
Repurposing Verapamil to Enhance Killing of T-ALL Cells by the mTOR Inhibitor Everolimus

 , ,
, ,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. T-ALL Cell Lines and Primary Samples
2.2. RNA-seq and Bioinformatic Analysis
2.3. Cell Death Assay
2.4. Synergy Study
2.5. Mass Spectrometry and Quantification of Intracellular Levels of Everolimus
2.6. Hoechst 33342 Exclusion Assay
2.7. Analysis of ROS Production in Living Cells
2.8. Lipid Peroxidation Assay
2.9. Immunoblotting and Protein Oxidation Status
2.10. In Vivo Experiments
2.11. Statistical Analysis and Graphics
3. Results
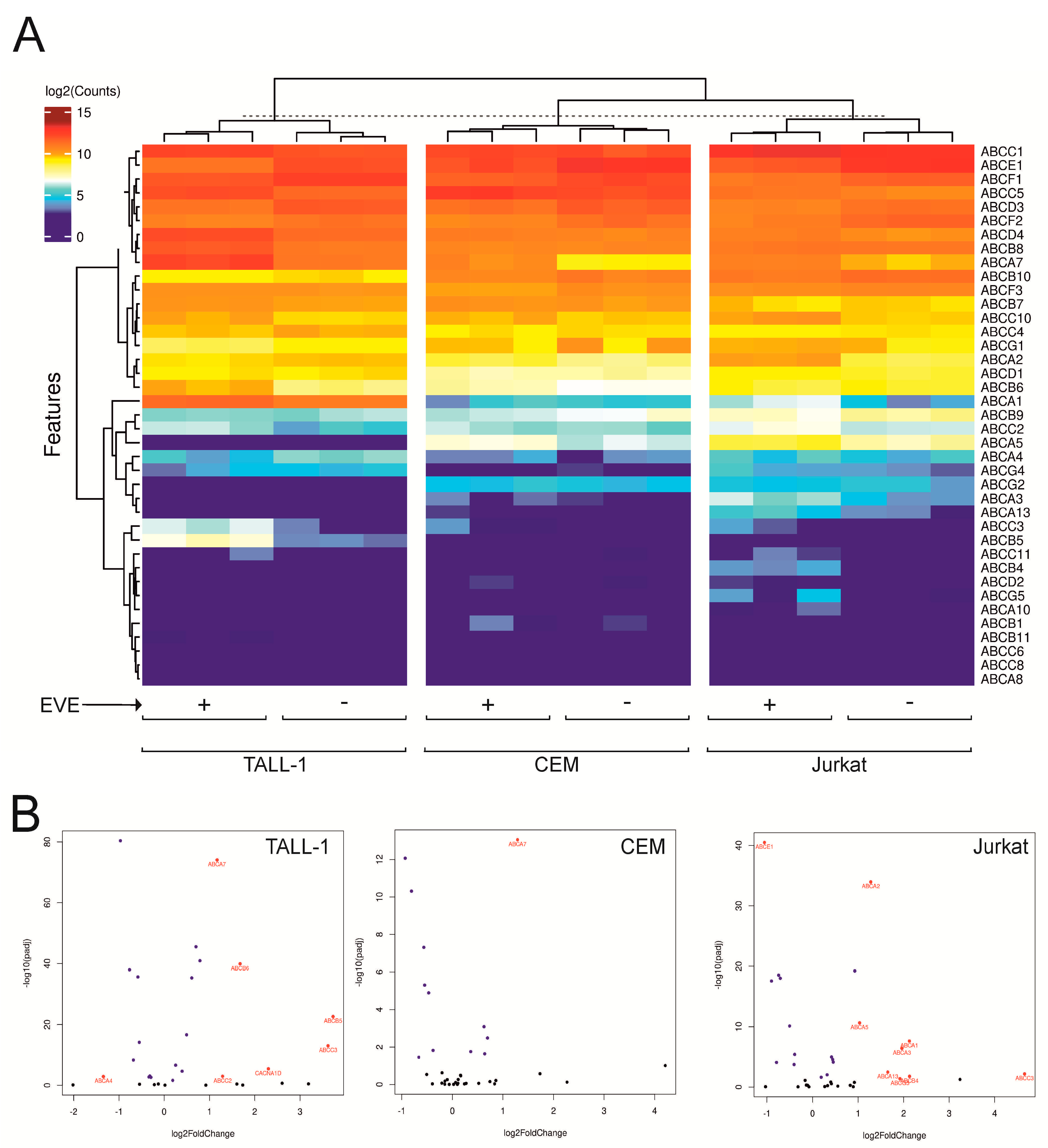
3.1. Expression of ABC Transporters in T-ALL Cells
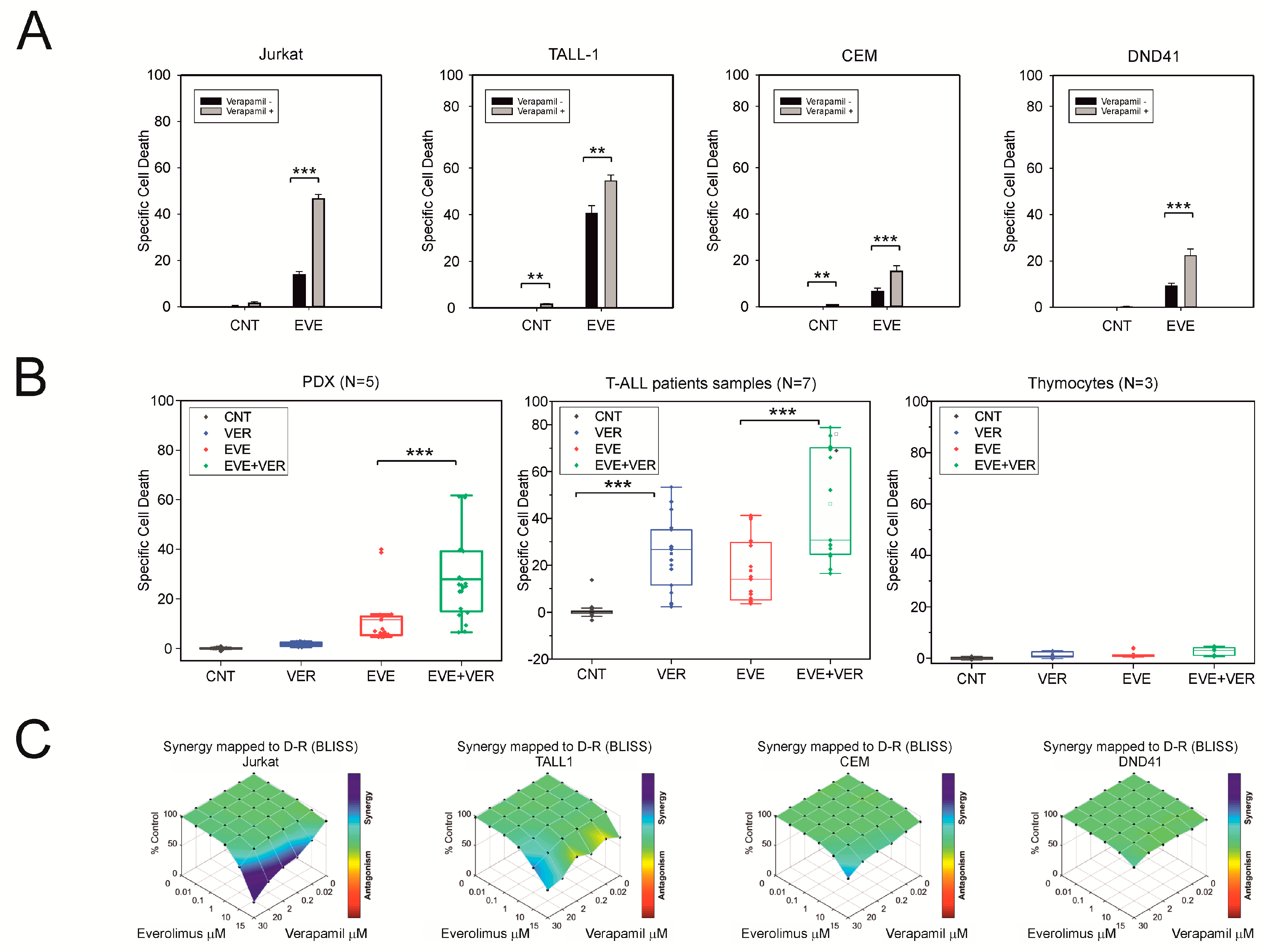
3.2. Verapamil Enhances the Killing Effect of Everolimus in T-ALL Cells
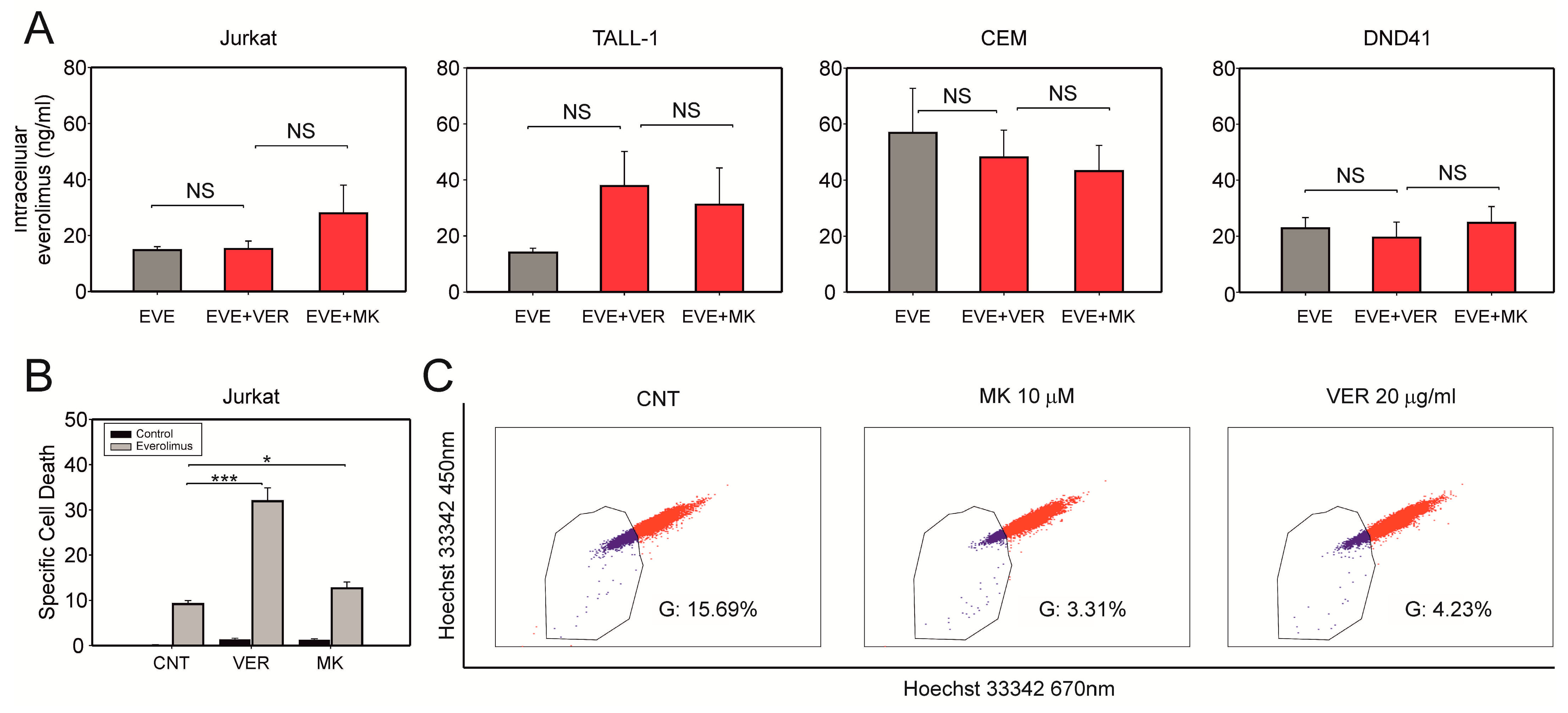
3.3. Verapamil does Not Significantly Change the Intracellular Concentration of Everolimus
3.4. Verapamil Enhances the Effects of Everolimus on Redox Homeostasis
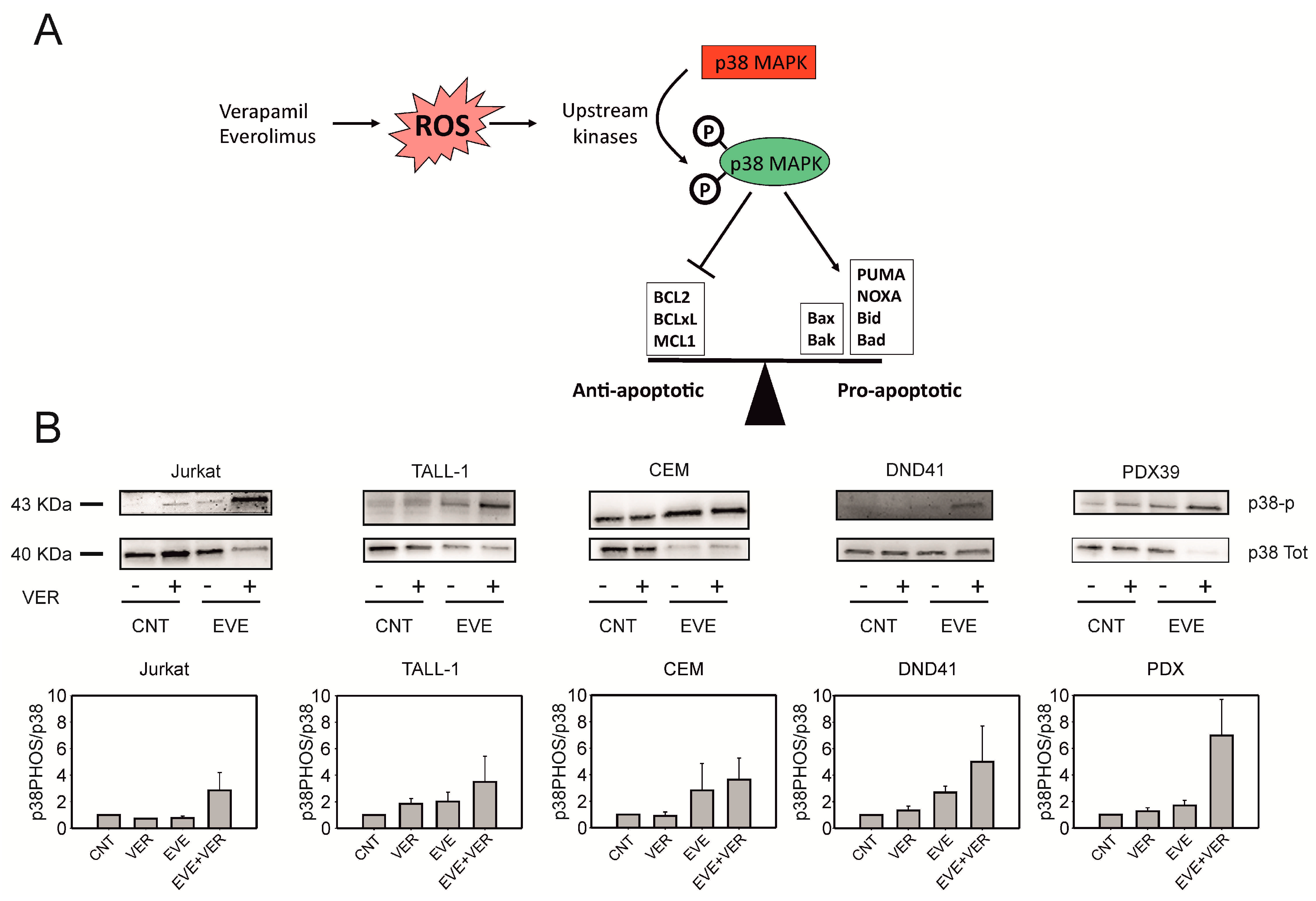
3.5. Verapamil and Everolimus Induce Phosphorylation of p38 MAPK
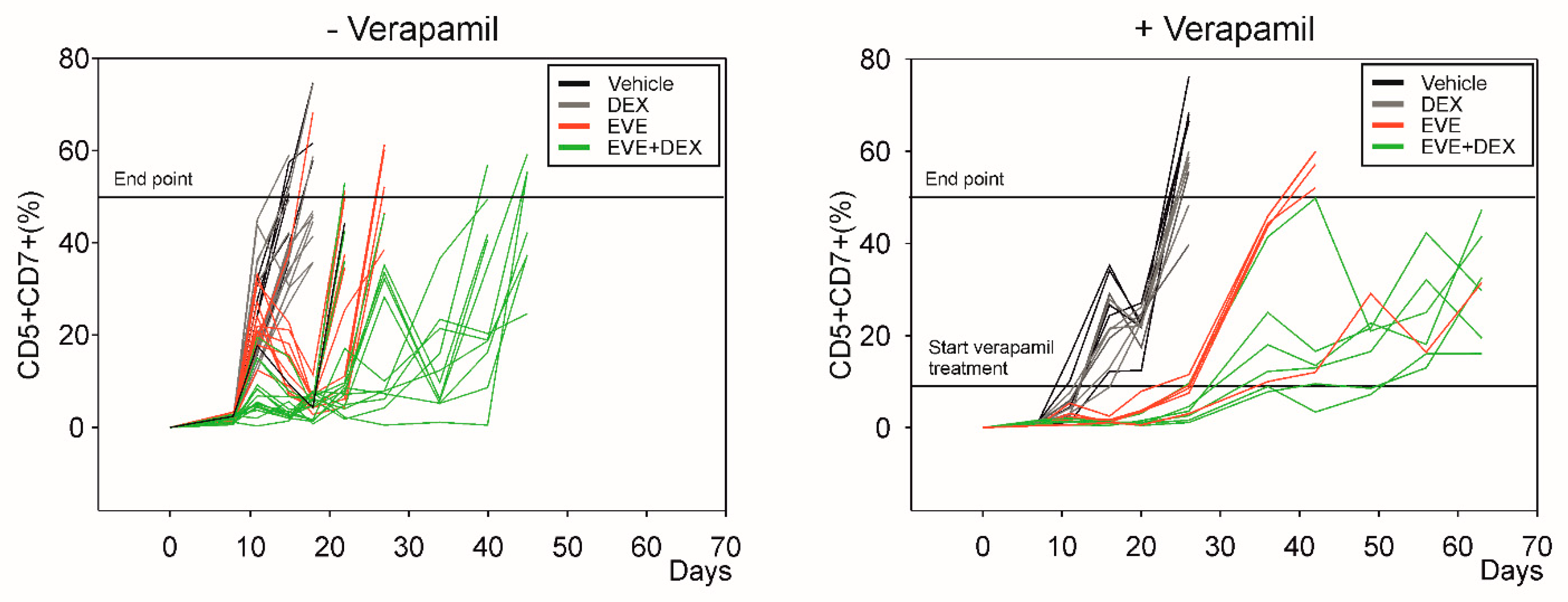
3.6. Verapamil Potentiates the Effects of Everolimus In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Karrman, K.; Johansson, B. Pediatric T-cell acute lymphoblastic leukemia. Genes Chromosomes Cancer 2017, 56, 89–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhadri, V.A.; Trahair, T.N.; Lock, R.B. Glucocorticoid resistance in paediatric acute lymphoblastic leukaemia. J. Paediatr. Child. Health 2012, 48, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.; Yunes, J.A.; Cardoso, B.A.; Martins, L.R.; Jotta, P.Y.; Abecasis, M.; Nowill, A.E.; Leslie, N.R.; Cardoso, A.A.; Barata, J.T. PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell leukemia viability. J. Clin. Investig. 2008, 118, 3762–3774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, A.; Girio, A.; Cebola, I.; Santos, C.I.; Antunes, F.; Barata, J.T. Intracellular reactive oxygen species are essential for PI3K/Akt/mTOR-dependent IL-7-mediated viability of T-cell acute lymphoblastic leukemia cells. Leukemia 2011, 25, 960–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silic-Benussi, M.; Cavallari, I.; Vajente, N.; Vidali, S.; Chieco-Bianchi, L.; Di Lisa, F.; Saggioro, D.; D’Agostino, D.M.; Ciminale, V. Redox regulation of T-cell turnover by the p13 protein of human T-cell leukemia virus type 1: Distinct effects in primary versus transformed cells. Blood 2010, 116, 54–62. [Google Scholar] [CrossRef]
- Silic-Benussi, M.; Cannizzaro, E.; Venerando, A.; Cavallari, I.; Petronilli, V.; La Rocca, N.; Marin, O.; Chieco-Bianchi, L.; Di Lisa, F.; D’Agostino, D.M.; et al. Modulation of mitochondrial K(+) permeability and reactive oxygen species production by the p13 protein of human T-cell leukemia virus type 1. Biochim. Biophys. Acta 2009, 1787, 947–954. [Google Scholar] [CrossRef]
- Silic-Benussi, M.; Sharova, E.; Ciccarese, F.; Cavallari, I.; Raimondi, V.; Urso, L.; Corradin, A.; Kotler, H.; Scattolin, G.; Buldini, B.; et al. mTOR inhibition downregulates glucose-6-phosphate dehydrogenase and induces ROS-dependent death in T-cell acute lymphoblastic leukemia cells. Redox Biol. 2022, 51, 102268. [Google Scholar] [CrossRef]
- Evangelisti, C.; Chiarini, F.; McCubrey, J.A.; Martelli, A.M. Therapeutic Targeting of mTOR in T-Cell Acute Lymphoblastic Leukemia: An Update. Int. J. Mol. Sci. 2018, 19, 1878. [Google Scholar] [CrossRef] [Green Version]
- Gottesman, M.M.; Pastan, I.H. The Role of Multidrug Resistance Efflux Pumps in Cancer: Revisiting a JNCI Publication Exploring Expression of the MDR1 (P-glycoprotein) Gene. J. Natl. Cancer Inst. 2015, 107, 452. [Google Scholar] [CrossRef] [Green Version]
- Tsuruo, T.; Iida, H.; Tsukagoshi, S.; Sakurai, Y. Overcoming of vincristine resistance in P388 leukemia in vivo and in vitro through enhanced cytotoxicity of vincristine and vinblastine by verapamil. Cancer Res. 1981, 41, 1967–1972. [Google Scholar]
- Agnusdei, V.; Minuzzo, S.; Frasson, C.; Grassi, A.; Axelrod, F.; Satyal, S.; Gurney, A.; Hoey, T.; Seganfreddo, E.; Basso, G.; et al. Therapeutic antibody targeting of Notch1 in T-acute lymphoblastic leukemia xenografts. Leukemia 2014, 28, 278–288. [Google Scholar] [CrossRef]
- Silic-Benussi, M.; Scattolin, G.; Cavallari, I.; Minuzzo, S.; Del Bianco, P.; Francescato, S.; Basso, G.; Indraccolo, S.; D’Agostino, D.M.; Ciminale, V. Selective killing of human T-ALL cells: An integrated approach targeting redox homeostasis and the OMA1/OPA1 axis. Cell Death Dis. 2018, 9, 822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Society. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Vasiliou, V.; Vasiliou, K.; Nebert, D.W. Human ATP-binding cassette (ABC) transporter family. Hum. Genom. 2009, 3, 281–290. [Google Scholar] [CrossRef]
- Di Veroli, G.Y.; Fornari, C.; Wang, D.; Mollard, S.; Bramhall, J.L.; Richards, F.M.; Jodrell, D.I. Combenefit: An interactive platform for the analysis and visualization of drug combinations. Bioinformatics 2016, 32, 2866–2868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Telford, W.G.; Bradford, J.; Godfrey, W.; Robey, R.W.; Bates, S.E. Side population analysis using a violet-excited cell-permeable DNA binding dye. Stem. Cells 2007, 25, 1029–1036. [Google Scholar] [CrossRef]
- Whitaker, R.H.; Cook, J.G. Stress Relief Techniques: p38 MAPK Determines the Balance of Cell Cycle and Apoptosis Pathways. Biomolecules 2021, 11, 1444. [Google Scholar] [CrossRef]
- Hofmann, W.K.; Trumpp, A.; Müller-Tidow, C. Therapy resistance mechanisms in hematological malignancies. Int. J. Cancer 2023, 152, 340–347. [Google Scholar] [CrossRef]
- Plasschaert, S.L.; de Bont, E.S.; Boezen, M.; vander Kolk, D.M.; Daenen, S.M.; Faber, K.N.; Kamps, W.A.; de Vries, E.G.; Vellenga, E. Expression of multidrug resistance-associated proteins predicts prognosis in childhood and adult acute lymphoblastic leukemia. Clin. Cancer Res. 2005, 11, 8661–8668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinbach, D.; Wittig, S.; Cario, G.; Viehmann, S.; Mueller, A.; Gruhn, B.; Haefer, R.; Zintl, F.; Sauerbrey, A. The multidrug resistance-associated protein 3 (MRP3) is associated with a poor outcome in childhood ALL and may account for the worse prognosis in male patients and T-cell immunophenotype. Blood 2003, 102, 4493–4498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowe, A.; Lemaire, M. In vitro and in situ absorption of SDZ-RAD using a human intestinal cell line (Caco-2) and a single pass perfusion model in rats: Comparison with rapamycin. Pharm. Res. 1998, 15, 1666–1672. [Google Scholar] [CrossRef] [PubMed]
- Cullen, K.V.; Davey, R.A.; Davey, M.W. Verapamil-stimulated glutathione transport by the multidrug resistance-associated protein (MRP1) in leukaemia cells. Biochem. Pharmacol. 2001, 62, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Trompier, D.; Chang, X.B.; Barattin, R.; du Moulinet D’Hardemare, A.; Di Pietro, A.; Baubichon-Cortay, H. Verapamil and its derivative trigger apoptosis through glutathione extrusion by multidrug resistance protein MRP1. Cancer Res. 2004, 64, 4950–4956. [Google Scholar] [CrossRef] [Green Version]
- Pluchino, K.M.; Hall, M.D.; Goldsborough, A.S.; Callaghan, R.; Gottesman, M.M. Collateral sensitivity as a strategy against cancer multidrug resistance. Drug Resist. Updat. 2012, 15, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Aguanno, D.; Board, M.; Callaghan, R. Exploiting the metabolic energy demands of drug efflux pumps provides a strategy to overcome multidrug resistance in cancer. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129915. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Giorgi, C.; Siviero, R.; Zecchini, E.; Rizzuto, R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 2008, 27, 6407–6418. [Google Scholar] [CrossRef] [Green Version]
- Erdogmus, S.; Concepcion, A.R.; Yamashita, M.; Sidhu, I.; Tao, A.Y.; Li, W.; Rocha, P.; Huang, B.; Garippa, R.; Lee, B.; et al. Cavβ1 regulates T cell expansion and apoptosis independently of voltage-gated Ca2+ channel function. Nat. Commun. 2022, 13, 2033. [Google Scholar] [CrossRef]
- Pancrazio, J.J.; Viglione, M.P.; Kleiman, R.J.; Kim, Y.I. Verapamil-induced blockade of voltage-activated K+ current in small-cell lung cancer cells. J. Pharmacol. Exp. Ther. 1991, 257, 184–191. [Google Scholar]
- Chandy, K.G.; Wulff, H.; Beeton, C.; Pennington, M.; Gutman, G.A.; Cahalan, M.D. K+ channels as targets for specific immunomodulation. Trends Pharmacol. Sci. 2004, 25, 280–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cahalan, M.D.; Chandy, K.G. The functional network of ion channels in T lymphocytes. Immunol. Rev. 2009, 231, 59–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leanza, L.; Henry, B.; Sassi, N.; Zoratti, M.; Chandy, K.G.; Gulbins, E.; Szabò, I. Inhibitors of mitochondrial Kv1.3 channels induce Bax/Bak-independent death of cancer cells. EMBO Mol. Med. 2012, 4, 577–593. [Google Scholar] [CrossRef] [PubMed]
- Szabo, I.; Zoratti, M.; Biasutto, L. Targeting mitochondrial ion channels for cancer therapy. Redox Biol. 2021, 42, 101846. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [Green Version]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silic-Benussi, M.; Sharova, E.; Corradin, A.; Urso, L.; Raimondi, V.; Cavallari, I.; Buldini, B.; Francescato, S.; Minuzzo, S.A.; D’Agostino, D.M.; et al. Repurposing Verapamil to Enhance Killing of T-ALL Cells by the mTOR Inhibitor Everolimus. Antioxidants 2023, 12, 625. https://doi.org/10.3390/antiox12030625
Silic-Benussi M, Sharova E, Corradin A, Urso L, Raimondi V, Cavallari I, Buldini B, Francescato S, Minuzzo SA, D’Agostino DM, et al. Repurposing Verapamil to Enhance Killing of T-ALL Cells by the mTOR Inhibitor Everolimus. Antioxidants. 2023; 12(3):625. https://doi.org/10.3390/antiox12030625
Chicago/Turabian StyleSilic-Benussi, Micol, Evgeniya Sharova, Alberto Corradin, Loredana Urso, Vittoria Raimondi, Ilaria Cavallari, Barbara Buldini, Samuela Francescato, Sonia A. Minuzzo, Donna M. D’Agostino, and et al. 2023. "Repurposing Verapamil to Enhance Killing of T-ALL Cells by the mTOR Inhibitor Everolimus" Antioxidants 12, no. 3: 625. https://doi.org/10.3390/antiox12030625
APA StyleSilic-Benussi, M., Sharova, E., Corradin, A., Urso, L., Raimondi, V., Cavallari, I., Buldini, B., Francescato, S., Minuzzo, S. A., D’Agostino, D. M., & Ciminale, V. (2023). Repurposing Verapamil to Enhance Killing of T-ALL Cells by the mTOR Inhibitor Everolimus. Antioxidants, 12(3), 625. https://doi.org/10.3390/antiox12030625