.; Mach, F.; Odermatt, B.; Gersbach, M.; Camici, G.G.; Stähli, B.E.; et al. PARP1 is required for adhesion molecule expression in atherogenesis. Cardiovasc. Res. 2007, 78, 158–166. [Google Scholar] [CrossRef]
- Oumouna-Benachour, K.; Hans, C.P.; Suzuki, Y.; Naura, A.; Datta, R.; Belmadani, S.; Fallon, K.; Woods, C.; Boulares, A.H. Poly(ADP-ribose) polymerase inhibition reduces atherosclerotic plaque size and promotes factors of plaque stability in apolipoprotein E-deficient mice: Effects on macrophage recruitment, nuclear factor-kappaB nuclear translocation, and foam cell death. Circulation 2007, 115, 2442–2450. [Google Scholar] [CrossRef]
- Ha, H.C.; Hester, L.D.; Snyder, S.H. Poly(ADP-ribose) polymerase-1 dependence of stress-induced transcription factors and associated gene expression in glia. Proc. Natl. Acad. Sci. USA 2002, 99, 3270–3275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soriano, F.G.; Virág, L.; Jagtap, P.; Szabó, E.; Mabley, J.G.; Liaudet, L.; Marton, A.; Hoyt, D.G.; Murthy, K.G.K.; Salzman, A.L.; et al. Diabetic endothelial dysfunction: The role of poly(ADP-ribose) polymerase activation. Nat. Med. 2001, 7, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Szabó, C.; Zanchi, A.; Komjáti, K.; Pacher, P.; Krolewski, A.S.; Quist, W.C.; LoGerfo, F.W.; Horton, E.S.; Veves, A. Poly(ADP-Ribose) Polymerase Is Activated in Subjects at Risk of Developing Type 2 Diabetes and Is Associated with Impaired Vascular Reactivity. Circulation 2002, 106, 2680–2686. [Google Scholar] [CrossRef] [PubMed]
- Conlon, N.; Ford, D. A systems-approach to NAD+ restoration. Biochem. Pharmacol. 2022, 198, 114946. [Google Scholar] [CrossRef] [PubMed]
- Roberti, A.; Fernández, A.F.; Fraga, M.F. Nicotinamide N-methyltransferase: At the crossroads between cellular metabolism and epigenetic regulation. Mol. Metab. 2021, 45, 101165. [Google Scholar] [CrossRef]
- Neelakantan, H.; Vance, V.; Wetzel, M.D.; Wang, H.L.; McHardy, S.F.; Finnerty, C.C.; Hommel, J.D.; Watowich, S.J. Selective and membrane-permeable small molecule inhibitors of nicotinamide N-methyltransferase reverse high fat diet-induced obesity in mice. Biochem. Pharmacol. 2018, 147, 141–152. [Google Scholar] [CrossRef]
- Pozzi, V.; Campagna, R.; Sartini, D.; Emanuelli, M. Nicotinamide N-Methyltransferase as Promising Tool for Management of Gastrointestinal Neoplasms. Biomolecules 2022, 12, 1173. [Google Scholar] [CrossRef]
- Campagna, R.; Pozzi, V.; Spinelli, G.; Sartini, D.; Milanese, G.; Galosi, A.B.; Emanuelli, M. The Utility of Nicotinamide N-Methyltransferase as a Potential Biomarker to Predict the Oncological Outcomes for Urological Cancers: An Update. Biomolecules 2021, 11, 1214. [Google Scholar] [CrossRef]
- Li, X.-Y.; Pi, Y.-N.; Chen, Y.; Zhu, Q.; Xia, B.-R. Nicotinamide N-Methyltransferase: A Promising Biomarker and Target for Human Cancer Therapy. Front. Oncol. 2022, 12, 2377. [Google Scholar] [CrossRef]
- Wang, W.; Yang, C.; Wang, T.; Deng, H. Complex roles of nicotinamide N-methyltransferase in cancer progression. Cell Death Dis. 2022, 13, 267. [Google Scholar] [CrossRef]
- Togni, L.; Mascitti, M.; Sartini, D.; Campagna, R.; Pozzi, V.; Salvolini, E.; Offidani, A.; Santarelli, A.; Emanuelli, M. Nicotinamide N-Methyltransferase in Head and Neck Tumors: A Comprehensive Review. Biomolecules 2021, 11, 1594. [Google Scholar] [CrossRef]
- Campagna, R.; Pozzi, V.; Sartini, D.; Salvolini, E.; Brisigotti, V.; Molinelli, E.; Campanati, A.; Offidani, A.; Emanuelli, M. Beyond Nicotinamide Metabolism: Potential Role of Nicotinamide N-Methyltransferase as a Biomarker in Skin Cancers. Cancers 2021, 13, 4943. [Google Scholar] [CrossRef]
- Campagna, R.; Mateuszuk, L.; Wojnar-Lason, K.; Kaczara, P.; Tworzydło, A.; Kij, A.; Bujok, R.; Mlynarski, J.; Wang, Y.; Sartini, D.; et al. Nicotinamide N-methyltransferase in endothelium protects against oxidant stress-induced endothelial injury. Biochim. Biophys. Acta Mol. Cell. Res. 2021, 1868, 119082. [Google Scholar] [CrossRef]
- Dymkowska, D.; Wrzosek, A.; Zablocki, K. Atorvastatin and pravastatin stimulate nitric oxide and reactive oxygen species generation, affect mitochondrial network architecture and elevate nicotinamide N-methyltransferase level in endothelial cells. J. Appl. Toxicol. 2021, 41, 1076–1088. [Google Scholar] [CrossRef]
- Stępińska, O.; Dymkowska, D.; Mateuszuk, L.; Zabłocki, K. Lipopolysaccharide affects energy metabolism and elevates nicotinamide N-methyltransferase level in human aortic endothelial cells (HAEC). Int. J. Biochem. Cell Biol. 2022, 151, 106292. [Google Scholar] [CrossRef]
- Zhou, Z.Y.; Shi, W.T.; Zhang, J.; Zhao, W.R.; Xiao, Y.; Zhang, K.Y.; Ma, J.; Tang, J.Y.; Wang, Y. Sodium tanshinone IIA sulfonate protects against hyperhomocysteine-induced vascular endothelial injury via activation of NNMT/SIRT1-mediated NRF2/HO-1 and AKT/MAPKs signaling in human umbilical vascular endothelial cells. Biomed. Pharmacother. 2022, 158, 114137. [Google Scholar] [CrossRef]
- Watała, C.; Kaźmierczak, P.; Dobaczewski, M.; Przygodzki, T.; Bartuś, M.; Łomnicka, M.; Slominska, E.M.; Duračkova, Z.; Chlopicki, S. Anti-diabetic effects of 1-methylnicotinamide (MNA) in streptozocin-induced diabetes in rats. Pharmacol. Rep. 2009, 61, 86–98. [Google Scholar] [CrossRef]
- Przygodzki, T.; Kazmierczak, P.; Sikora, J.; Watala, C. 1-methylnicotinamide effects on the selected markers of endothelial function, inflammation and haemostasis in diabetic rats. Eur. J. Pharmacol. 2010, 640, 157–162. [Google Scholar] [CrossRef]
- Rygula, A.; Pacia, M.Z.; Mateuszuk, L.; Kaczor, A.; Kostogrys, R.B.; Chlopicki, S.; Baranska, M. Identification of a biochemical marker for endothelial dysfunction using Raman spectroscopy. Analyst 2015, 140, 2185–2189. [Google Scholar] [CrossRef]
- Chlopicki, S.; Swies, J.; Mogielnicki, A.; Buczko, W.; Bartus, M.; Lomnicka, M.; Adamus, J.; Gebicki, J. 1-Methylnicotinamide (MNA), a primary metabolite of nicotinamide, exerts anti-thrombotic activity mediated by a cyclooxygenase-2/prostacyclin pathway. Br. J. Pharmacol. 2007, 152, 230–239. [Google Scholar] [CrossRef]
- Reiss, A.B.; Edelman, S.D. Recent insights into the role of prostanoids in atherosclerotic vascular disease. Curr. Vasc. Pharmacol. 2006, 4, 395–408. [Google Scholar]
- Biedroń, R.; Ciszek, M.; Tokarczyk, M.; Bobek, M.; Kurnyta, M.; Słominska, E.M.; Smoleński, R.T.; Marcinkiewicz, J. 1-Methylnicotinamide and nicotinamide: Two related anti-inflammatory agents that differentially affect the functions of activated macrophages. Arch. Immunol. Ther. Exp. 2008, 56, 127–134. [Google Scholar] [CrossRef] [Green Version]
- Mateuszuk, L.; Jasztal, A.; Maslak, E.; Gasior-Glogowska, M.; Baranska, M.; Sitek, B.; Kostogrys, R.; Zakrzewska, A.; Kij, A.; Walczak, M.; et al. Antiatherosclerotic Effects of 1-Methylnicotinamide in Apolipoprotein E/Low-Density Lipoprotein Receptor-Deficient Mice: A Comparison with Nicotinic Acid. J. Pharmacol. Exp. Ther. 2016, 356, 514–524. [Google Scholar] [CrossRef]
- Menon, R.M.; Adams, M.H.; González, M.A.; Tolbert, D.S.; Leu, J.H.; Cefali, E.A. Plasma and urine pharmacokinetics of niacin and its metabolites from an extended-release niacin formulation. Int. J. Clin. Pharmacol. Ther. 2007, 45, 448–454. [Google Scholar] [CrossRef]
- Lukasova, M.; Hanson, J.; Tunaru, S.; Offermanns, S. Nicotinic acid (niacin): New lipid-independent mechanisms of action and therapeutic potentials. Trends Pharmacol. Sci. 2011, 32, 700–707. [Google Scholar] [CrossRef]
- Zou, L.; Liang, B.; Gao, Y.; Ye, T.; Li, M.; Zhang, Y.; Lu, Q.; Hu, X.; Li, H.; Yuan, Y.; et al. Nicotinic Acid Riboside Regulates Nrf-2/P62-Related Oxidative Stress and Autophagy to Attenuate Doxorubicin-Induced Cardiomyocyte Injury. Biomed. Res. Int. 2022, 2022, 6293329. [Google Scholar] [CrossRef]
- Pissios, P. Nicotinamide N-Methyltransferase: More Than a Vitamin B3 Clearance Enzyme. Trends Endocrinol. Metab. 2017, 28, 340–353. [Google Scholar] [CrossRef]
- Kannt, A.; Rajagopal, S.; Hallur, M.S.; Swamy, I.; Kristam, R.; Dhakshinamoorthy, S.; Czech, J.; Zech, G.; Schreuder, H.; Ruf, S. Novel Inhibitors of Nicotinamide-N-Methyltransferase for the Treatment of Metabolic Disorders. Molecules 2021, 26, 991. [Google Scholar] [CrossRef]
- Barrows, R.D.; Jeffries, D.E.; Vishe, M.; Tukachinsky, H.; Zheng, S.-L.; Li, F.; Ma, Z.; Li, X.; Jin, S.; Song, H.; et al. Potent Uncompetitive Inhibitors of Nicotinamide N-Methyltransferase (NNMT) as In Vivo Chemical Probes. J. Med. Chem. 2022, 65, 14642–14654. [Google Scholar] [CrossRef]
- van Haren, M.J.; Gao, Y.; Buijs, N.; Campagna, R.; Sartini, D.; Emanuelli, M.; Mateuszuk, L.; Kij, A.; Chlopicki, S.; Escude Martinez de Castilla, P.; et al. Esterase-Sensitive Prodrugs of a Potent Bisubstrate Inhibitor of Nicotinamide N-Methyltransferase (NNMT) Display Cellular Activity. Biomolecules 2021, 11, 1357. [Google Scholar] [CrossRef]
- Ruf, S.; Rajagopal, S.; Kadnur, S.V.; Hallur, M.S.; Rani, S.; Kristam, R.; Swaminathan, S.; Zope, B.R.; Gondrala, P.K.; Swamy, I.; et al. Novel tricyclic small molecule inhibitors of Nicotinamide N-methyltransferase for the treatment of metabolic disorders. Sci. Rep. 2022, 12, 15440. [Google Scholar] [CrossRef]
- van Haren, M.J.; Zhang, Y.; Thijssen, V.; Buijs, N.; Gao, Y.; Mateuszuk, L.; Fedak, F.A.; Kij, A.; Campagna, R.; Sartini, D.; et al. Macrocyclic peptides as allosteric inhibitors of nicotinamide N-methyltransferase (NNMT). RSC Chem. Biol. 2021, 2, 1546–1555. [Google Scholar] [CrossRef] [PubMed]
- Sabnis, R.W. Novel Pyrimidine-5-carboxamide Compounds as NNMT Inhibitors for Treating Diabetes. ACS Med. Chem. Lett. 2021, 12, 538–539. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; van Haren, M.J.; Buijs, N.; Innocenti, P.; Zhang, Y.; Sartini, D.; Campagna, R.; Emanuelli, M.; Parsons, R.B.; Jespers, W.; et al. Potent Inhibition of Nicotinamide N-Methyltransferase by Alkene-Linked Bisubstrate Mimics Bearing Electron Deficient Aromatics. J. Med. Chem. 2021, 64, 12938–12963. [Google Scholar] [CrossRef]
- Policarpo, R.L.; Decultot, L.; May, E.; Kuzmič, P.; Carlson, S.; Huang, D.; Chu, V.; Wright, B.A.; Dhakshinamoorthy, S.; Kannt, A.; et al. High-Affinity Alkynyl Bisubstrate Inhibitors of Nicotinamide N-Methyltransferase (NNMT). J. Med. Chem. 2019, 62, 9837–9873. [Google Scholar] [CrossRef] [Green Version]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campagna, R.; Vignini, A. NAD+ Homeostasis and NAD+-Consuming Enzymes: Implications for Vascular Health. Antioxidants 2023, 12, 376. https://doi.org/10.3390/antiox12020376
Campagna R, Vignini A. NAD+ Homeostasis and NAD+-Consuming Enzymes: Implications for Vascular Health. Antioxidants. 2023; 12(2):376. https://doi.org/10.3390/antiox12020376
Chicago/Turabian StyleCampagna, Roberto, and Arianna Vignini. 2023. "NAD+ Homeostasis and NAD+-Consuming Enzymes: Implications for Vascular Health" Antioxidants 12, no. 2: 376. https://doi.org/10.3390/antiox12020376