
Neuroprotection of NRF2 against Ferroptosis after Traumatic Brain Injury in Mice
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Model Handling and Sample Collection
2.3. Immunofluorescence, TUNEL, and Fluoro-Jade C Staining
2.4. Nissl Staining and Perl’s Staining
2.5. Iron Content Determination
2.6. Western Blotting Analysis
2.7. Glutathione Detection
2.8. Transmission Electron Microscopy
2.9. Quantitative Real-Time PCR
2.10. Assessment of Neuronal Function
2.11. Statistical Analysis
3. Results
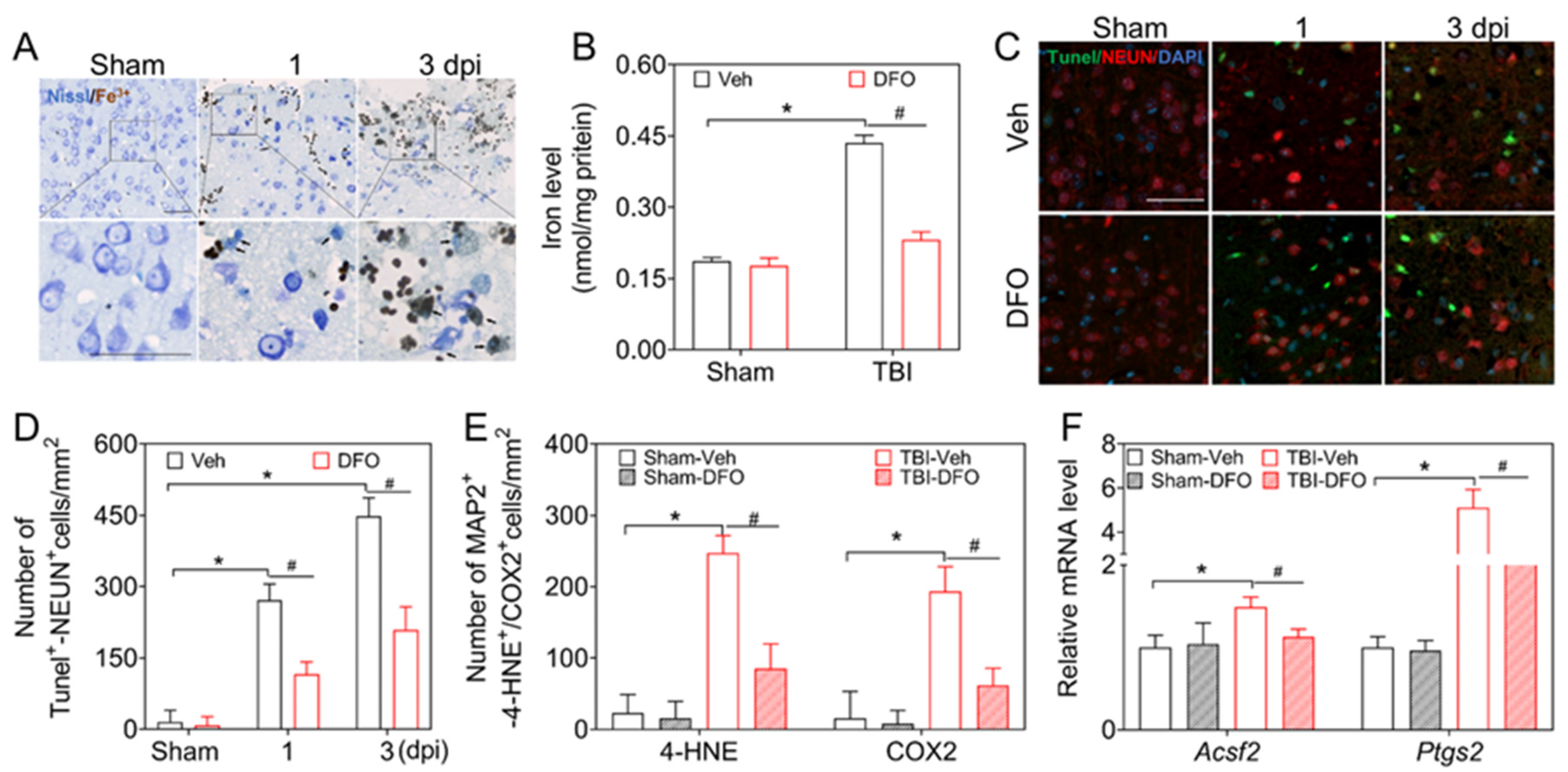
3.1. Ferroptosis Is Related to Neuronal Damage and Dysfunction after TBI
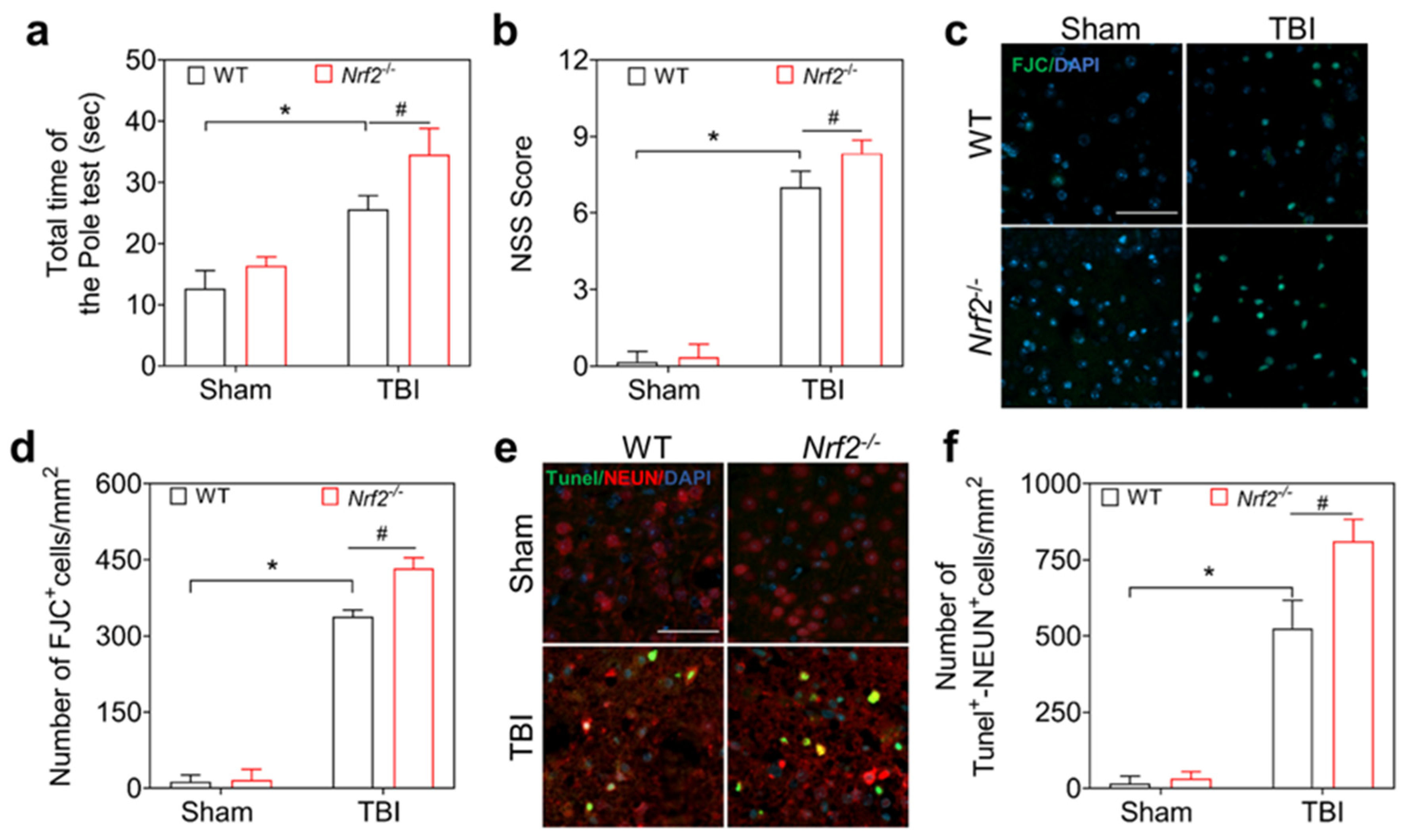
3.2. Nrf2 Deficiency Aggravates Neurological Dysfunction and Neuronal Damage after TBI in Mice
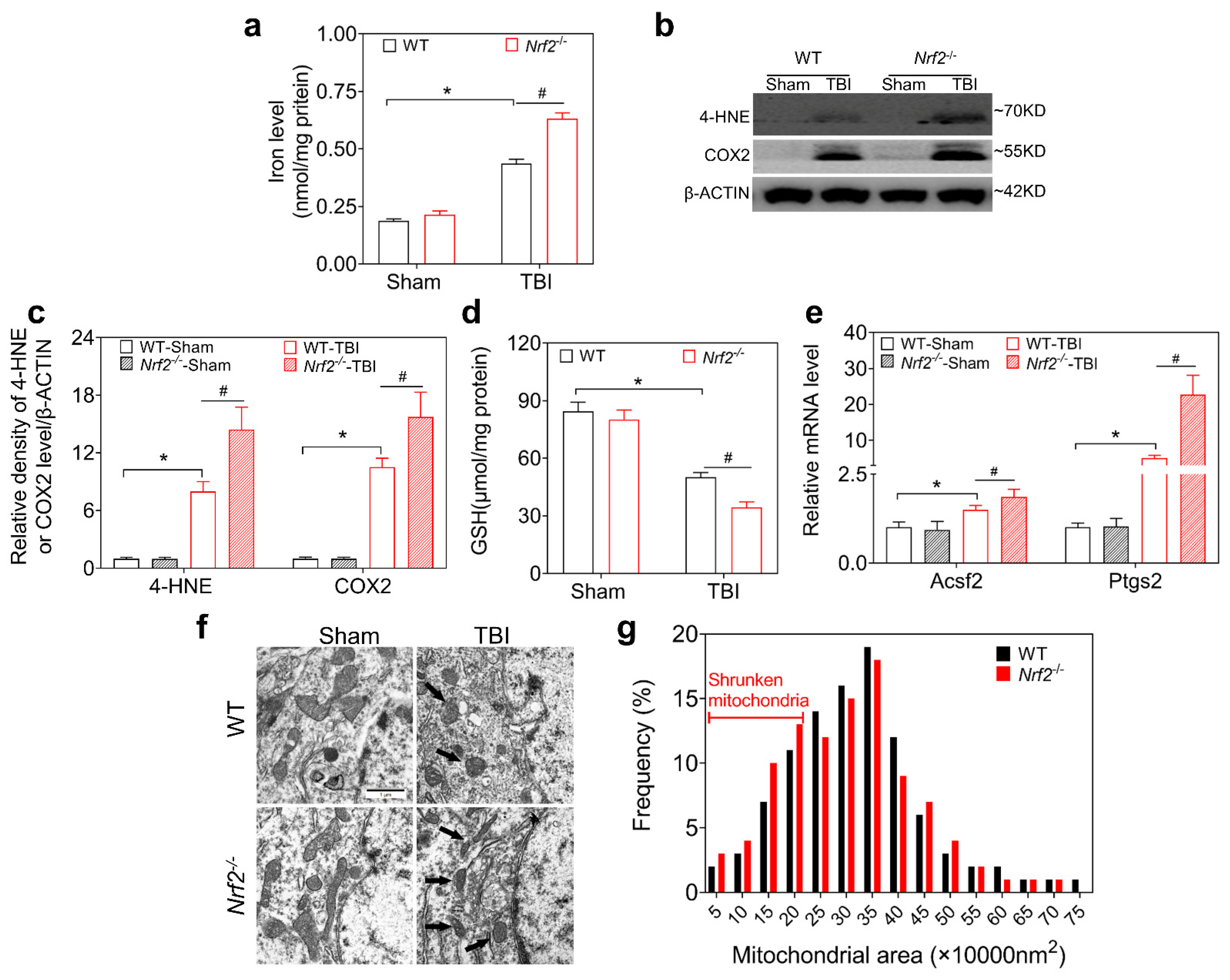
3.3. NRF2 Deletion Aggravates TBI-Induced Ferroptosis
3.4. DMF Mitigates TBI-Induced Ferroptosis and Neurological Deficits
3.5. NRF2 Regulates Ferroptosis through FTH-FTL, xCT-GPX4, and FSP1 after TBI
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maas, A.I.R.; Menon, D.K.; Adelson, P.D.; Andelic, N.; Bell, M.J.; Belli, A.; Bragge, P.; Brazinova, A.; Büki, A.; Chesnut, R.M.; et al. Traumatic brain injury: Integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 2017, 16, 987–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iaccarino, C.; Carretta, A.; Nicolosi, F.; Morselli, C. Epidemiology of severe traumatic brain injury. J. Neurosurg. Sci. 2018, 62, 535–541. [Google Scholar] [CrossRef]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal Cell Death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-J.; Chen, T.-H.; Yang, L.-Y.; Shih, C.-M. Resveratrol protects astrocytes against traumatic brain injury through inhibiting apoptotic and autophagic cell death. Cell Death Dis. 2014, 5, e1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wehn, A.C.; Khalin, I.; Duering, M.; Hellal, F.; Culmsee, C.; Vandenabeele, P.; Plesnila, N.; Terpolilli, N.A. RIPK1 or RIPK3 deletion prevents progressive neuronal cell death and improves memory function after traumatic brain injury. Acta Neuropathol. Commun. 2021, 9, 138. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.-Y.; Greig, N.H.; Tweedie, D.; Jung, Y.J.; Chiang, Y.-H.; Hoffer, B.J.; Miller, J.P.; Chang, K.-H.; Wang, J.-Y. The p53 inactivators pifithrin-mu and pifithrin-alpha mitigate TBI-induced neuronal damage through regulation of oxidative stress, neuroinflammation, autophagy and mitophagy. Exp. Neurol. 2020, 324, 113135. [Google Scholar] [CrossRef]
- Garton, T.; Keep, R.F.; Hua, Y.; Xi, G. Brain iron overload following intracranial haemorrhage. Stroke Vasc. Neurol. 2016, 1, 172–184. [Google Scholar] [CrossRef]
- Xiong, X.-Y.; Wang, J.; Qian, Z.-M.; Yang, Q.-W. Iron and intracerebral hemorrhage: From mechanism to translation. Transl. Stroke Res. 2014, 5, 429–441. [Google Scholar] [CrossRef]
- Pontel, L.B.; Bueno-Costa, A.; Morellato, A.E.; Santos, J.C.; Roué, G.; Esteller, M. Acute lymphoblastic leukemia necessitates GSH-dependent ferroptosis defenses to overcome FSP1-epigenetic silencing. Redox Biol. 2022, 55, 102408. [Google Scholar] [CrossRef]
- Yang, M.; Tsui, M.G.; Tsang, J.K.W.; Goit, R.K.; Yao, K.-M.; So, K.-F.; Lam, W.-C.; Lo, A.C.Y. Involvement of FSP1-CoQ10-NADH and GSH-GPx-4 pathways in retinal pigment epithelium ferroptosis. Cell Death Dis. 2022, 13, 468. [Google Scholar] [CrossRef]
- Kenny, E.M.; Fidan, E.; Yang, Q.; Anthonymuthu, T.S.; New, L.A.; Meyer, E.A.; Wang, H.; Kochanek, P.M.; Dixon, C.E.; Kagan, V.E.; et al. Ferroptosis contributes to neuronal death and functional outcome after traumatic brain injury. Crit. Care Med. 2019, 47, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Jiang, Y.; Liang, W.; Wang, Y.; Cao, S.; Yan, H.; Gao, L.; Zhang, L. miR-212-5p attenuates ferroptotic neuronal death after traumatic brain injury by targeting Ptgs2. Mol. Brain 2019, 12, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.; Wang, N.; Ma, X.; Wang, P.; Dong, W.; Chen, Z.; Wu, M.; Wang, Z.; Wang, L.; Guan, D.; et al. Spatial-temporal changes of iron deposition and iron metabolism after traumatic brain injury in mice. Front. Mol. Neurosci. 2022, 15, 949573. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Weiland, A.; Chen, X.; Lan, X.; Han, X.; Durham, F.; Liu, X.; Wan, J.; Ziai, W.C.; Hanley, D.F.; et al. Ultrastructural characteristics of neuronal death and white matter injury in mouse brain tissues after intracerebral hemorrhage: Coexistence of ferroptosis, autophagy, and necrosis. Front. Neurol. 2018, 9, 581. [Google Scholar] [CrossRef] [Green Version]
- Yao, X.; Zhang, Y.; Hao, J.; Duan, H.-Q.; Zhao, C.-X.; Sun, C.; Li, B.; Fan, B.-Y.; Li, W.-X.; Fu, X.-H.; et al. Deferoxamine promotes recovery of traumatic spinal cord injury by inhibiting ferroptosis. Neural Regen. Res. 2019, 14, 532–541. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Liu, S.; Li, J.; Li, Z.; Quan, J.; Liu, X.; Tang, Y.; Liu, B. The latest view on the mechanism of ferroptosis and its research progress in spinal cord injury. Oxidative Med. Cell. Longev. 2020, 2020, 6375938. [Google Scholar] [CrossRef]
- Dixon, S.J. Ferroptosis: Bug or feature? Immunol. Rev. 2017, 277, 150–157. [Google Scholar] [CrossRef]
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting ferroptosis to iron out cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef]
- de la Ossa, N.P.; Sobrino, T.; Silva, Y.; Blanco, M.; Millán, M.; Gomis, M.; Agulla, J.; Araya, P.; Reverté, S.; Serena, J.; et al. Iron-related brain damage in patients with intracerebral hemorrhage. Stroke 2010, 41, 810–813. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Hu, W.; Tang, Q.; Wang, L.; Sun, X.-G.; Chen, Y.; Yin, Y.; Xue, F.; Sun, Z. Effect comparison of both iron chelators on outcomes, iron deposit, and iron transporters after intracerebral hemorrhage in rats. Mol. Neurobiol. 2016, 53, 3576–3585. [Google Scholar] [CrossRef]
- Zille, M.; Karuppagounder, S.S.; Chen, Y.; Gough, P.J.; Bertin, J.; Finger, J.; Milner, T.A.; Jonas, E.A.; Ratan, R.R.; X, Z.; et al. Neuronal death after hemorrhagic stroke in vitro and in vivo shares features of ferroptosis and necroptosis. Stroke 2017, 48, 1033–1043. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Gao, P.; Chen, H.; Zhou, X.; Ou, Y.; He, Y. The role of iron, its metabolism and ferroptosis in traumatic brain injury. Front. Cell. Neurosci. 2020, 14, 590789. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by lipid peroxidation. Trends. Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arosio, P.; Ingrassia, R.; Cavadini, P. Ferritins: A family of molecules for iron storage, antioxidation and more. Biochim. Biophys. Acta 2009, 1790, 589–599. [Google Scholar] [CrossRef]
- Thompson, K.; Menzies, S.; Muckenthaler, M.; Torti, F.M.; Wood, T.; Torti, S.V.; Hentze, M.; Beard, J.; Connor, J. Mouse brains deficient in H-ferritin have normal iron concentration but a protein profile of iron deficiency and increased evidence of oxidative stress. J. Neurosci. Res. 2003, 71, 46–63. [Google Scholar] [CrossRef]
- Rui, T.; Wang, H.; Li, Q.; Cheng, Y.; Gao, Y.; Fang, X.; Ma, X.; Chen, G.; Gao, C.; Gu, Z.; et al. Deletion of ferritin H in neurons counteracts the protective effect of melatonin against traumatic brain injury-induced ferroptosis. J. Pineal. Res. 2021, 70, e12704. [Google Scholar] [CrossRef] [PubMed]
- Maccarinelli, F.; Pagani, A.; Cozzi, A.; Codazzi, F.; Di Giacomo, G.; Capoccia, S.; Rapino, S.; Finazzi, D.; Politi, L.S.; Cirulli, F.; et al. A novel neuroferritinopathy mouse model (FTL 498InsTC) shows progressive brain iron dysregulation, morphological signs of early neurodegeneration and motor coordination deficits. Neurobiol. Dis. 2015, 81, 119–133. [Google Scholar] [CrossRef] [Green Version]
- Ding, H.; Wang, X.; Wang, H.; Zhu, L.; Wang, Q.; Jia, Y.; Wei, W.; Zhou, C.; Wu, H.; Ding, K. Nrf2-ARE signaling provides neuroprotection in traumatic brain injury via modulation of the ubiquitin proteasome system. Neurochem. Int. 2017, 111, 32–44. [Google Scholar] [CrossRef]
- Huang, Y.; Wu, H.; Hu, Y.; Zhou, C.; Wu, J.; Wu, Y.; Wang, H.; Lenahan, C.; Huang, L.; Nie, S.; et al. Puerarin attenuates oxidative stress and ferroptosis via AMPK/PGC1alpha/Nrf2 pathway after subarachnoid hemorrhage in rats. Antioxidants 2022, 11, 1259. [Google Scholar] [CrossRef] [PubMed]
- Ge, M.; Tian, H.; Mao, L.; Li, D.; Lin, J.; Hu, H.; Huang, S.; Zhang, C.; Mei, X. Zinc attenuates ferroptosis and promotes functional recovery in contusion spinal cord injury by activating Nrf2/GPX4 defense pathway. CNS Neurosci. Ther. 2021, 27, 1023–1040. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Wang, H.; Yan, W.; Zhu, L.; Hu, Z.; Ding, Y.; Tang, K. Role of Nrf2 in protection against traumatic brain injury in mice. J. Neurotrauma 2009, 26, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Yang, B.; Wang, L.; Li, B.; Guo, X.; Zhang, M.; Jiang, Z.; Fu, J.; Pi, J.; Guan, D.; et al. Curcumin plays neuroprotective roles against traumatic brain injury partly via Nrf2 signaling. Toxicol. Appl. Pharmacol. 2018, 346, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.M.; Singh, I.N.; Wang, J.A.; Hall, E.D. Nrf2–ARE activator carnosic acid decreases mitochondrial dysfunction, oxidative damage and neuronal cytoskeletal degradation following traumatic brain injury in mice. Exp. Neurol. 2015, 264, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Dash, P.K.; Zhao, J.; Orsi, S.A.; Zhang, M.; Moore, A.N. Sulforaphane improves cognitive function administered following traumatic brain injury. Neurosci. Lett. 2009, 460, 103–107. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.Y.; Wang, H.D.; Xu, J.G.; Ding, K.; Li, T. Pretreatment with tert-butylhydroquinone attenuates cerebral oxidative stress in mice after traumatic brain injury. J. Surg. Res. 2013, 188, 206–212. [Google Scholar] [CrossRef]
- Okada, K.; Warabi, E.; Sugimoto, H.; Horie, M.; Tokushige, K.; Ueda, T.; Harada, N.; Taguchi, K.; Hashimoto, E.; Itoh, K.; et al. Nrf2 inhibits hepatic iron accumulation and counteracts oxidative stress-induced liver injury in nutritional steatohepatitis. J. Gastroenterol. 2012, 47, 924–935. [Google Scholar] [CrossRef]
- Jiang, T.; Cheng, H.; Su, J.; Wang, X.; Wang, Q.; Chu, J.; Li, Q. Gastrodin protects against glutamate-induced ferroptosis in HT-22 cells through Nrf2/HO-1 signaling pathway. Toxicol. Vitr. 2020, 62, 104715. [Google Scholar] [CrossRef]
- Gao, Q.; Zhang, G.; Zheng, Y.; Yang, Y.; Chen, C.; Xia, J.; Liang, L.; Lei, C.; Hu, Y.; Cai, X.; et al. SLC27A5 deficiency activates NRF2/TXNRD1 pathway by increased lipid peroxidation in HCC. Cell Death Differ. 2020, 27, 1086–1104. [Google Scholar] [CrossRef]
- Nicolay, J.P.; Albrecht, J.D.; Assaf, C.; Dippel, E.; Stadler, R.; Wehkamp, U.; Wobser, M.; Guelow, K.; Goerdt, S.; Krammer, P.H. Dimethyl fumarate (DMF) therapy in CTCL: Results from a clinical phase II study. Eur. J. Cancer 2021, 156 (Suppl. S1), S21–S22. [Google Scholar] [CrossRef]
- Xue, P.; Hou, Y.; Chen, Y.; Yang, B.; Fu, J.; Zheng, H.; Yarborough, K.; Woods, C.G.; Liu, D.; Yamamoto, M.; et al. Adipose deficiency of Nrf2 in ob/ob mice results in severe metabolic syndrome. Diabetes 2013, 62, 845–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Pan, K.; Chen, L.; Ning, J.-L.; Li, X.; Yang, T.; Terrando, N.; Gu, J.; Tao, G. Deferoxamine regulates neuroinflammation and iron homeostasis in a mouse model of postoperative cognitive dysfunction. J. Neuroinflamm. 2016, 13, 268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krämer, T.; Grob, T.; Menzel, L.; Hirnet, T.; Griemert, E.; Radyushkin, K.; Thal, S.; Methner, A.; Schaefer, M.K.E. Dimethyl fumarate treatment after traumatic brain injury prevents depletion of antioxidative brain glutathione and confers neuroprotection. J. Neurochem. 2017, 143, 523–533. [Google Scholar] [CrossRef]
- Casili, G.; Campolo, M.; Paterniti, I.; Lanza, M.; Filippone, A.; Cuzzocrea, S.; Esposito, E. Dimethyl fumarate attenuates neuroinflammation and neurobehavioral deficits induced by experimental traumatic brain injury. J. Neurotrauma 2018, 35, 1437–1451. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.H.; Martens, K.M.; Bashir, A.; Cheung, H.; Stukas, S.; Gibbs, E.; Namjoshi, D.R.; Button, E.B.; Wilkinson, A.; Barron, C.J.; et al. CHIMERA repetitive mild traumatic brain injury induces chronic behavioural and neuropathological phenotypes in wild-type and APP/PS1 mice. Alzheimers Res. Ther. 2019, 11, 6. [Google Scholar] [CrossRef] [Green Version]
- Leconte, C.; Benedetto, M.C.; Lentini, M.F.; Simon, M.K.; Ouaazizi, C.; Taib, T.; Cho, A.H.; Plotkine, M.; Mongeau, R.; Marchand-Leroux, C.; et al. Histological and behavioral evaluation after traumatic brain injury in mice: A ten months follow-up study. J. Neurotrauma 2020, 37, 1342–1357. [Google Scholar] [CrossRef]
- Wang, H.; Zhou, X.-M.; Wu, L.-Y.; Liu, G.-J.; Xu, W.-D.; Zhang, X.-S.; Gao, Y.-Y.; Tao, T.; Zhou, Y.; Lu, Y.; et al. Aucubin alleviates oxidative stress and inflammation via Nrf2-mediated signaling activity in experimental traumatic brain injury. J. Neuroinflamm. 2020, 17, 188. [Google Scholar] [CrossRef]
- Wu, A.-G.; Yong, Y.-Y.; Pan, Y.-R.; Zhang, L.; Wu, J.-M.; Zhang, Y.; Tang, Y.; Wei, J.; Yu, L.; Law, B.Y.-K.; et al. Targeting Nrf2-mediated oxidative stress response in traumatic brain injury: Therapeutic perspectives of phytochemicals. Oxid. Med. Cell Longev. 2022, 2022, 1015791. [Google Scholar] [CrossRef]
- Dong, W.; Sun, Y.; Cheng, H.; Yang, B.; Wang, L.; Jiang, Z.; Li, B.; Wen, S.; Guo, X.; Guan, D.; et al. Dynamic cell type-specific expression of Nrf2 after traumatic brain injury in mice. Eur. J. Neurosci. 2019, 50, 1981–1993. [Google Scholar] [CrossRef]
- Kim, S.; Kang, S.-W.; Joo, J.; Han, S.H.; Shin, H.; Nam, B.Y.; Park, J.; Yoo, T.-H.; Kim, G.; Lee, P.; et al. Characterization of ferroptosis in kidney tubular cell death under diabetic conditions. Cell Death Dis. 2021, 12, 160. [Google Scholar] [CrossRef]
- Wang, F.; Li, J.; Zhao, Y.; Guo, D.; Liu, D.; Chang, S.; Qiao, H.; Li, J.; Yang, Y.; Zhang, C.; et al. miR-672-3p Promotes Functional Recovery in Rats with Contusive Spinal Cord Injury by Inhibiting Ferroptosis Suppressor Protein 1. Oxid. Med. Cell Longev. 2022, 2022, 6041612. [Google Scholar] [CrossRef]
- Peeples, E.S.; Genaro-Mattos, T.C. Ferroptosis: A promising therapeutic target for neonatal hypoxic-ischemic brain injury. Int. J. Mol. Sci. 2022, 23, 7420. [Google Scholar] [CrossRef]
- de Souza, I.; Monteiro, L.K.S.; Guedes, C.B.; Silva, M.M.; Andrade-Tomaz, M.; Contieri, B.; Latancia, M.T.; Mendes, D.; Porchia, B.F.M.M.; Lazarini, M.; et al. High levels of NRF2 sensitize temozolomide-resistant glioblastoma cells to ferroptosis via ABCC1/MRP1 upregulation. Cell Death Dis. 2022, 13, 591. [Google Scholar] [CrossRef]
- Pang, Q.; Zheng, L.; Ren, Z.; Xu, H.; Guo, H.; Shan, W.; Liu, R.; Gu, Z.; Wang, T. Mechanism of ferroptosis and its relationships with other types of programmed cell death: Insights for potential therapeutic benefits in traumatic brain injury. Oxidative Med. Cell. Longev. 2022, 2022, 1274550. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hu, R.; Li, M.; Li, F.; Meng, H.; Zhu, G.; Lin, J.; Feng, H. Deferoxamine attenuates iron-induced long-term neurotoxicity in rats with traumatic brain injury. Neurol. Sci. 2012, 34, 639–645. [Google Scholar] [CrossRef]
- Xie, B.S.; Wang, Y.; Lin, Y.; Mao, Q.; Feng, J.; Gao, G.; Jiang, J. Inhibition of ferroptosis attenuates tissue damage and improves long—Term outcomes after traumatic brain injury in mice. CNS Neurosci. Ther. 2019, 25, 465–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Miranda Ramos, V.; Zanotto-Filho, A.; de Bittencourt Pasquali, M.A.; Klafke, K.; Gasparotto, J.; Dunkley, P.; Moreira, J.C.F. NRF2 Mediates Neuroblastoma Proliferation and Resistance to Retinoic Acid Cytotoxicity in a Model of In Vitro Neuronal Differentiation. Mol. Neurobiol. 2016, 53, 6124–6135. [Google Scholar] [CrossRef]
- Zhang, D.; Chang, R.; Ren, Y.; He, Y.; Guo, S.; Guan, F.; Yao, M. Injectable and reactive oxygen species-scavenging gelatin hydrogel promotes neural repair in experimental traumatic brain injury. Int. J. Biol. Macromol. 2022, 219, 844–863. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.-J.; Zhu, H.-Y.; Wang, X.-L.; Lu, X.-W.; Pan, C.-L.; Xu, L.; Liu, X.; Xu, N.; Zhang, Z.-Y. Oridonin ameliorates traumatic brain injury-induced neurological damage by improving mitochondrial function and antioxidant capacity and suppressing neuroinflammation through the Nrf2 pathway. J. Neurotrauma 2022, 39, 530–543. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, S.; D’Mello, V.; Caruso, D.; Abdul-Muneer, P.M. Traumatic brain injury-induced downregulation of Nrf2 activates inflammatory response and apoptotic cell death. J. Mol. Med. 2019, 97, 1627–1641. [Google Scholar] [CrossRef]
- Hower, V.; Mendes, P.; Torti, F.M.; Laubenbacher, R.; Akman, S.; Shulaev, V.; Torti, S.V. A general map of iron metabolism and tissue-specific subnetworks. Mol. Biosyst. 2009, 5, 422–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajarabille, N.; Latunde-Dada, G.O. Programmed cell-death by ferroptosis: Antioxidants as mitigators. Int. J. Mol. Sci. 2019, 20, 4968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, G.; Arosio, P.; Chasteen, N.D. Iron(II) and hydrogen peroxide detoxification by human h-chain ferritin. An EPR spin-trapping study. Biochemistry 2006, 45, 3429–3436. [Google Scholar] [CrossRef]
- Arosio, P.; Levi, S. Cytosolic and mitochondrial ferritins in the regulation of cellular iron homeostasis and oxidative damage. Biochim. Biophys. Acta 2010, 1800, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Duarte, T.L.; Talbot, N.P.; Drakesmith, H. NRF2 and hypoxia-inducible factors: Key players in the redox control of systemic iron homeostasis. Antioxid. Redox Signal. 2021, 35, 433–452. [Google Scholar] [CrossRef]
- Cairo, G.; Tacchini, L.; Pogliaghi, G.; Anzon, E.; Tomasi, A.; Bernelli-Zazzera, A. Induction of ferritin synthesis by oxidative stress. J. Biol. Chem. 1995, 270, 700–703. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, Y.; Ayaki, H.; Whitman, S.P.; Morrow, C.S.; Torti, S.V.; Torti, F.M. Coordinate transcriptional and translational regulation of ferritin in response to oxidative stress. Mol. Cell. Biol. 2000, 20, 5818–5827. [Google Scholar] [CrossRef] [Green Version]
- Wasserman, W.W.; Fahl, W.E. Functional antioxidant responsive elements. Proc. Natl. Acad. Sci. USA 1997, 94, 5361–5366. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Tamba, M.; Kuriyama-Matsumura, K.; Okuno, S.; Bannai, S. Molecular cloning and expression of human xCT, the light chain of amino acid transport system xc. Antioxid. Redox. Signal. 2000, 2, 665–671. [Google Scholar] [CrossRef]
- Kobayashi, S.; Sato, M.; Kasakoshi, T.; Tsutsui, T.; Sugimoto, M.; Osaki, M.; Okada, F.; Igarashi, K.; Hiratake, J.; Homma, T.; et al. Cystathionine is a novel substrate of cystine/glutamate transporter: Implications for immune function. J. Biol. Chem. 2015, 290, 8778–8788. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, H.; Stockwell, B.R. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018, 16, e2006203. [Google Scholar] [CrossRef] [PubMed]
- Gaschler, M.M.; Andia, A.A.; Liu, H.; Csuka, J.M.; Hurlocker, B.; Vaiana, C.A.; Heindel, D.W.; Zuckerman, D.S.; Bos, P.H.; Reznik, E.; et al. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat. Chem. Biol. 2018, 14, 507–515. [Google Scholar] [CrossRef]
- Hambright, W.S.; Fonseca, R.S.; Chen, L.; Na, R.; Ran, Q. Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 2017, 12, 8–17. [Google Scholar] [CrossRef]
- Lewerenz, J.; Albrecht, P.; Tien, M.-L.T.; Henke, N.; Karumbayaram, S.; Kornblum, H.I.; Wiedau-Pazos, M.; Schubert, D.; Maher, P.; Methner, A. Induction of Nrf2 and xCT are involved in the action of the neuroprotective antibiotic ceftriaxone in vitro. J. Neurochem. 2009, 111, 332–343. [Google Scholar] [CrossRef]
- Habib, E.; Linher-Melville, K.; Lin, H.-X.; Singh, G. Expression of xCT and activity of system xc(-) are regulated by NRF2 in human breast cancer cells in response to oxidative stress. Redox Biol. 2015, 5, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Proneth, B.; Conrad, M. Ferroptosis and necroinflammation, a yet poorly explored link. Cell Death Differ. 2019, 26, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osburn, W.O.; Wakabayashi, N.; Misra, V.; Nilles, T.; Biswal, S.; Trush, M.A.; Kensler, T.W. Nrf2 regulates an adaptive response protecting against oxidative damage following diquat-mediated formation of superoxide anion. Arch. Biochem. Biophys. 2006, 454, 7–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef] [Green Version]
- Chorley, B.N.; Campbell, M.R.; Wang, X.; Karaca, M.; Sambandan, D.; Bangura, F.; Xue, P.; Pi, J.; Kleeberger, S.R.; Bell, D.A. Identification of novel NRF2-regulated genes by ChIP-Seq: Influence on retinoid X receptor alpha. Nucleic Acids Res. 2012, 40, 7416–7429. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, H.; Wang, P.; Wang, N.; Dong, W.; Chen, Z.; Wu, M.; Wang, Z.; Yu, Z.; Guan, D.; Wang, L.; et al. Neuroprotection of NRF2 against Ferroptosis after Traumatic Brain Injury in Mice. Antioxidants 2023, 12, 731. https://doi.org/10.3390/antiox12030731
Cheng H, Wang P, Wang N, Dong W, Chen Z, Wu M, Wang Z, Yu Z, Guan D, Wang L, et al. Neuroprotection of NRF2 against Ferroptosis after Traumatic Brain Injury in Mice. Antioxidants. 2023; 12(3):731. https://doi.org/10.3390/antiox12030731
Chicago/Turabian StyleCheng, Hao, Pengfei Wang, Ning Wang, Wenwen Dong, Ziyuan Chen, Mingzhe Wu, Ziwei Wang, Ziqi Yu, Dawei Guan, Linlin Wang, and et al. 2023. "Neuroprotection of NRF2 against Ferroptosis after Traumatic Brain Injury in Mice" Antioxidants 12, no. 3: 731. https://doi.org/10.3390/antiox12030731