Abstract
Disappointing results from the POLAR A and M phase III trials involving colorectal cancer patients on chemotherapy with FOLFOX6 in curative (A) and palliative (M) settings have been reported by the principal investigators and the sponsor (PledPharma AB/Egetis Therapeutics AB). FOLFOX6, oxaliplatin in combination with 5-fluorouracil (5-FU), possesses superior tumoricidal activity in comparison to 5-FU alone, but suffers seriously from dose-limiting platinum-associated Chemotherapy-Induced Peripheral Neuropathy (CIPN). The aim of the POLAR trials was to demonstrate that PledOx [calmangafodipir; Ca4Mn(DPDP)5] reduced the incidence of persistent CIPN from 40% to 20%. However, this assumption was based on “explorative” data in the preceding PLIANT phase II trial, which did not mirror the expected incidence of unwanted toxicity in placebo patients. In POLAR A and M, the assessment of PledOx efficacy was conducted in patients that received at least six cycles of FOLFOX6, enabling analyses of efficacy in 239 A and 88 M patients. Instead of a hypothesized improvement from 40% to 20% incidence of persistent CIPN in the PledOx group, i.e., a 50% improvement, the real outcome was the opposite, i.e., an about 50% worsening in this bothersome toxicity. Mechanisms that may explain the disastrous outcome, with a statistically significant number of patients being seriously injured after having received PledOx, indicate interactions between two redox active metal cations, Pt2+ (oxaliplatin) and Mn2+ (PledOx). A far from surprising causal relationship that escaped prior detection by the study group and the sponsor. Most importantly, recently published data (ref 1) unequivocally indicate that the PLIANT study was not suited to base clinical phase III studies on. In conclusion, the POLAR and PLIANT trials show that PledOx and related manganese-containing compounds are unsuited for co-treatment with platinum-containing compounds. For use as a therapeutic adjunct in rescue treatment, like in ischemia-reperfusion of the heart or other organs, or in acetaminophen (paracetamol)-associated liver failure, there is little or nothing speaking against the use of PledOx or other PLED compounds. However, this must be thoroughly documented in more carefully designed clinical trials.
1. POLAR A and M Clinical Phase III Trials
The POLAR A phase III trial was conducted in colorectal cancer patients going through curative FOLFOX6 chemotherapy with the intention to prove that co-treatment with PledOx [calmangafodipir; Ca4Mn(DPDP)5] lowers the incidence of persistent oxaliplatin-associated chemotherapy-induced sensory neuropathy (CIPN) by 50% [1]. The POLAR M phase III trial was more or less an identical trial but included palliative patients instead of curative patients. On 6 April 2020, these trials were terminated due to “allergic” hypersensitivity reactions in PledOx-treated patients, first reported by Qvortrup et al., 2021 [2] to equal twelve in the PledOx group and later reported by Pfeiffer et al., 2022 [1] to equal nine. At that timepoint, the POLAR M trial was already put on hold since 1 March, 2021 by the FDA and the responsible regulatory authority in France because of suspected manganese-related toxicity in the PledOx group. At closure, the POLAR A study was fully recruited and enabled efficacy assessments of 5 µmol/kg PledOx and placebo in 120 and 119 patients, respectively (ClinicalTrials.gov; NCT04034355). The recruitment in the POLAR M trial laid behind the POLAR A and enabled efficacy assessment of 2 µmol/kg PledOx, 5 µmol/kg PledOx and placebo in 31, 27 and 25 patients, respectively (ClinicalTrials.gov; NCT03654729).
2. Therapeutic Efficacy of PLED Compounds and Importance of Scrutinizing What Went Wrong in the POLAR Trials
The MRI contrast agent mangafodipir (MnDPDP; Teslascan) and derivatives thereof, known as PLED (diPyridoxyl EthylDiamine) compounds (Figure 1), have shown therapeutic efficacy in a wide range of serious pathological condition preclinically as well as clinically [3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19].
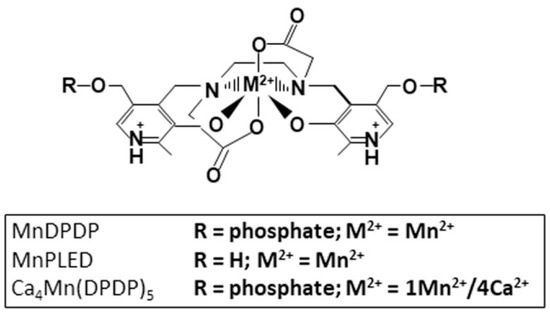
Figure 1.
Chemical structure of MnDPDP (manganese dipyridoxyl diphosphate; generic name mangafodipir), MnPLED (manganese dipyridoxyl ethyldiamine), and Ca4Mn(DPDP)5 [tetracalcium monomanganese penta(dipyridoxyl diphosphate); calmangafodipir].
However, highly negative results were reported from the POLAR A and M phase III clinical trials. Instead of an anticipated 50% decrease in the incidence of persistent oxaliplatin-related CIPN in cancer patients treated with PledOx, a diametrically opposite outcome was obtained, namely an about 50% exacerbation of this bothersome toxicity in the POLAR A trial [1]. The incidence of CIPN in PledOx-treated patients was lower in the POLAR M trial but was still higher than in the placebo group.
As founders of PledPharma, a company founded to develop PLED substances into approved rescue drugs for treatment of life-threatening conditions, such as acute myocardial infarction, acetaminophen (paracetamol)-induced liver failure and fibril neutropenia, we find it important to discuss what might have gone wrong in the above-described trials, and what kind of mechanisms may explain the disastrous outcome, where a statistically significant number of patients have been seriously injured after having received PledOx. Such discussions may hopefully help to avoid similar negative outcomes in future clinical trials.
Although, the principal investigators (IPs) have entered an agreement with the sponsor that restricts the PIs’ rights to discuss or publish trial results (see ClinicalTrials.gov; NCT04034355 and NCT NCT03654729), we nevertheless look forward to receiving their comments.
3. Reported Results from the POLAR A and M Trials
The core reported results by Pfeiffer et al., 2022 [1], confirmed those previously disclosed by Qvortrup et al., 2021 [2], showing that PledOx caused a 37% (p = 0.0445) increase in the incidence of persistent CIPN in A and M pooled patients. The exacerbating effect was even larger in the POLAR A study alone where PledOx increased the incidence by 52% (p = 0.028) [1]. In the preceding power analyses, both studies were estimated to display a decrease in the incidence from 40% to 20% in PledOx treated patients, i.e., a 50% improvement [1]. However, in the POLAR A study, PledOx in fact increased the incidence of persistent CIPN from roughly 40% to 60% (see ClinicalTrials.gov; NCT04034355), i.e., a 50% exacerbation. To characterize such an outcome as “failure to meet primary endpoint” as the authors do [1,2] is misleading.
Furthermore, seeking a genetic explanation for the failure, the authors refer to data supported by p-values of about 0.5. However, the most obvious explanation of the failure was the premature decision to advance PledOx into phase III based on the PLIANT phase II study [20].
4. What Caused the POLAR Failure?
A common aim of a clinical phase III trial is to confirm the main result obtained in a preceding phase II trial. This result defines the primary endpoint and forms the statistical prerequisites for the subsequent phase III study, from which a so-called power analysis is done to determine a good sample size for a particular effect. Such analysis demands that a certain effect of the test drug has been obtained, compared to a placebo drug. However, the overriding problem with the PLIANT trial was the exceedingly small number of oxaliplatin-related adverse events (AEs) in placebo patients, including oxaliplatin-induced peripheral neuropathy [20]. Neither the original primary endpoint, i.e., neutropenia, nor any other endpoint was reached in the PLIANT trial.
It is essential that a clinical trial aimed to test whether a particular treatment reduces AEs, in fact roughly mirrors the expected frequency of AEs in the placebo group—which PLIANT did not. Karlsson and Jynge [21] raised serious criticism in a Letter to the Editor of Acta Oncologica (where the PLIANT study was published) against the decision to advance PledOx into phase III based on the PLIANT trial. Karlsson and Jynge characterized the decision as a “hazardous” one. However, three of the authors of PLIANT publication maintained in their reply that the decision was based on “trustworthy” data [22].
The selected primary endpoint in the POLAR studies, i.e., patient-reported persistent CIPN, according to the FACT/GOG-NTX-13 subscale questionnaire, targeting numbness, tingling, or discomfort in hands and/or feet. This questionnaire was not included in the PLIANT trial but was based on another patient-reported questionnaire, according to the Leonard scale, targeting “mean sensory score of the average sum of tingling, numbness and burning pain to cold in hands and feet”, as presented in Figure 3c in the PLIANT publication [20]. Although this figure seemingly displays some real neurotoxic effects of oxaliplatin, presenting the results as the average sum of several variables, where the maximum effect for each of them equals 10 on a 0–10 scale, artificially enlarges the mean effect. In the POLAR trials, the four used variables were instead analysed out from the criterion “in at least 1 of the first 4 items”. The PLIANT results were furthermore based on pooled data from three different dose groups, which of course, further belittles the value of the results. Furthermore, presenting the results as mean ± SEM (SEM = SD/ with sample sizes around 30 gives much smaller SEM than the true variabilities, SD, within the samples. Multiplying SEM with 1.96 gives an approximate 95% confidence interval, which in turn, reveals a huge overlap in these intervals, as presented in Figure 3c in Glimelius et al., 2017 [20]. The true incidence of persistent CIPN still appears exceedingly small in both the placebo and PledOx groups in the PLIANT trial.
Furthermore, physician-judged CIPN-incidence under eight cycles of FOLFOX chemotherapy in both placebo patients and PledOx patients in the PLIANT trial, as shown in Figure 2, Glimelius et al., 2017 [20], was also exceedingly small. Taken in consideration that the original primary endpoint of the PLIANT actually was grade 3/4 neutropenia, it should also be noted that this incidence was similarly low, 12% instead of the expected 40%, and more in line with what you expect from 5-FU alone. Hence, the profile of AEs corresponded much better to 5-FU alone than that of the combination of oxaliplatin and 5-FU, as noted by Karlsson and Jynge already in 2017 [21].
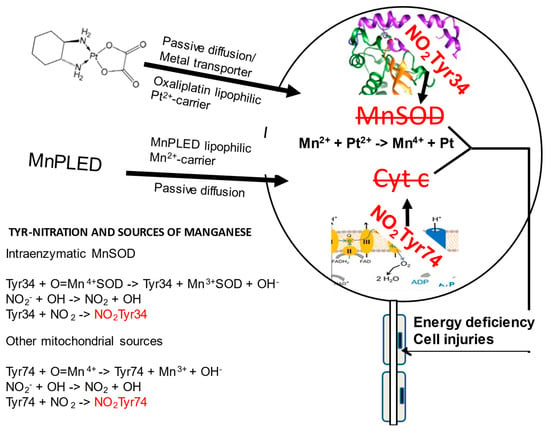
Figure 2.
Schematic illustration of a DRG neuron showing two tyrosine nitration reactions taking place during high oxidative stress, relevant to co-administration of manganese-containing PledOx and platinum-containing oxaliplatin. High oxidative stress due the presence of Pt2+ causes oxidation of Mn2+/Mn3+ to Mn4+ and simultaneously a reaction between superoxide (O2−) and nitric oxide (NO) forming highly toxic peroxynitrite (ONOO−). In presence of Mn4+, ONOO− nitrates two mitochondrial tyrosine residues, Tyr34 in the hMnSOD and Tyr74 in the cytochrome c, resulting in irreversible enzyme inactivation and disturbed electron transport, as illustrated to the right. DRG influx of lipophilic oxaliplatin and the MnDPDP metabolite MnPLED is illustrated to the left, and below that illustration, manganese sources together with tyrosine nitration chemistry are shown.
PLIANT authors presented at the MASCC meeting in Copenhagen, 2015, and at the ASCO meeting in Chicago, 2016, an objective response rate (ORR) of 27% in the placebo group of the PLIANT trial. The reported ORR of 27% in the placebo group is identical to what is expected from 5-FU alone (cf. 18). Addition of oxaliplatin to 5-FU has increased the ORR from about 27% to 45%. Karlsson and Jynge notified the sponsor on the huge discrepancy between the expected tumoricidal efficacy and of that obtained in the PLIANT trial. Before the PLIANT trial was published in Acta Oncologica [20], Karlsson was informed by Glimelius that the ORR was incorrect due to a miscalculation done by the clinical contract research organisation, PCG Clinical Services AB in Uppsala. After recalculation, the ORR was changed from 27% to 43% in the placebo group, a rather expected figure for FOLFOX6, but the progression-free survival (PFS) was still no longer than 7 months and the incidence of AEs remained exceedingly low [21].
There was nothing, whatsoever, in the PLIANT trial supporting the decision to advance PledOx into phase III. Neither was there evidence in the PLIANT trial supporting the power analysis of the POLAR trials, anticipating a 40% incidence of persistent CIPN in the placebo groups and a 20% in the PledOx group.
5. Redox Interaction between Mn2+-Containing PledOx and Pt2+-Containing Oxaliplatin
In vivo mixing of two metal complexes (oxaliplatin and PledOx) with inherent redox properties may lead to devastating drug interactions. In a recent opinion article in Antioxidants [23], we discussed possible mechanisms behind the POLAR failure, suggesting that it is explained by intravenous administrations of PledOx and oxaliplatin being too close in time and, thereby, causing unfavorable redox interactions between Mn2+ and Pt2+. In that discussion, we probably proceeded from an incorrect assumption of an acute interaction between Pt2+ and Mn2+. A more plausible explanation may be a persistent interaction, where these redox active cations co-accumulate in the dorsal root ganglion (DRG) and where Pt2+ oxidizes Mn2+ into Mn4+, and where Mn4+ drives the devastating protein nitration, as schematically shown in Figure 2.
6. Involvement of Manganese in Mitochondrial Protein Nitration
During high cellular oxidative stress, nitric oxide (NO) readily reacts with O2− and this forms peroxynitrite (ONOO−), which is in the presence of oxometal complexes, e.g., O = Fe4+X and O = Mn4+X [18,24]. These two species are able to oxidize and nitrate Tyr-residues, including site specific nitration of human Tyr34, located about 5Å from the active site of the mitochondrial manganese superoxide dismutase [24]. These are conditions that irreversibly inactivate this critical enzyme. Furthermore, Tyr74 of cytochrome c is another mitochondrial target for Mn4+-driven nitration, triggering a conformational change, resulting in an alternative conformation lacking its normal electron transport capacity. Altogether, this will result in a critically exacerbated situation. Protein tyrosine nitration has for many years been known to occur in several pathologies, such as cardiovascular disease, neurodegeneration, inflammation, and cancer [25].
7. Plausible Mechanism behind PledOx-Induced CIPN Exacerbation
The clinical trial by Coriat et al., 2014 [16] is of particular interest when it comes to persistent CIPN, suggesting that co-treatment with MnDPDP (Teslascan) not only protects the patients from CIPN, but in fact also cured ongoing oxaliplatin-related CIPN. We have in a previous Antioxidants publication [23] discussed the importance of distancing the administrations of PledOx and oxaliplatin in order to avoid negative redox interactions. In the Coriat trial, MnDPDP was administered immediately upon the oxaliplatin infusion, which may indicate that the distance between administrations does not really matter. However, the CIPN exacerbation in the POLAR trials was observed 9 months after the start of chemotherapy, and we are not aware of such long follow-ups in the Coriat trial. Furthermore, asserting that such huge exacerbation should be caused by an acute and transient redox interaction between platinum and manganese is less plausible. A more plausible explanation seems to be co-accumulation and retention of both these metals in the DRG. Similar arguments are also viable when it comes the 6 mM content of the antioxidant ascorbic acid in the ready-to-use formulation of MnDPDP (Teslascan), i.e., it will not have any crucial effect on the final outcome.
There are good reasons to anticipate that intravenously administrated organic platinum as well as manganese will accumulate in nerve tissue, particularly in the DRG (Figure 2), located outside the blood-brain barrier and lacking a draining lymph system and cerebrospinal fluid. This makes potentially dangerous substances, such as chemotherapy drugs and toxic metals, to accumulate in the peripheral nerve system and causes oxidative stress and detrimental nerve injuries (cf. 18). Furthermore, oxaliplatin is a small (Mw 397) and highly lipophilic compound with a distribution volume of almost 600 L [26]. Oxaliplatin will therefore readily get intracellular access to the DRG cells. Similarly, the MnDPDP metabolite MnPLED is a small and lipophilic compound that is expected to readily get intracellular access to DRG neurons.
Platinum(II), with its high reduction potential, oxidizes Mn2+ into highly toxic Mn4+, which in turn drives nitration of tyrosine residues as described above, leaving the mitochondrion without antioxidant protection. This is of course extremely critical for neurons, such as the DRG neurons. Similar negative interactions may also explain the “allergic” hypersensitivity reactions and the manganese toxicity seen in the POLAR trials, as notified by us in a previous paper in Antioxidants [23].
Manganese is an essential metal present in nerve tissue, e.g., in the active site of the mitochondrial MnSOD enzyme, where it disarms devastating superoxide radicals (O2−) [3]. Intravenously administered Pt2+ (associated to oxaliplatin) is taken up by DRG cells, either via metal transporters or through passive diffusion. This may in turn drive oxidation of endogenous Mn2+ into Mn4+, resulting in an irreversible inactivation of both mitochondrial MnSOD and cytochrome c (Figure 2). Interestingly, such devastating reactions may in fact be a plausible mechanism behind oxaliplatin-associated persistent CIPN presence, driven by oxidation of endogenous Mn2+/Mn3+. Interestingly, protein nitration is implicated in the development of diabetic peripheral neuropathy [27].
Importantly, the new publication by Pfeiffer et al., 2022 [1] discloses the true timepoint for administration of PledOx, which previously has been defined as “on top of modified FOLFOX6” (2; ClinicalTrials.gov; NCT03654729 and NCT04034355). However, as the POLAR study reported a closely similar administration regimen as that used in the preceding phase II PLIANT study, i.e., an intravenous PledOx infusion 10 min ahead of oxaliplatin, it clearly indicates that the literally negative effect of PledOx should have been easily detected in a properly executed and monitored phase II study. That is, a study displaying the expected incidence of oxaliplatin-induced CIPN in placebo patients, similar to the CIPN incidence displayed in the POLAR A and M studies.
8. An Alternative and Manganese-Independent Possibility to Treat Platinum-Associated CIPN
As noted above, the clinical trial by Coriat and co-workers 2014 [16] suggested that co-treatment with MnDPDP not only protected the patients from CIPN, but in fact also cured ongoing oxaliplatin-related CIPN. The authors maintained that these effects were due to the MnSOD-mimetic activity of MnDPDP. However, this explanation is questionable from a pharmacokinetic perspective, where the MnSOD mimetic activity is only expected to last a couple of hours. Another but until recently undisclosed possibility is that Pt2+ has a high enough formation constant (10logKML) to outcompete Mn2+/Ca2+/Zn2+ for binding to DPDP, or its dephosphorylated metabolite PLED, and transforms toxic Pt2+ into a non-toxic complex, which can be excreted from the body by a process known as chelation therapy. As opposed to the MnSOD mimetic alternative, chelation therapy may solve the underlying problem, namely retention of Pt2+ in the DRG. Electron paramagnetic resonance (EPR)-guided competition experiments between MnDPDP (10logKML ≈ 15) and K2PtCl4, and between MnDPDP and ZnCl2 (10logKML ≈ 19), respectively, from which an estimate of the 10logKML of PtDPDP was obtained. This estimate clearly suggested that DPDP has high enough affinity for acting as a chelation drug [28].
The study by Canta et al., 2020 [29], utilizing a mouse model of oxaliplatin-induced peripheral sensory neuropathy, is of central interest. In this study, histopathological findings, after eight weeks of follow up, demonstrated significant neuroprotective efficacy of PledOx. When it came to the mechanistic part of the conclusion, it was expressed as “the current study lends mechanistic support and insights to the findings that the iron chelator and SOD-mimetic calmangafodipir and the related mangafodipir have demonstrated clinical efficacy against OHP-induced CIPN”. Although, both iron chelation and MnSOD mimetic activity in the first glance may appear trustworthy. Platinum-associated CIPN is in our eyes caused by long retention of Pt2+ in the body. Significantly increased levels in the body can be detected 10 to 20 years after chemotherapy, causing a long-lasting oxidative and nitrosative stress and chronic toxicity. From a pharmacokinetic perspective, a few hours MnSOD mimetic activity in conjunction with chemotherapy is probably of negligible help [28]. Similar arguments could also be used against iron-chelation. Interestingly, an Electron Paramagnetic Resonance (EPR) study has demonstrated that Pt2+ is binding to the DPDP-chelator with high affinity [28]. A more plausible mechanism than those described by Canta et al., is that DPDP (or its metabolite PLED) binds Pt2+ and forms a low molecular weight complex that can be excreted through the kidneys.
According to available information, the Canta et al. study [29] also included paclitaxel, a chemotherapy drug causing similar peripheral neuropathy as that of oxaliplatin. However, the etiology differs between these two forms of neuropathy; in the case of oxaliplatin, it is caused by platinum retention, and in the case of paclitaxel, it is caused by binding of paclitaxel to microtubules and a consequent inhibition of axonal transport. Since the result of paclitaxel is not reported, we presume that the result was negative, i.e., PledOx did not offer any neuroprotective efficacy against paclitaxel. The presumed negative result in turn suggests that the neuroprotective efficacy of PledOx, as that seen in mice, is a property independent of Mn2+, through a so-called chelation therapeutic mechanism of action.
9. Conclusions
A costly lesson to be learned from POLAR trials, for both participating patients and shareholders, is that PledOx should not be used in combination with platinum-containing drugs, such as oxaliplatin and cisplatin. This should have been obvious already during a well conducted and monitored phase II trial.
Author Contributions
Writing—review and editing: J.O.G.K., P.J. and L.J.I. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Karlsson-Tuner Invest AS, Norway.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
J.O.G.K., P.J. and L.J.I. are founders of PledPharma AB.; J.O.G.K. is a former employee of PledPharma; L.J.I. and P.J. are former scientific advisors to PledPharma; J.O.G.K. is the first inventor on two granted patent families (e.g., US8377969, US8633174, and US9187509) covering the therapeutic use of calmangafodipir, which are owned by PledPharma/Egetis; J.O.G.K. and P.J. are, furthermore, inventors on a granted patent family (e.g., US10888553) covering the therapeutic use of fodipir to treat platinum-induced peripheral sensory neuropathy, which is owned by Karlsson-Tuner Invest AS; P.J. is scientific advisor to and owns shares in IC Targets AS, a company developing mangafodipir as a contrast agent for MRI.
References
- Pfeiffer, P.; Lustberg, M.; Näsström, J.; Carlsson, S.; Persson, A.; Nagahama, F.; Cavaletti, G.; Glimelius, B.; Muro, K. Calmangafodipir for Prevention of Oxaliplatin-Induced Peripheral Neuropathy: Two Placebo-Controlled, Randomized Phase 3 Studies (POLAR-A/POLAR-M). JNCI Cancer Spectr. 2022, 6, pkac075. [Google Scholar] [CrossRef] [PubMed]
- Qvortrup, C.; Muro, K.; Lustberg, M.; Persson, A.; Näsström, J.; Carlsson, S.; Nagahama, F.; Pfeiffer, P. SO-17 The global POLAR program: Top-line results of placebo-controlled studies of calmangafodipir on top of modified FOLFOX6 to prevent chemotherapy-induced peripheral neuropathy. Ann. Oncol. 2021, 32 (Suppl. S3), S209–S210. [Google Scholar] [CrossRef]
- Karlsson, J.O.; Ignarro, L.J.; Lundström, I.; Jynge, P.; Almén, T. Calmangafodipir [Ca4Mn(DPDP)5], mangafodipir (MnDPDP) and MnPLED with special reference to their SOD mimetic and therapeutic properties. Drug Discov. Today 2015, 20, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Brurok, H.; Ardenkjaer-Larsen, J.H.; Hansson, G.; Skarra, S.; Berg, K.; Karlsson, J.O.; Laursen, I.; Jynge, P. Manganese dipyridoxyl diphosphate: MRI contrast agent with antioxidative and cardioprotective properties? Biochem. Biophys. Res. Commun. 1999, 254, 768–772. [Google Scholar] [CrossRef]
- Karlsson, J.O.; Brurok, H.; Eriksen, M.; Towart, R.; Toft, K.G.; Moen, O.; Engebretsen, B.; Jynge, P.; Refsum, H. Cardioprotective effects of the MR contrast agent MnDPDP and its metabolite MnPLED upon reperfusion of the ischemic porcine myocardium. Acta Radiol. 2001, 42, 540–547. [Google Scholar] [CrossRef]
- Smith, H.J. Contrast-enhanced MR imaging in the diagnosis and preservation of cardiac viability. Acta Radiol. 2001, 42, 539. [Google Scholar] [CrossRef]
- Bedda, S.; Laurent, A.; Conti, F.; Chéreau, C.; Tran, A.; Tran-Van Nhieu, J.; Jaffray, P.; Soubrane, O.; Goulvestre, C.; Calmus, Y.; et al. Mangafodipir prevents liver injury induced by acetaminophen in the mouse. J. Hepatol. 2003, 39, 765–772. [Google Scholar] [CrossRef]
- Laurent, A.; Nicco, C.; Chéreau, C.; Goulvestre, C.; Alexandre, J.; Alves, A.; Lévy, E.; Goldwasser, F.; Panis, Y.; Soubrane, O.; et al. Controlling tumor growth by modulating endogenous production of reactive oxygen species. Cancer Res. 2005, 65, 948–956. [Google Scholar] [CrossRef]
- Alexandre, J.; Nicco, C.; Chéreau, C.; Laurent, A.; Weill, B.; Goldwasser, F.; Batteux, F. Improvement of the therapeutic index of anticancer drugs by the superoxide dismutase mimic mangafodipir. J. Natl. Cancer Inst. 2006, 98, 236–244. [Google Scholar] [CrossRef]
- Doroshow, J.H. Redox modulation of chemotherapy-induced tumor cell killing and normal tissue toxicity. J. Natl. Cancer Inst. 2006, 98, 223–225. [Google Scholar] [CrossRef]
- Laskar, A.; Miah, S.; Andersson, R.G.; Li, W. Prevention of 7β-hydroxycholesterol-induced cell death by mangafodipir is mediated through lysosomal and mitochondrial pathways. Eur. J. Pharmacol. 2010, 640, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Coriat, R.; Leconte, M.; Kavian, N.; Bedda, S.; Nicco, C.; Chereau, C.; Goulvestre, C.; Weill, B.; Laurent, A.; Batteux, F. Mangafodipir protects against hepatic ischemia-reperfusion injury in mice. PLoS ONE 2011, 6, e27005. [Google Scholar] [CrossRef]
- Karlsson, J.O.; Kurz, T.; Flechsig, S.; Näsström, J.; Andersson, R.G. Superior therapeutic index of calmangafodipir in comparison to mangafodipir as a chemotherapy adjunct. Transl. Oncol. 2012, 5, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, J.O.; Adolfsson, K.; Thelin, B.; Jynge, P.; Andersson, R.G.; Falkmer, U.G. First clinical experience with the magnetic resonance imaging contrast agent and superoxide dismutase mimetic mangafodipir as an adjunct in cancer chemotherapy-a translational study. Transl. Oncol. 2012, 5, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Kurz, T.; Grant, D.; Andersson, R.G.; Towart, R.; De Cesare, M.; Karlsson, J.O. Effects of MnDPDP and ICRF-187 on Doxorubicin-Induced Cardiotoxicity and Anticancer Activity. Transl. Oncol. 2012, 5, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Coriat, R.; Alexandre, J.; Nicco, C.; Quinquis, L.; Benoit, E.; Chéreau, C.; Lemaréchal, H.; Mir, O.; Borderie, D.; Tréluyer, J.M.; et al. Treatment of oxaliplatin-induced peripheral neuropathy by intravenous mangafodipir. J. Clin. Investig. 2014, 124, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, J.E.; El-Saadi, W.; Ali, M.; Puskar, W.; Skogvard, P.; Engvall, J.E.; Andersson, R.G.; Maret, E.; Jynge, P. Mangafodipir as a cardioprotective adjunct to reperfusion therapy: A feasibility study in patients with ST-segment elevation myocardial infarction. Eur. Heart J. Cardiovasc. Pharmacother. 2015, 1, 39–45. [Google Scholar] [CrossRef]
- Karlsson, J.O.G.; Andersson, R.G.; Jynge, P. Mangafodipir a Selective Cytoprotectant—With Special Reference to Oxaliplatin and Its Association to Chemotherapy-Induced Peripheral Neuropathy (CIPN). Transl. Oncol. 2017, 10, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Morrison, E.E.; Oatey, K.; Gallagher, B.; Grahamslaw, J.; O’Brien, R.; Black, P.; Oosthuyzen, W.; Lee, R.J.; Weir, C.J.; Henriksen, D.; et al. Principal results of a randomised open label exploratory, safety and tolerability study with calmangafodipir in patients treated with a 12 h regimen of N-acetylcysteine for paracetamol overdose (POP trial). EBioMedicine 2019, 46, 423–430. [Google Scholar] [CrossRef]
- Glimelius, B.; Manojlovic, N.; Pfeiffer, P.; Mosidze, B.; Kurteva, G.; Karlberg, M.; Mahalingam, D.; Buhl Jensen, P.; Kowalski, J.; Bengtson, M.; et al. Persistent prevention of oxaliplatin-induced peripheral neuropathy using calmangafodipir (PledOx®): A placebo-controlled randomised phase II study (PLIANT). Acta Oncol. 2018, 57, 393–402. [Google Scholar] [CrossRef]
- Karlsson, J.O.G.; Jynge, P. Is it possible to draw firm conclusions from the PLIANT trial? Acta Oncol. 2018, 57, 862–864. [Google Scholar] [CrossRef]
- Glimelius, B.; Kowalski, J.; Näsström, J. The PLIANT trial gives trustworthy data. Acta Oncol. 2018, 57, 864–866. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, J.O.G.; Jynge, P.; Ignarro, L.J. The Damaging Outcome of the POLAR Phase III Trials Was Due to Avoidable Time-Dependent Redox Interaction between Oxaliplatin and PledOx. Antioxidants 2021, 10, 1937. [Google Scholar] [CrossRef] [PubMed]
- Radi, R. Protein tyrosine nitration: Biochemical mechanisms and structural basis of functional effects. Acc. Chem. Res. 2013, 46, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Koppenol, W.H. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and ugly. Am. J. Physiol. 1996, 271 Pt 1, C1424–C1437. [Google Scholar] [CrossRef] [PubMed]
- Graham, M.A.; Lockwood, G.F.; Greenslade, D.; Brienza, S.; Bayssas, M.; Gamelin, E. Clinical pharmacokinetics of oxaliplatin: A critical review. Clin. Cancer Res. 2000, 6, 1205–1218. [Google Scholar] [PubMed]
- Stavniichuk, R.; Shevalye, H.; Lupachyk, S.; Obrosov, A.; Groves, J.T.; Obrosova, I.G.; Yorek, M.A. Peroxynitrite and protein nitration in the pathogenesis of diabetic peripheral neuropathy. Diabetes Metab. Res. Rev. 2014, 30, 669–678. [Google Scholar] [CrossRef]
- Stehr, J.E.; Lundström, I.; Karlsson, J.O.G. Evidence that fodipir (DPDP) binds neurotoxic Pt2+ with a high affinity: An electron paramagnetic resonance study. Sci. Rep. 2019, 9, 15813. [Google Scholar] [CrossRef]
- Canta, A.; Chiorazzi, A.; Pozzi, E.; Fumagalli, G.; Monza, L.; Meregalli, C.; Carozzi, V.A.; Rodriguez-Menendez, V.; Oggioni, N.; Näsström, J.; et al. Calmangafodipir Reduces Sensory Alterations and Prevents Intraepidermal Nerve Fibers Loss in a Mouse Model of Oxaliplatin Induced Peripheral Neurotoxicity. Antioxidants 2020, 9, 594. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).