1. Introduction
Thioredoxin 1 (Trx1) is an oxido-reductase that was first described for its ability to reduce disulfide bonds and protect against oxidative stress through a mechanism that involves its reduced C
32xxC
35 cysteine (Cys) active center. As such, it is one of the most critical regulators of cellular redox and a key player in the antioxidant defense system in cells [
1]. We previously showed that soluble guanylyl cyclase (GC1) could form a complex with Trx1, which, in turn, enhanced the NO-stimulated catalytic activity of GC1 in cells and in vitro [
2]. The hundred-fold stimulation of cGMP production by NO requires the α and β subunits of GC1 to form a heterodimer. A Trx1-trap system and mutational analysis suggest that a mixed disulfide between C
32 of the Trx1 active site and Cys 610 (C
610) of the α subunit of GC1 is necessary for a Trx1-dependent increase of GC1 in response to NO stimulation [
2,
3]. We previously showed that the NO-stimulated activity of GC1 was reduced by the S-nitrosation of several Cys [
4,
5,
6]. S-nitrosation (addition of a NO moiety to a Cys) is a posttranslational modification (PTM) that changes the properties of the proteins, including activity, localization, and interaction [
7]. Interestingly, Trx1 can catalyze denitrosation, which is the removal of the nitroso group (SNO) from the Cys of a protein through a mechanism that involves its reduced C
32xxC
35 active center, as for disulfide reduction. As such, the mechanism of Trx1-dependent increases in GC1 activity could involve a reduction in oxidized thiols and/or the protection of GC1 from desensitization to NO by decreasing S-nitrosation of GC1 [
2].
Trx1 is involved in the regulation of cellular S-nitrosation, not only via de-nitrosation but also via transnitrosation reactions. Transnitrosation by protein–protein interactions is the direct transfer of the nitroso group from the thiol on one protein to a target thiol on another protein, thus supporting a high degree of specificity for this PTM and its relevance as another form of NO signaling [
8]. Trx1 transnitrosation requires Trx1 to be oxidized (oTrx1) with the formation of a disulfide bond between C
32–C
35 of its active site, while Cys73 (C
73) is the main SNO donor, i.e., transferring its SNO to the thiol of the targeted protein [
9]. We recently reported that the α subunit of GC1, in the absence of an active cGMP-forming α/β heterodimer, can itself transnitrosate more than 200 proteins under oxidative/nitrosative stress [
10]. Remarkably, this high number of GC1’ SNO-targets is partially due to GC1’s ability to interact with and use oTrx1 as a mediator of the S-nitrosothiols transfer. We demonstrated that the SNO-GC1→oTrx1 transnitrosation reaction was unilateral, did not take place if Trx1 was in its reduced form (rTrx1), and that the SNO transfer involved C
610 of the GC1-α subunit to C
73 of oTrx1 [
10]. As an example of the biological relevance of the transnitrosation reaction, we specifically identified a transnitrosation cascade initiated by SNO-GC1 that required oTrx1 and led to S-nitrosation of RhoA and the inhibition of its activity in cells.
Together, these findings suggest that the GC1/Trx1 complex differentially supports NO signaling, that is, cGMP production and nitrosation, as a function of the redox of cells. In addition, we showed that αC610 was involved in both the reduced and oxidized GC1/Trx1 complex formation and function. Our previous studies used mutants of Trx1 or an inhibitor of Trx1-reductase, thus altering the entire redox state of the cells and limiting the interpretation of the role of the GC1/Trx1 complex in cells. Likewise, these previous studies involved the deletion of the α subunit of GC1 or replacement of Cys 610 with a Ser (αC610S) with the caveat that such a mutation could affect the structure and activity of GC1. In order to (a) understand how a different GC1 and Trx1 interaction takes place under reduced and oxidative conditions, (b) characterize the complex properties, and (c) seek their biological relevance, we designed a set of mimetic peptides of the αC610 region to disrupt the two complexes without affecting the integrity of Trx1 and GC1 under both reduced and oxidized conditions.
2. Materials and Methods
2.1. Reagents
The purified recombinant human GC1 protein was purchased from Enzo life sciences (ALX-201-177-C010), and the Thioredoxin 1 (Trx1) protein was from Fitzgerald industries (30R-2985). FBS was from VWR (97068-085), the GC1 α antibody was from ThermoFisher (MA517086, Waltham, MA, USA), the GC1 β antibody was from Cayman chemical company (160897), Trx1 antibodies were from cell signaling (2298S and human-specific: 2285S). The caspase 3 antibody was purchased from Novus biologicals (NB100-56708), and the biotin antibody from Abcam (ab1227). The following reagents were purchased from Sigma (St. Louis, MI, USA): DTT (D0632), diamide (D3648), Amicon Ultra-0.5 centrifugal filter (UFC500324), Bradford reagent (B6916), PMSF (P7626), protease inhibitor cocktail (P8340), Etoposide (E1383), and the Caspase 3 activity assay kit (CASP3C-1KT). [α-32P]-GTP was from PerkinElmer (NEG006H250UC), S-nitrosoglutathione from Calbiochem (487920). The jetPRIME transfection kit was from Polyplus transfection (101000015), Diethylamine NONOate (DEA-NO) from Enzo life sciences (ALX-430-034-5005) and biotin-HPDP from ThermoFisher (21341). The Jurkat T cell line, clone E6-1 was purchased from ATCC (TIB-152).
2.2. Peptides Synthesis
Two sets of peptides were synthesized. The first set of three 11-amino acid peptides, synthesized by Epoch Life science, mimicked the sequence of amino acids in the proximity of, or overlapping, Cysteine 610 of the α subunit of GC1. A fourth scramble peptide was synthesized as a control. The sequences are provided in
Figure S1 of the Supplementary Information. As the 3 peptides were efficient at disrupting the GC1/Trx1 complex in vitro, peptide 3 (inhibitory peptide) and the scramble peptide were modified (Abclonal Science, Wuhan, China) for cell penetration through the addition of a tat/penetrating hybrid peptide and FITC (
Figure S2) [
11].
2.3. Cell Culture and Transfection
COS-7 cells (ATCC) were cultured in a 6-well plate with a complete medium DMEM from Corning (10-013-CV, Somerville, MA, USA) supplemented with 10% FBS (heat inactivated) and Pen-Strep (ThermoFisher, 15140122) at 37 °C with 5% CO
2. The cells were transfected with the wild-type GC1 α and β subunits from rats subcloned in a pCMV5 vector [
2], using the jetPRIME kit. After 4 h, the medium was replaced with a complete medium supplemented with 10%FBS + P/S. After 24 h of transfection, the cells were washed twice and cultured in a serum-free medium for peptide treatment.
Jurkat cells were cultured in a 6-well plate at the density of 0.25 × 106/mL in complete medium RPMI 1640 from Corning (10-040-CV) supplemented with 10% FBS and Pen-Strep and incubated at 37 °C with 5% CO2 for 24 h. The cells were collected by centrifuging at 1000× g for 5 min, washed twice with serum-free medium, and re-suspended in a 2 mL serum-free medium prior to peptide treatment.
2.4. Peptides Treatment and Assay of GC1 Activity in a Purified System
Each peptide from set 1 (
Figure S1) was dissolved in 70% DMSO to obtain a 10 mM stock solution and was further diluted with H
2O to have a 250 μM working stock. In each reaction tube, 0.25 μg of human recombinant GC1 was mixed with 0.15 μg of Trx1 (molar ratio GC1:Trx1 ~ 1:7) or not (to control the effect of peptides on GC1 activity). In each tube of the GC1 and Trx1 mixture or GC1 alone, 5 μL of each peptide was added in a 50 μL final volume of 50 mM HEPES, pH 8.0. The final concentration of each peptide was 25 μM. The molar ratio of GC1 to each peptide was 1:800. The tubes were incubated on ice for 20 min in the dark. After incubation, the samples were subjected to a GC1 activity assay under basal and NO-stimulated conditions. The GC1 activity was measured by the conversion of [α-
32P]-cGMP from [α-
32P]-GTP, as previously described [
12]. A fifty μL reaction mix containing 50 mM HEPES, pH 8.0, 5 mM MgCl
2, and 0.5 mM GTP was added to each sample and incubated at 30 °C for 5 min. DEA-NO (1 μM final concentration) was used as the NO donor to stimulate GC1 activity. The reaction was stopped by adding 500 μL of 120 mM zinc acetate, followed by the addition of 500 μL of 144 mM sodium carbonate. [α-
32P]-cGMP was separated using alumina columns. The recovery rate was calculated using 100 μL of cGMP-
3H. This specific activity was first expressed in nmol cGMP·min
−1·mg
−1 (
Figure S3); then, these values were expressed as a fold change normalized to GC1 NO-stimulated activity alone.
n = 4 independent experiments were performed in duplicate.
2.5. Peptides Treatment and Assay of GC1 Activity in COS-7 Cells
After 24 h of transfection with WT GC1, COS-7 cells were either treated with 70% DMSO (0.35% final), the selected inhibitory peptide or scramble peptide at 37 °C for 1 h in a serum-free medium. The final concentration of each peptide was 5–10 μM (sequences are provided in
Figure S2a). After 1 h incubation, the cells were washed twice and incubated in the complete medium at 37 °C for 24 h. After 24 h incubation, the cells were washed 3 times with ice-cold PBS and sonicated in homogenization buffer containing 50 mM HEPES, pH 8.0 and 150 mM NaCl supplemented with PMSF and a protease inhibitor cocktail. The protein concentration of each sample was measured using the Bradford method. A total of 20 μg of protein in each sample was used for GC1 activity assay under both basal and NO-stimulated conditions. The GC1 activity assay was conducted as above. Prior to lysis, coverslips were collected at 12 h and 24 h to visualize FITC fluorescence and assess peptide penetration (
Figure S2b). Conversely, the expression of GC1 α and β subunits in COS-7 cells and the endogenous expression of Trx1 were assayed by Western blot (
Figure S2c).
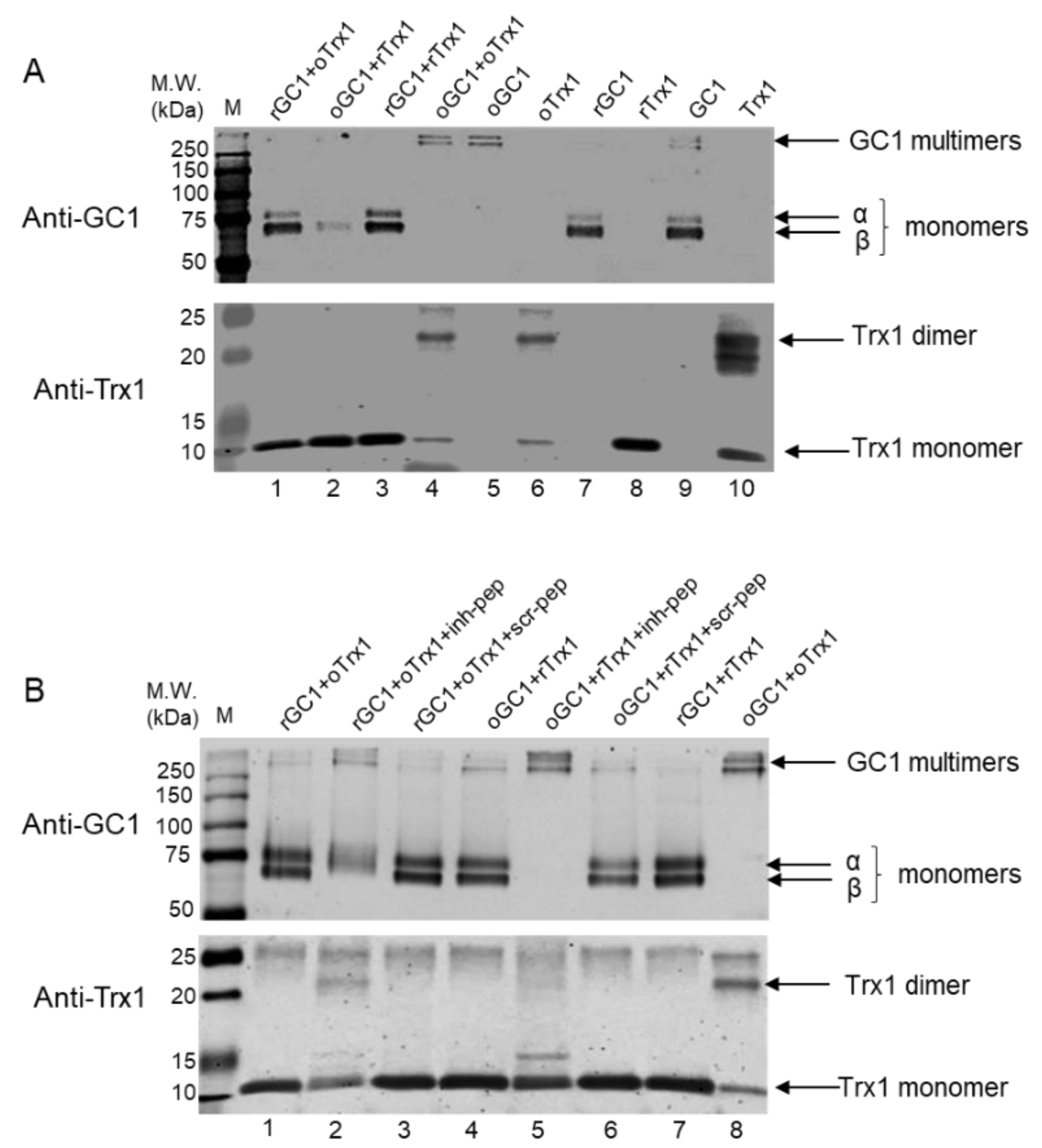
2.6. Peptide Treatments and Assays of Reduction in Oxidized GC1 by Reduced Trx1 and of Oxidized Trx1 by Reduced GC1, in a Purified System
One μg of recombinant human GC1 was treated with 10 mM DTT at 37 °C for 30 min in the dark or with 100 μM diamide on ice for 30 min in the dark to generate reduced (rGC1) and oxidized GC1 (oGC1), respectively. Four hundred ng of recombinant human Trx1 was treated with 10 mM DTT at 37 °C for 30 min in the dark or with 100 μM diamide on ice for 30 min in the dark to generate reduced (rTrx1) and oxidized Trx1 (oTrx1), respectively. The proteins were transferred onto Amicon Ultra-0.5 centrifugal filters, centrifuged at 14,000× g for 10 min, and washed 3 times with a buffer containing 50 mM HEPES, pH 8.0, and 5 mM EDTA to remove excess reagent. The proteins were recovered by flipping the filters and centrifuging at 1000× g for 2 min. Various combinations of reduced or oxidized forms of GC1 and Trx1 with or without peptides were mixed and incubated at room temperature for 30 min in the dark. In each tube, a GC1:Trx1 molar ratio was estimated to be 1:5; the concentration of peptides, if added, was 125 μM final. The samples were analyzed by Western blots: gel (12% SDS-PAGE) electrophoresis was conducted under reducing and non-reducing conditions, transferred on nitrocellulose membrane, and then probed for either Trx1 or GC1.
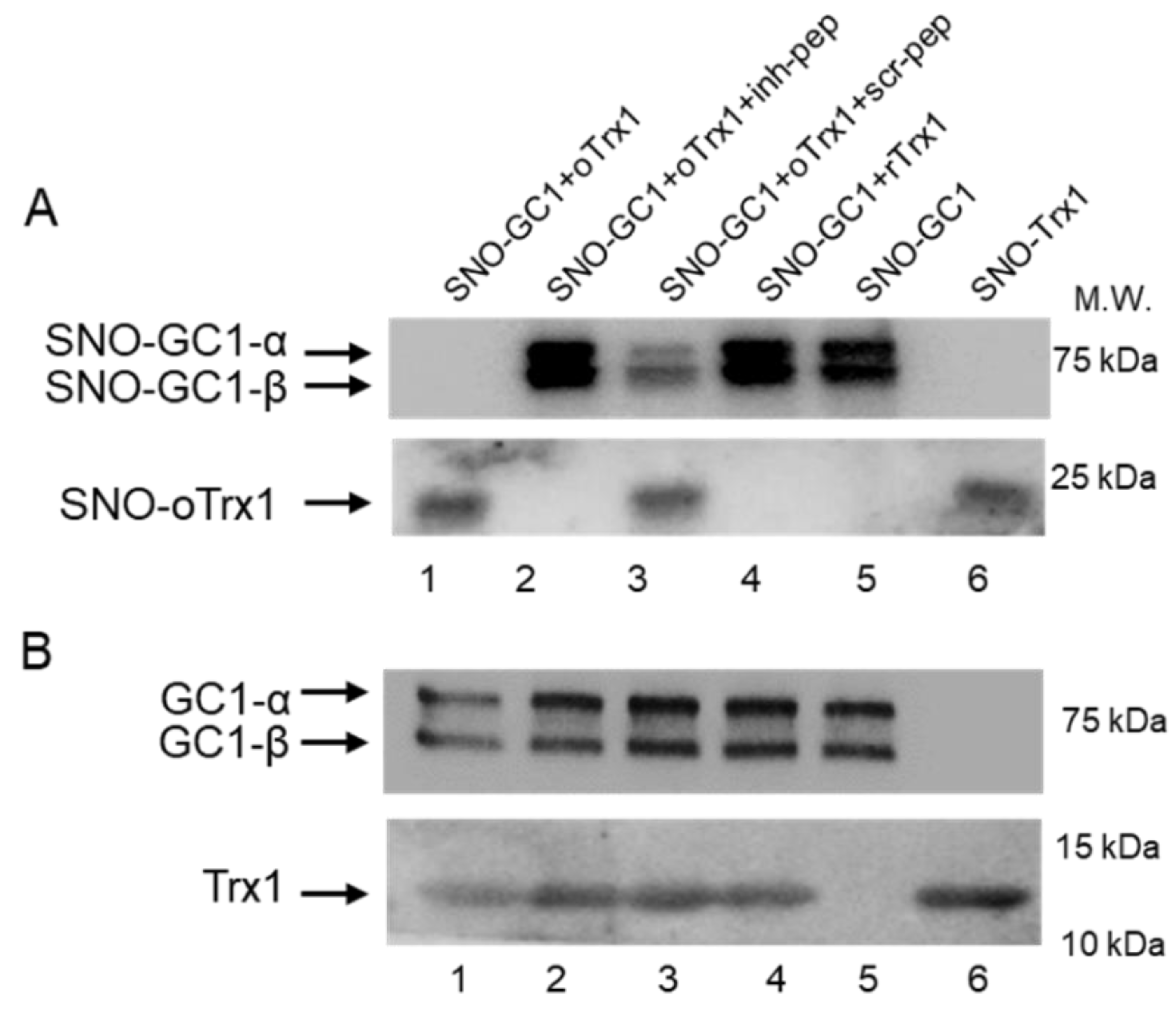
2.7. Peptides Treatment of GC1-Trx1 Transnitrosation Reaction in a Purified System
Recombinant human GC1 was treated with 100 μM S-nitrosoglutathione (GSNO) at 37 °C for 30 min in the dark to generate SNO-GC1, as previously described [
10]. Recombinant human Trx1 was treated with 10 mM DTT at 37 °C for 30 min in the dark or with 100 μM diamide on ice for 30 min in the dark to generate rTrx1 and oTrx1, respectively, and excess reagents were removed as above. oTrx1 was treated with 100 μM GSNO at 37 °C for 30 min in the dark to generate SNO-oTrx1, as previously described [
10]. The proteins were transferred onto Amicon Ultra-0.5 centrifugal filters, centrifuged at 14,000×
g for 10 min, and washed 3 times with a buffer containing 50 mM HEPES, pH 8.0, and 5 mM EDTA to remove excess reagent. The proteins were recovered by flipping the filters and centrifuging at 1000×
g for 2 min. Various combinations of SNO-GC1, oTrx1, or rTrx1 with or without peptides were mixed (GC1:Trx1 molar ratio was estimated at 1:5, and the final concentration of each peptide was 125 μM) and incubated at room temperature for 30 min in the dark. The samples were analyzed by biotin or biotin/avidin switch assays as previously described [
10], followed by Western blotting. To detect biotinylated proteins, the samples were electrophoresed under non-reducing conditions and probed with the anti-biotin antibody (1:3000, Abcam, Cambridge, UK); the inputs (starting material) were electrophoresed under reducing conditions and probed for Trx1 and GC1.
2.8. Biotin Switch Assay and Avidin Enrichment
Free thiols of proteins were blocked in a lysis-blocking buffer containing 50 mM Tris, pH 7.5, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 2% SDS, 0.1 mM neocuproine, 0.2 mM PSMF, and 40 mM N-ethylmaleimide (NEM) at 50 °C for 30 min in the dark. Excess NEM was removed using cold acetone precipitation. The protein pellets were generated by centrifuging at 14,000× g for 10 min at 4 °C and washing 3 times using cold acetone. The pellets were resuspended in a 300 μL buffer containing 25 mM HEPES, pH 7.7, 1 mM EDTA, and 1% SDS, supplemented with 0.1 mM biotin-HPDP, 10 mM ascorbate, and 1 μM CuCl, at room temperature for 1 h in the dark. The negative controls were the blocked samples without ascorbate. Excess biotin-HPDP was removed, and biotinylated proteins were precipitated using cold acetone at −20 °C for 1 h, followed by centrifugation at 5000× g for 20 min at 4 °C. The pellets were then dissolved in a HENS buffer. For the detection of biotinylated proteins, resuspended samples were mixed with a 4× Laemmli buffer without β-Mercaptoethanol.
For avidin enrichment, the biotinylated samples were diluted in 700 μL PBS and mixed with 100 μL streptavidin-agarose beads. The mixture was incubated for 1 h at room temperature with regular agitation. The beads were pelleted by centrifuging at 5000× g for 10 min and were washed 3 times with 1 mL PBS. The washed beads were mixed with 100 μL of a 1× Laemmli loading buffer with 10% β-Mercaptoethanol and heated at 85 °C for 5 min. The proteins released from the beads were then subjected to Western blotting.
2.9. Jurkat T Cells Treatment with Peptides and Etoposide-Induced Apoptosis
Cells in suspension in a 2 mL serum-free medium were treated with either 70% DMSO (20 μL, 0.7% final), scramble peptide, or the inhibitory peptide at a final concentration of 10 μM and incubated at 37 °C for 1 h. After incubation, the cells were washed twice and incubated in a complete medium at 37 °C for 8 h. The cells were then treated with (or without) etoposide (ETO) at 8 μM final at 37 °C for 16 h.
For biotin-avidin switch assays: after 16 h of incubation, the cells were washed twice with cold PBS and lysed in a lysis-blocking buffer containing 50 mM Tris, pH 7.5, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 2% SDS, 0.1 mM neocuproine, and 40 mM NEM supplemented with PMSF and a protease inhibitor cocktail. The protein concentration was measured using the Bradford method. Fifty μg of each lysate were analyzed by Western blot as the starting material (inputs), and 200 μg of proteins from each sample were subjected to the biotin switch and avidin assays, as described above. Blots under non-reducing conditions were probed with anti-biotin. Under reducing conditions following avidin assays, blots were probed with anti-GC1 α and β (1:1000), anti-Caspase 3 (1:500), and anti-Trx1 (1:500).
For Caspase-3 activity assays: after 16 h incubation, the cells were washed twice with cold PBS and lysed in the supplier’s 1x lysis buffer (at 10μL per 106 cells). After 15 min incubation on ice, the lysates were centrifuged at 16,000× g for 15 min at 4 °C, and the supernatant was collected. Caspase-3 activity was measured following the supplier’s instructions (CASP3C-1KT, Sigma, St. Louis, MI, USA) after a 2 and 3 h incubation.
2.10. Statistical Analysis
Value outputs are expressed as the average ± SEM with n ≥ 3. The comparison studies with GC1 NO-stimulated activity were conducted with a two-tailed Student’s t-test, an α level of 0.05 in the purified system, and the use of one-way ANOVA followed by Tuckey’s posthoc test in the cell studies. Caspase-3 activity was measured in three independent experiments (n = 3), and each measurement was in duplicate. The means and standard deviation of each measurement were calculated, and the variance was compared using one-way ANOVA followed by Sidak’s multiple comparison tasks with p < 0.05 using GraphPad Prism v9.4.1. p < 0.05 was considered statistically significant.
4. Discussion
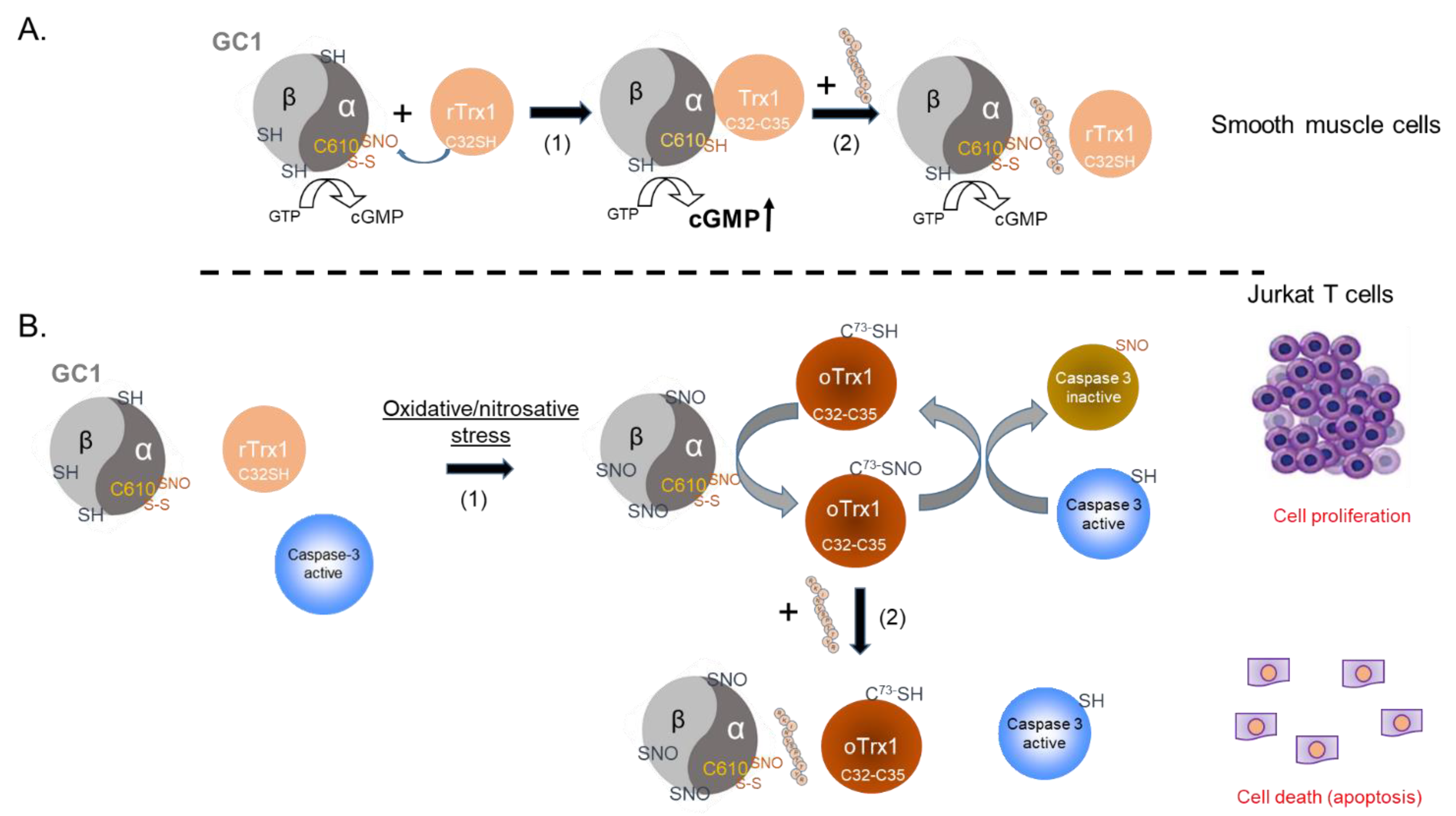
The NO-cGMP pathway is a crucial component of vasorelaxation, regulation of blood pressure, cardiac protection, and platelets aggregation, and we showed that the interaction between GC1 and Trx1 supported NO-stimulated GC1 activity and, thus, could be essential for cardiovascular homeostasis. On the other hand, we recently identified the formation of a thiol-oxidized SNO-GC1/oTrx1 complex, which carries transnitrosation cascades. Some of the targets of this oxidized complex are known to be regulated by the NO-cGMP pathway. As oxidative conditions led to the disruption of the canonical NO-GC1-cGMP pathway via heme oxidation and extensive S-nitrosation of GC1, we proposed that the transnitrosation activity of SNO-GC1, amplified by oTrx1, provides an adaptive response to oxidative stress [
10].
The chemistry of the interaction between Trx1 and GC1 under oxidized and reduced conditions is different. Under reduced conditions, our previous mutational analyses showed that the association between GC1 and Trx1 involved a mixed disulfide, potentially between the C
32 of Trx1’s reduced active site and C
610 of the α subunit of the GC1 heterodimer [
2]. By contrast, under diamide-oxidized conditions, oTrx1 was detected as a dimer, as previously observed [
13,
14]. Previous mass spectrometry (MS) analyses showed that oTrx1’ Cys active site formed a disulfide and C
73 was amenable to S-nitrosation [
18], indicating that oTrx1 lost its reductase activity but gained a transnitrosation activity. We successfully characterized the interaction between oTrx1 and SNO-GC1, revealing a unilateral transfer of S-nitrosothiols from SNO-GC1 to oTrx1 and identifying the C
610 of the α subunit as a major “SNO donor” (to the Trx1-C
73 SNO acceptor). In this oxidized complex, the interaction with Trx1 could not involve a mixed disulfide with the C
32 of Trx1, as C
32 was engaged in a disulfide with C
35. To gain insight into these various complexes and their relevance, we used an inhibitory peptide for the interaction. This allowed us to avoid the potential activity/conformational alterations generated by mutations of GC1 or Trx1. The downside of not using Trx1 mutants as previously [
2] meant that our ability to “trap” this complex was lost, as was our analysis via the pull-down of this transient complex. The peptides were designed around C
610 of the α subunit to theoretically block both the mixed disulfide of the GC1/Trx1 reduced complex and the S-nitrosothiol transfer between SNO-C
610 and C
73 in the oxidized complex.
We showed in vitro that these C
610-mimetic peptides blunt the Trx1-dependent enhancement of NO-stimulated GC1 activity without directly affecting NO-stimulated GC1 activity, indicating that the peptides did not alter the conformational/structural properties of the NO-responsive GC1 heterodimer. The efficiency of the peptides and their ability to block the formation of the reduced GC1/Trx1 complex was confirmed in cells after rendering one selected peptide cell permeable. We inferred that the selected peptide (RKINVSPTTYR), which did not contain a disulfide bond unlike the targets of Trx1 reductase activity should not be able to compete with these targets or interfere with the thiol redox regulatory function of Trx1 and the cellular redox state. On the other hand, C
610 is in a solvent-exposed region of the α subunit of the GC1 heterodimer and has the potential to interact with other proteins. One of these proteins could be a protein disulfide isomerase (PDI), as we previously showed that GC1 interacts with PDI via a mixed disulfide [
19]. The consequences of potentially blocking this interaction or others with the inhibitory peptide need to be addressed in future studies.
The assays of the reductase activity of Trx1 and GC1 in the absence and presence of the inhibitory peptide revealed that GC1 has the ability, in vitro, to reduce the Trx1-oxidized dimer. As this reaction was blocked by the inhibitory peptide, it strongly suggests that the disulfide exchange involves αC
610. In the absence of MS analysis, we did not know what Cys was involved in the Trx1 homodimer. Because of the difference in experimental conditions, the Cys engaged in the homodimer was not unequivocally identified and could be C
69, C
73, or/and C
32 of the active site [
13,
14,
20,
21]. As C
610 is involved in a mixed disulfide with C
32 of Trx1, and the reduction of Trx1-homodimer by GC1 is blocked by the inhibitory peptide, we speculate that C
610 could reduce disulfide bonds involving C
32 in both the active site (C
32–C
35) and homodimerization.
GC1 in vivo is found as a dimer, which is sensitive to reducing agents [
22], and we proposed that disulfides could have a role in GC1 response to NO [
3]. While the diamide-induced multimers of GC1 might not reproduce physiological conditions, our results indicate that rTrx1 can recognize these GC1 multimers and convert them to monomers in an efficient way. Because this Trx1-dependent reduction of GC1 multimers is completely blocked by the inhibitory peptide, this suggests that the multimers formation involves C
610 of the α subunit, including a potential C
610 disulfide bond between two GC1 heterodimers. By contrast, we previously showed [
10] and confirmed herein that rTrx1 cannot reduce the thiol oxidation of S-nitrosated GC1 (SNO-GC1). Together these results suggest that rTrx1 is not able to interact with SNO-GC1 in vitro.
On the other hand, we confirmed that oTrx1 interacts with SNO-GC1 and demonstrated that the transfer of S-nitrosothiols from SNO-GC1 to oTrx1 is blocked by the inhibitory peptide. We previously identified in vitro other Cys of GC1 that drive a nitrosothiol transfer [
10]. The bands of biotinylated SNO-GC1 mixed with oTrx1 are undetectable on the blot of
Figure 3, as expected if there is a complete transfer of nitrosothiols; however, in the presence of the inhibitory peptide, the intensity of SNO-GC1 was similar to the controls SNO-GC1, and SNO-GC1 + rTrx1, as if no SNO transfer occurred. This could suggest that the inhibitory peptide blocks the formation of the complex SNO-GC1/oTrx1 rather than the specific SNO-αC
610 to Trx1-C
73 transnitrosation reaction. MS analysis will be conducted, in the future, to assess the exact mechanism (due to the transient property of the interaction, we have not been able to do pull-downs).
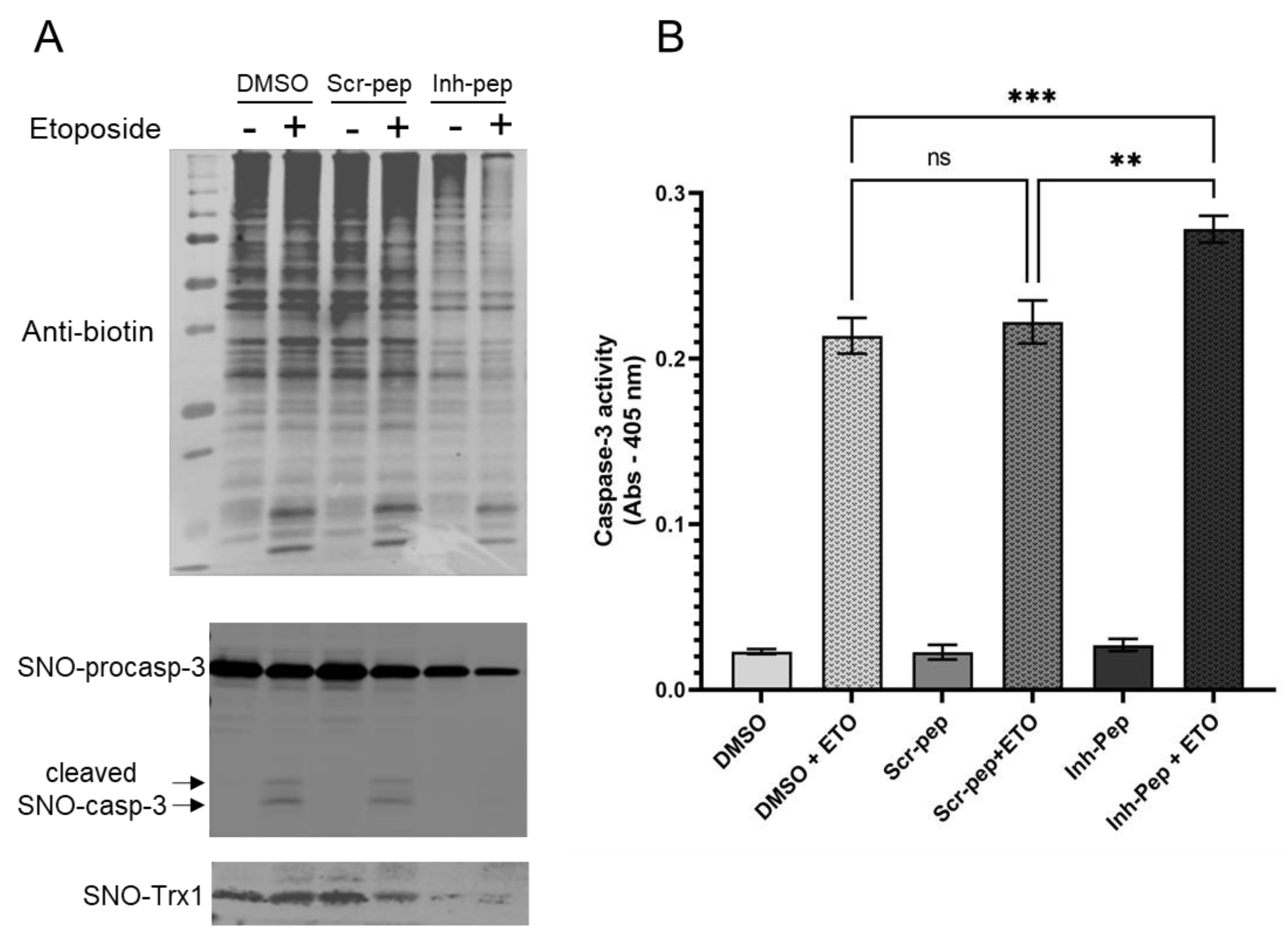
The addition of the inhibitory peptide to cells under reducing conditions showed that the enhancement of a Trx1-dependent NO-cGMP pathway could be blunted (
Figure 5A). This provides us with a tool to directly assess, in the future, the significance of this complex in the vasorelaxation of blood vessels under homeostatic conditions. Conversely, to test the biological, potentially therapeutic relevance of this inhibitory peptide, we thought of blocking GC1/Trx1-dependent transnitrosation under specific oxidative pathophysiological conditions (
Figure 5B). We focused on the known link between cancer cell proliferation and the inhibition of caspases activity by S-nitrosation [
23,
24]. Specifically, we used Jurkat T cells for which Trx1-dependent transnitrosation was shown to inhibit the apoptotic caspase-3 pathway [
15,
16]. We observed that in the absence or presence of etoposide (ETO), which induces apoptosis, baseline levels of global S-nitrosation were elevated in the controls (DMSO and scramble peptide) and correlated with strong S-nitrosation of procaspase-3 and detectable SNO-Trx1. However, in the presence of the inhibitory peptide, the basal S-nitrosated procaspase-3 levels were drastically reduced, and SNO-Trx1 was barely detectable. When ETO was added, SNO-procaspase 3 levels were slightly reduced with the controls and the inhibitory peptide, suggesting that ETO reduced S-nitrosation of procaspase-3 to some extent but not as pronounced as that of Fas treatment [
24]. Conversely, ETO treatment increased significantly caspase-3 activity; importantly, caspase-3 activity was significantly higher in the cells treated with the inhibitory peptide. Together with the decreased S-nitrosation of procaspase-3 and Trx1, compared to the control DMSO and scramble peptide, the increase in caspase-3 activity indicated that the inhibitory peptide efficiently blocked the transfer of nitrosothiol from GC1 to Trx1 and consequently decreased the transnitrosation from Trx1 to procaspase 3. The disruption of the SNO-GC1→Trx1→procaspase3 transnitrosation cascade confirmed that SNO-GC1 was a major source of SNO-Trx1, yet not unique, and importantly, that this mimetic peptide could be a valuable tool to potentially decrease the tumor development associated with excess S-nitrosation (
Figure 5B).
In fact, a direct correlation between Trx1 and S-nitrosation has been established in other cancer types [
26], including hepatocellular carcinoma [
27]. Another promising aspect of this peptide is that under these conditions of nitrosative/oxidative stress, SNO-GC1 cannot interact with rTrx1; thus, the peptide does not interfere with Trx1’s ability to regulate the redox state of the cells, including its denitrosation activity. This could be important considering the dichotomous effect of Trx1 on the development and inhibition of tumor growth in relation to Trx1’s ability to transnitrosate and denitrosate various caspases as a function of its redox state [
28]. Nonetheless, we cannot rule out that this peptide, by mimicking part of the α subunit’ Trx1-binding domain, could potentially interfere with other transnitrosation reactions driven by SNO-oTrx1 with its own targets. Interestingly, the GC1 α subunit, which is sufficient to initiate transnitrosation cascades [
10], was reported to mediate, independently of the β subunit or cGMP production, prostate and endometrial cancer cell proliferation [
29,
30]. Together, these reports and our study support the idea that the α subunit of GC1 is a key player in oncogenic processes and in association with oTrx1 and transnitrosation reactions.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




