
Oxidative Stress-Induced Cellular Senescence: Is Labile Iron the Connecting Link?

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Oxidative Stress
2.1. Oxygen: A Double-Edged Sword for Aerobes
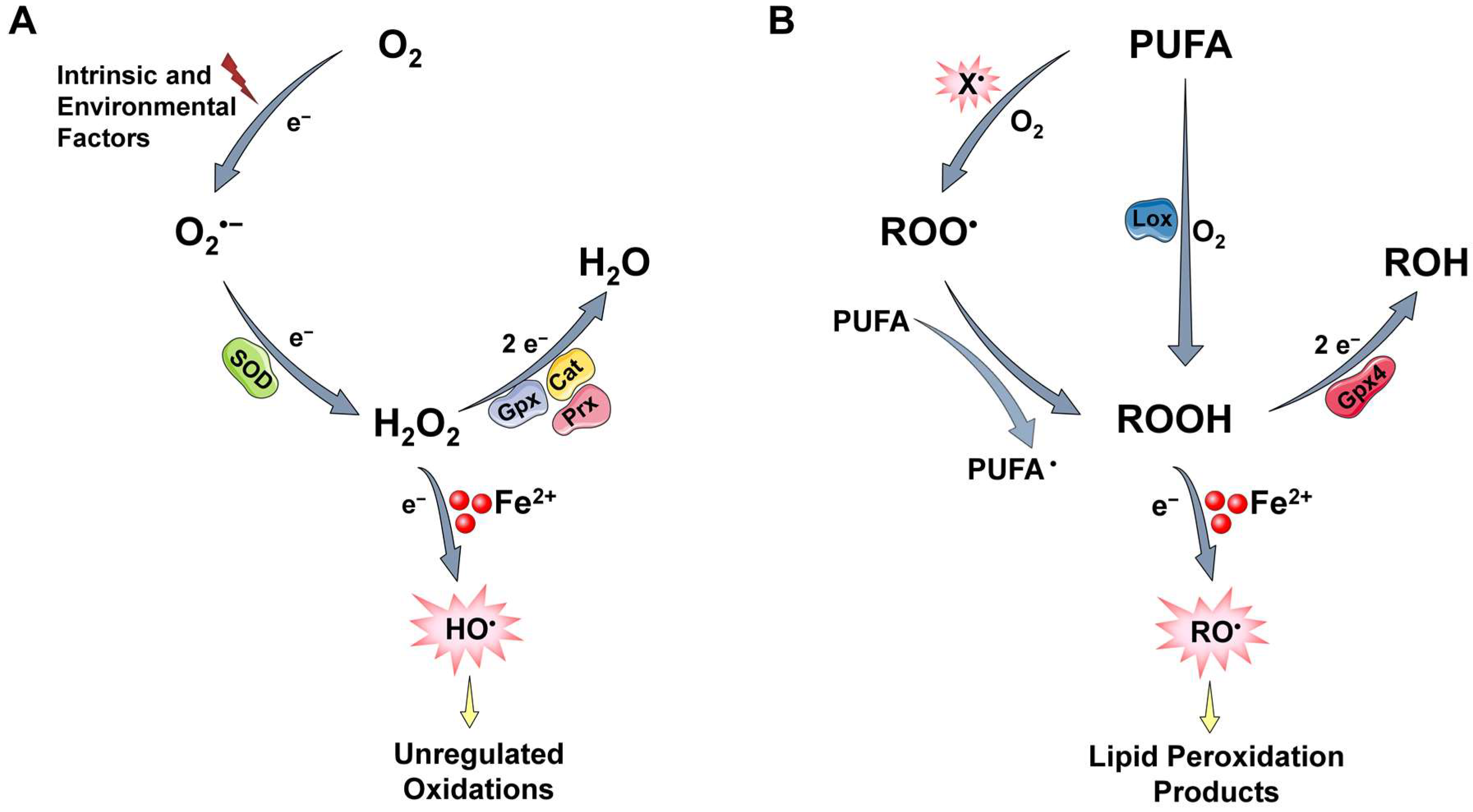
2.2. ROS Generation and Regulation
2.2.1. ROS Generation
2.2.2. Protective Mechanisms
2.3. Labile Iron and Its Key Role in ROS-Induced Toxicity
2.4. The Pleiotropic Effects of ROS on Normal Proliferating Cells
3. Cellular Senescence
3.1. A Brief Historical Overview and Some General Aspects of Cellular Senescence
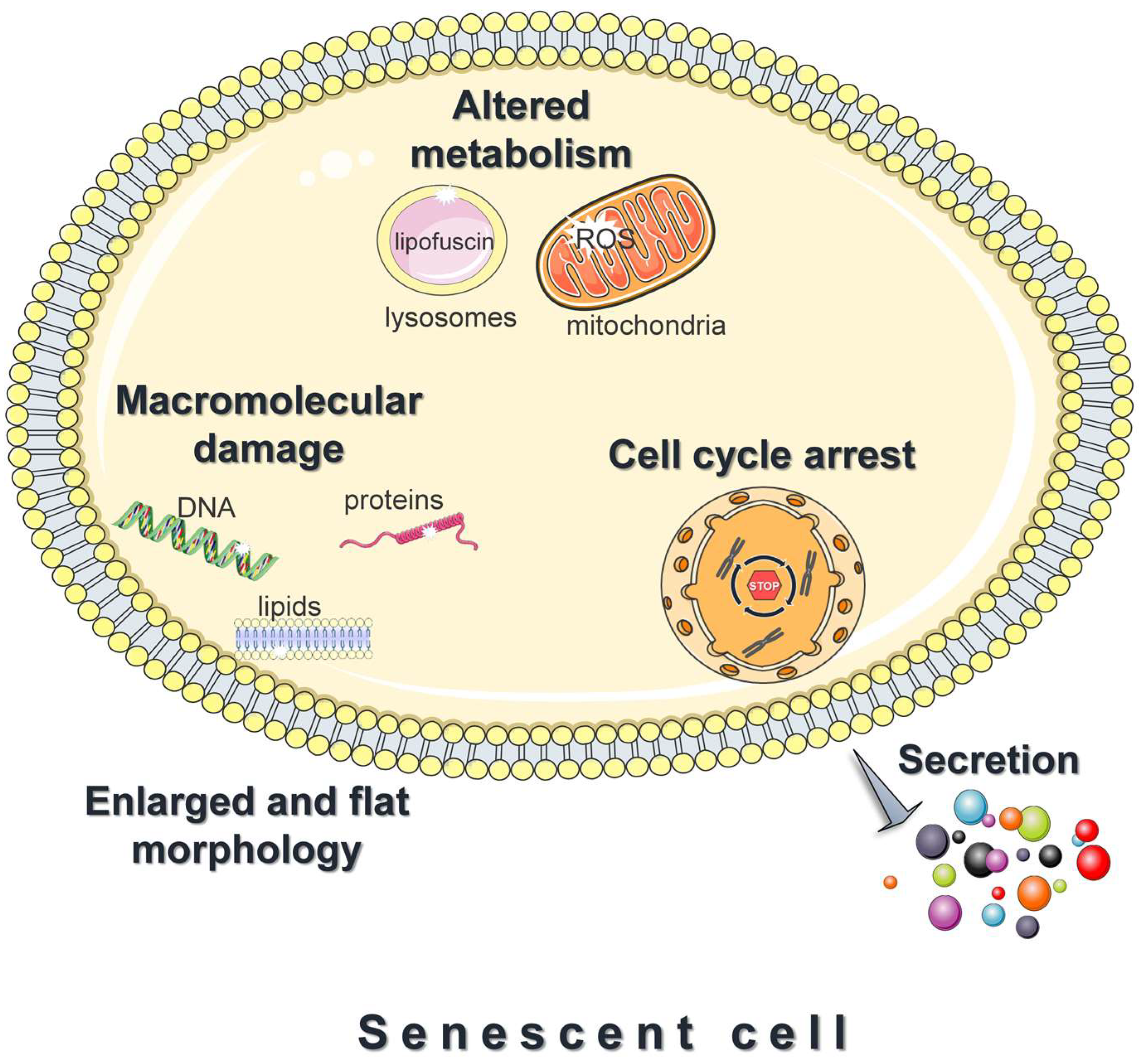
3.2. Features of Senescent Cells
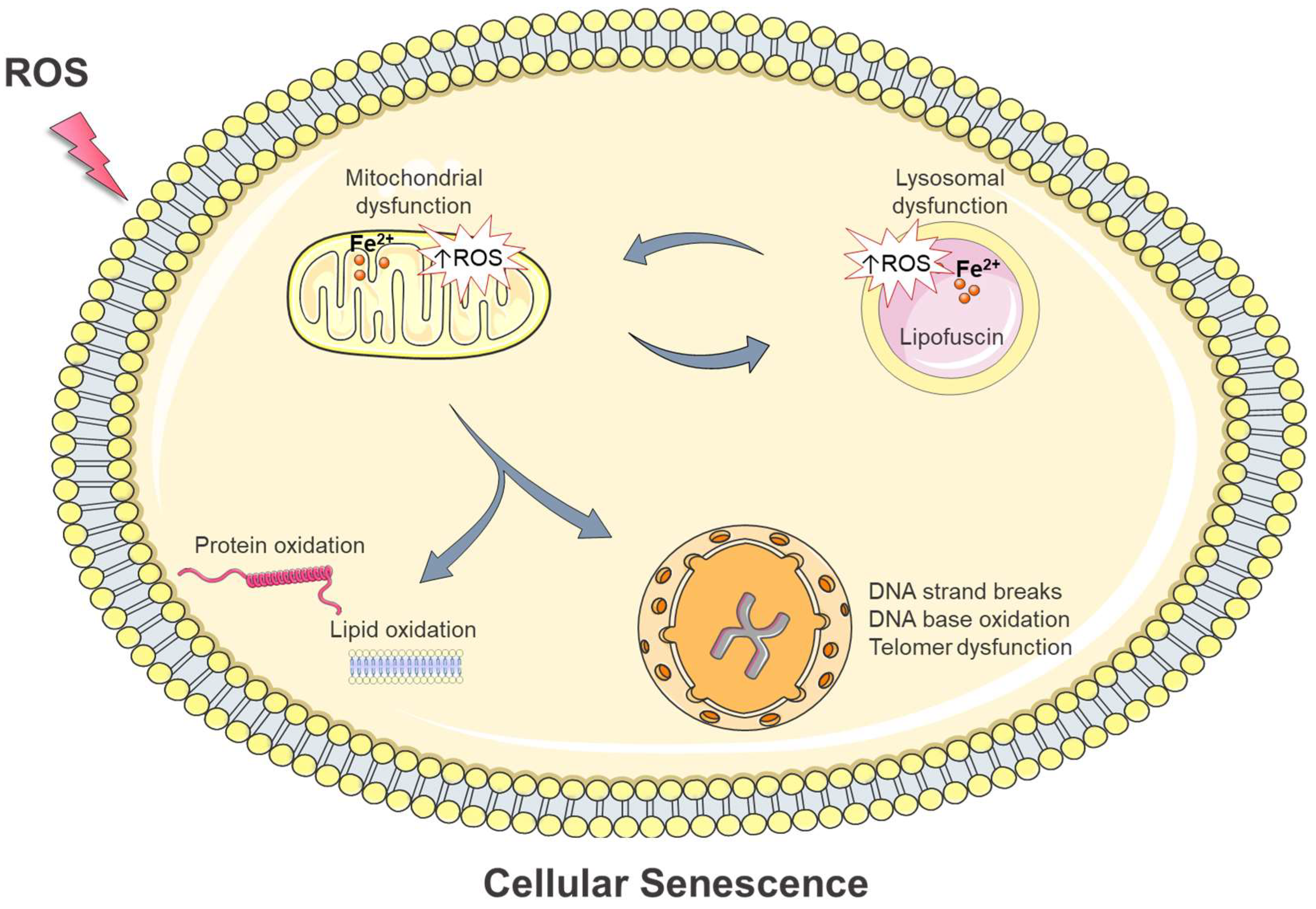
4. Intracellular Damage in Oxidative Stress-Induced Cellular Senescence: Is Iron Involved?
4.1. DNA Damage
4.1.1. Activation of the DNA Damage Response Pathway
4.1.2. Telomeres and how they signal senescence
4.1.3. New Insights into Oxidative Stress-Induced Telomere Shortening and/or Dysfunction
4.1.4. The Putative Role of Labile Iron in Oxidative Stress-Mediated DNA Damage and Cellular Senescence
4.2. Protein Oxidation
4.3. Lipid Oxidation
4.4. Lipofuscin Formation and Accumulation
4.4.1. Lipofuscin: A Non-Degradable Product That Accumulates in Postmitotic and Senescent Cells
4.4.2. Mechanisms of Lipofuscin Formation: The Role of Intracellular Iron Homeostasis and Oxidative Stress
4.5. Alterations in Mitochondria
4.6. Alterations in Lysosomes
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Espín, D.; Serrano, M. Cellular Senescence: From Physiology to Pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Campisi, J. Four Faces of Cellular Senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Hudgins, A.D.; Tazearslan, C.; Tare, A.; Zhu, Y.; Huffman, D.; Suh, Y. Age- and Tissue-Specific Expression of Senescence Biomarkers in Mice. Front. Genet. 2018, 9, 59. [Google Scholar] [CrossRef] [Green Version]
- Ogrodnik, M.; Salmonowicz, H.; Gladyshev, V.N. Integrating Cellular Senescence with the Concept of Damage Accumulation in Aging: Relevance for Clearance of Senescent Cells. Aging Cell 2019, 18, e12841. [Google Scholar] [CrossRef] [Green Version]
- Park, J.T.; Lee, Y.-S.; Cho, K.A.; Park, S.C. Adjustment of the Lysosomal-Mitochondrial Axis for Control of Cellular Senescence. Ageing Res. Rev. 2018, 47, 176–182. [Google Scholar] [CrossRef]
- Terman, A.; Kurz, T.; Navratil, M.; Arriaga, E.A.; Brunk, U.T. Mitochondrial Turnover and Aging of Long-Lived Postmitotic Cells: The Mitochondrial–Lysosomal Axis Theory of Aging. Antioxid. Redox Signal. 2010, 12, 503–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H.; Jones, D.P. Reactive Oxygen Species (ROS) as Pleiotropic Physiological Signalling Agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron Homeostasis and Oxidative Stress: An Intimate Relationship. Biochim. Biophys. Acta BBA—Mol. Cell Res. 2019, 1866, 118535. [Google Scholar] [CrossRef] [PubMed]
- Davies, K. Oxidative Stress, Antioxidant Defenses, and Damage Removal, Repair, and Replacement Systems. IUBMB Life 2001, 50, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Imlay, J.A.; Chin, S.M.; Linn, S. Toxic DNA Damage by Hydrogen Peroxide Through the Fenton Reaction in Vivo and in Vitro. Science 1988, 240, 640–642. [Google Scholar] [CrossRef] [PubMed]
- Galaris, D.; Pantopoulos, K. Oxidative Stress and Iron Homeostasis: Mechanistic and Health Aspects. Crit. Rev. Clin. Lab. Sci. 2008, 45, 1–23. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial Formation of Reactive Oxygen Species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleier, L.; Wittig, I.; Heide, H.; Steger, M.; Brandt, U.; Dröse, S. Generator-Specific Targets of Mitochondrial Reactive Oxygen Species. Free Radic. Biol. Med. 2015, 78, 1–10. [Google Scholar] [CrossRef]
- Soehnlein, O.; Lindbom, L. Phagocyte Partnership during the Onset and Resolution of Inflammation. Nat. Rev. Immunol. 2010, 10, 427–439. [Google Scholar] [CrossRef]
- Brandes, R.P.; Weissmann, N.; Schröder, K. Nox Family NADPH Oxidases: Molecular Mechanisms of Activation. Free Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef]
- Niedzwiecki, M.M.; Walker, D.I.; Vermeulen, R.; Chadeau-Hyam, M.; Jones, D.P.; Miller, G.W. The Exposome: Molecules to Populations. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 107–127. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conrad, M.; Friedmann Angeli, J.P. Glutathione Peroxidase 4 (Gpx4) and Ferroptosis: What’s so Special about It? Mol. Cell. Oncol. 2015, 2, e995047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the Ferroptosis Regulator Gpx4 Triggers Acute Renal Failure in Mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobo, V.; Patil, A.; Phatak, A.; Chandra, N. Free Radicals, Antioxidants and Functional Foods: Impact on Human Health. Pharmacogn. Rev. 2010, 4, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbouti, A.; Lagopati, N.; Veroutis, D.; Goulas, V.; Evangelou, K.; Kanavaros, P.; Gorgoulis, V.G.; Galaris, D. Implication of Dietary Iron-Chelating Bioactive Compounds in Molecular Mechanisms of Oxidative Stress-Induced Cell Ageing. Antioxidants 2021, 10, 491. [Google Scholar] [CrossRef]
- Barbouti, A.; Briasoulis, E.; Galaris, D. Protective Effects of Olive Oil Components Against Hydrogen Peroxide-Induced DNA Damage. In Olives and Olive Oil in Health and Disease Prevention; Elsevier: Amsterdam, The Netherlands, 2010; pp. 1103–1109. ISBN 978-0-12-374420-3. [Google Scholar]
- Melidou, M.; Riganakos, K.; Galaris, D. Protection against Nuclear DNA Damage Offered by Flavonoids in Cells Exposed to Hydrogen Peroxide: The Role of Iron Chelation. Free Radic. Biol. Med. 2005, 39, 1591–1600. [Google Scholar] [CrossRef]
- Nousis, L.; Doulias, P.-T.; Aligiannis, N.; Bazios, D.; Agalias, A.; Galaris, D.; Mitakou, S. DNA Protecting and Genotoxic Effects of Olive Oil Related Components in Cells Exposed to Hydrogen Peroxide. Free Radic. Res. 2005, 39, 787–795. [Google Scholar] [CrossRef]
- Kitsati, N.; Mantzaris, M.D.; Galaris, D. Hydroxytyrosol Inhibits Hydrogen Peroxide-Induced Apoptotic Signaling via Labile Iron Chelation. Redox Biol. 2016, 10, 233–242. [Google Scholar] [CrossRef] [Green Version]
- Gerogianni, P.S.; Chatziathanasiadou, M.V.; Diamantis, D.A.; Tzakos, A.G.; Galaris, D. Lipophilic Ester and Amide Derivatives of Rosmarinic Acid Protect Cells against H2O2-Induced DNA Damage and Apoptosis: The Potential Role of Intracellular Accumulation and Labile Iron Chelation. Redox Biol. 2018, 15, 548–556. [Google Scholar] [CrossRef]
- Kitsati, N.; Fokas, D.; Ouzouni, M.-D.; Mantzaris, M.D.; Barbouti, A.; Galaris, D. Lipophilic Caffeic Acid Derivatives Protect Cells against H2O2-Induced DNA Damage by Chelating Intracellular Labile Iron. J. Agric. Food Chem. 2012, 60, 7873–7879. [Google Scholar] [CrossRef]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [Green Version]
- Katsarou, A.; Pantopoulos, K. Basics and Principles of Cellular and Systemic Iron Homeostasis. Mol. Aspects Med. 2020, 75, 100866. [Google Scholar] [CrossRef] [PubMed]
- Cabantchik, Z.I. Labile Iron in Cells and Body Fluids: Physiology, Pathology, and Pharmacology. Front. Pharmacol. 2014, 5, 45. [Google Scholar] [CrossRef] [Green Version]
- Kakhlon, O.; Cabantchik, Z.I. The Labile Iron Pool: Characterization, Measurement, and Participation in Cellular Processes. Free Radic. Biol. Med. 2002, 33, 1037–1046. [Google Scholar] [CrossRef]
- Chevion, M. A Site-Specific Mechanism for Free Radical Induced Biological Damage: The Essential Role of Redox-Active Transition Metals. Free Radic. Biol. Med. 1988, 5, 27–37. [Google Scholar] [CrossRef]
- Sies, H. Oxidative Stress: A Concept in Redox Biology and Medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayflick, L.; Moorhead, P.S. The Serial Cultivation of Human Diploid Cell Strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. The Biology of Replicative Senescence. Eur. J. Cancer 1997, 33, 703–709. [Google Scholar] [CrossRef]
- Muller, H.J. The Remaking of Chromosomes. Collect. Net 1938, 8, 182–198. [Google Scholar]
- d’Adda di Fagagna, F.; Teo, S.-H.; Jackson, S.P. Functional Links between Telomeres and Proteins of the DNA-Damage Response. Genes Dev. 2004, 18, 1781–1799. [Google Scholar] [CrossRef] [Green Version]
- Shay, J.W.; Wright, W.E. Telomeres and Telomerase: Three Decades of Progress. Nat. Rev. Genet. 2019, 20, 299–309. [Google Scholar] [CrossRef]
- Watson, J.D. Origin of Concatemeric T7DNA. Nature. New Biol. 1972, 239, 197–201. [Google Scholar] [CrossRef]
- Olovnikov, A.M. A Theory of Marginotomy. J. Theor. Biol. 1973, 41, 181–190. [Google Scholar] [CrossRef]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres Shorten during Ageing of Human Fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Boonekamp, J.J.; Simons, M.J.P.; Hemerik, L.; Verhulst, S. Telomere Length Behaves as Biomarker of Somatic Redundancy Rather than Biological Age. Aging Cell 2013, 12, 330–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasco, M.A. Telomere Length, Stem Cells and Aging. Nat. Chem. Biol. 2007, 3, 640–649. [Google Scholar] [CrossRef]
- Armanios, M.; Alder, J.K.; Parry, E.M.; Karim, B.; Strong, M.A.; Greider, C.W. Short Telomeres Are Sufficient to Cause the Degenerative Defects Associated with Aging. Am. J. Hum. Genet. 2009, 85, 823–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera, E.; Samper, E.; Martín-Caballero, J.; Flores, J.M.; Lee, H.W.; Blasco, M.A. Disease States Associated with Telomerase Deficiency Appear Earlier in Mice with Short Telomeres. EMBO J. 1999, 18, 2950–2960. [Google Scholar] [CrossRef] [Green Version]
- Rudolph, K.L.; Chang, S.; Lee, H.-W.; Blasco, M.; Gottlieb, G.J.; Greider, C.; DePinho, R.A. Longevity, Stress Response, and Cancer in Aging Telomerase-Deficient Mice. Cell 1999, 96, 701–712. [Google Scholar] [CrossRef] [Green Version]
- Bernardes de Jesus, B.; Vera, E.; Schneeberger, K.; Tejera, A.M.; Ayuso, E.; Bosch, F.; Blasco, M.A. Telomerase Gene Therapy in Adult and Old Mice Delays Aging and Increases Longevity without Increasing Cancer. EMBO Mol. Med. 2012, 4, 691–704. [Google Scholar] [CrossRef] [PubMed]
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular Senescence and Its Effector Programs. Genes Dev. 2014, 28, 99–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence Is a Developmental Mechanism That Contributes to Embryonic Growth and Patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef] [Green Version]
- Barbouti, A.; Vasileiou, P.V.S.; Evangelou, K.; Vlasis, K.G.; Papoudou-Bai, A.; Gorgoulis, V.G.; Kanavaros, P. Implications of Oxidative Stress and Cellular Senescence in Age-Related Thymus Involution. Oxid. Med. Cell. Longev. 2020, 2020, 7986071. [Google Scholar] [CrossRef] [Green Version]
- Barbouti, A.; Evangelou, K.; Pateras, I.S.; Papoudou-Bai, A.; Patereli, A.; Stefanaki, K.; Rontogianni, D.; Muñoz-Espín, D.; Kanavaros, P.; Gorgoulis, V.G. In Situ Evidence of Cellular Senescence in Thymic Epithelial Cells (TECs) during Human Thymic Involution. Mech. Ageing Dev. 2019, 177, 88–90. [Google Scholar] [CrossRef]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef]
- Campisi, J. Aging, Cellular Senescence, and Cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [Green Version]
- Hamsanathan, S.; Gurkar, A.U. Lipids as Regulators of Cellular Senescence. Front. Physiol. 2022, 13, 796850. [Google Scholar] [CrossRef] [PubMed]
- Höhn, A.; Weber, D.; Jung, T.; Ott, C.; Hugo, M.; Kochlik, B.; Kehm, R.; König, J.; Grune, T.; Castro, J.P. Happily (n)Ever after: Aging in the Context of Oxidative Stress, Proteostasis Loss and Cellular Senescence. Redox Biol. 2017, 11, 482–501. [Google Scholar] [CrossRef]
- Höhn, A.; Grune, T. Lipofuscin: Formation, Effects and Role of Macroautophagy. Redox Biol. 2013, 1, 140–144. [Google Scholar] [CrossRef] [Green Version]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular Senescence in Aging and Age-Related Disease: From Mechanisms to Therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [Green Version]
- Brunk, U.T.; Terman, A. Lipofuscin: Mechanisms of Age-Related Accumulation and Influence on Cell Function. Free Radic. Biol. Med. 2002, 33, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Petrat, F.; Rauen, U.; de Groot, H. Determination of the Chelatable Iron Pool of Isolated Rat Hepatocytes by Digital Fluorescence Microscopy Using the Fluorescent Probe, Phen Green SK. Hepatology 1999, 29, 1171–1179. [Google Scholar] [CrossRef]
- Ma, Y.; de Groot, H.; Liu, Z.; Hider, R.C.; Petrat, F. Chelation and Determination of Labile Iron in Primary Hepatocytes by Pyridinone Fluorescent Probes. Biochem. J. 2006, 395, 49–55. [Google Scholar] [CrossRef] [Green Version]
- Höhn, A.; Jung, T.; Grimm, S.; Grune, T. Lipofuscin-Bound Iron Is a Major Intracellular Source of Oxidants: Role in Senescent Cells. Free Radic. Biol. Med. 2010, 48, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Brunk, U.T.; Terman, A. The Mitochondrial-Lysosomal Axis Theory of Aging: Accumulation of Damaged Mitochondria as a Result of Imperfect Autophagocytosis. Eur. J. Biochem. 2002, 269, 1996–2002. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Davies, K.J.A. Oxidative DNA Damage & Repair: An Introduction. Free Radic. Biol. Med. 2017, 107, 2–12. [Google Scholar] [CrossRef]
- Pateras, I.S.; Havaki, S.; Nikitopoulou, X.; Vougas, K.; Townsend, P.A.; Panayiotidis, M.I.; Georgakilas, A.G.; Gorgoulis, V.G. The DNA Damage Response and Immune Signaling Alliance: Is It Good or Bad? Nature Decides When and Where. Pharmacol. Ther. 2015, 154, 36–56. [Google Scholar] [CrossRef]
- d’Adda di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA Damage Checkpoint Response in Telomere-Initiated Senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef]
- Rossiello, F.; Jurk, D.; Passos, J.F.; d’Adda di Fagagna, F. Telomere Dysfunction in Ageing and Age-Related Diseases. Nat. Cell Biol. 2022, 24, 135–147. [Google Scholar] [CrossRef]
- Davalli, P.; Marverti, G.; Lauriola, A.; D’Arca, D. Targeting Oxidatively Induced DNA Damage Response in Cancer: Opportunities for Novel Cancer Therapies. Oxid. Med. Cell. Longev. 2018, 2018, 2389523. [Google Scholar] [CrossRef] [Green Version]
- Engeland, K. Cell Cycle Regulation: P53-P21-RB Signaling. Cell Death Differ. 2022, 29, 946–960. [Google Scholar] [CrossRef]
- de Lange, T. How Telomeres Solve the End-Protection Problem. Science 2009, 326, 948–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, R.P.; de Rosa, M.; Thosar, S.A.; Detwiler, A.C.; Roginskaya, V.; Van Houten, B.; Bruchez, M.P.; Stewart-Ornstein, J.; Opresko, P.L. Telomeric 8-Oxo-Guanine Drives Rapid Premature Senescence in the Absence of Telomere Shortening. Nat. Struct. Mol. Biol. 2022, 29, 639–652. [Google Scholar] [CrossRef]
- Lagnado, A.; Leslie, J.; Ruchaud-Sparagano, M.; Victorelli, S.; Hirsova, P.; Ogrodnik, M.; Collins, A.L.; Vizioli, M.G.; Habiballa, L.; Saretzki, G.; et al. Neutrophils Induce Paracrine Telomere Dysfunction and Senescence in ROS-dependent Manner. EMBO J. 2021, 40, e106048. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, W.; Lingner, J. Impact of Oxidative Stress on Telomere Biology. Differentiation 2018, 99, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Griffith, J.D.; Comeau, L.; Rosenfield, S.; Stansel, R.M.; Bianchi, A.; Moss, H.; de Lange, T. Mammalian Telomeres End in a Large Duplex Loop. Cell 1999, 97, 503–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Lange, T. Shelterin: The Protein Complex That Shapes and Safeguards Human Telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [Green Version]
- Herbig, U.; Jobling, W.A.; Chen, B.P.C.; Chen, D.J.; Sedivy, J.M. Telomere Shortening Triggers Senescence of Human Cells through a Pathway Involving ATM, P53, and P21CIP1, but Not P16INK4a. Mol. Cell 2004, 14, 501–513. [Google Scholar] [CrossRef]
- von Zglinicki, T. Oxidative Stress Shortens Telomeres. Trends Biochem. Sci. 2002, 27, 339–344. [Google Scholar] [CrossRef]
- von Zglinicki, T.; Pilger, R.; Sitte, N. Accumulation of Single-Strand Breaks Is the Major Cause of Telomere Shortening in Human Fibroblasts. Free Radic. Biol. Med. 2000, 28, 64–74. [Google Scholar] [CrossRef]
- Passos, J.F.; Saretzki, G.; Ahmed, S.; Nelson, G.; Richter, T.; Peters, H.; Wappler, I.; Birket, M.J.; Harold, G.; Schaeuble, K.; et al. Mitochondrial Dysfunction Accounts for the Stochastic Heterogeneity in Telomere-Dependent Senescence. PLoS Biol. 2007, 5, e110. [Google Scholar] [CrossRef] [Green Version]
- von Zglinicki, T.; Saretzki, G.; Döcke, W.; Lotze, C. Mild Hyperoxia Shortens Telomeres and Inhibits Proliferation of Fibroblasts: A Model for Senescence? Exp. Cell Res. 1995, 220, 186–193. [Google Scholar] [CrossRef]
- Kawanishi, S.; Oikawa, S. Mechanism of Telomere Shortening by Oxidative Stress. Ann. N. Y. Acad. Sci. 2004, 1019, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Saretzki, G.; Murphy, M.P.; von Zglinicki, T. MitoQ Counteracts Telomere Shortening and Elongates Lifespan of Fibroblasts under Mild Oxidative Stress. Aging Cell 2003, 2, 141–143. [Google Scholar] [CrossRef] [PubMed]
- Reichert, S.; Stier, A. Does Oxidative Stress Shorten Telomeres in Vivo? A Review. Biol. Lett. 2017, 13, 20170463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fumagalli, M.; Rossiello, F.; Clerici, M.; Barozzi, S.; Cittaro, D.; Kaplunov, J.M.; Bucci, G.; Dobreva, M.; Matti, V.; Beausejour, C.M.; et al. Telomeric DNA Damage Is Irreparable and Causes Persistent DNA-Damage-Response Activation. Nat. Cell Biol. 2012, 14, 355–365. [Google Scholar] [CrossRef] [Green Version]
- Kruk, P.A.; Rampino, N.J.; Bohr, V.A. DNA Damage and Repair in Telomeres: Relation to Aging. Proc. Natl. Acad. Sci. USA 1995, 92, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Bombarde, O.; Boby, C.; Gomez, D.; Frit, P.; Giraud-Panis, M.-J.; Gilson, E.; Salles, B.; Calsou, P. TRF2/RAP1 and DNA–PK Mediate a Double Protection against Joining at Telomeric Ends. EMBO J. 2010, 29, 1573–1584. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, G.; Jurk, D.; Marques, F.D.M.; Correia-Melo, C.; Hardy, T.; Gackowska, A.; Anderson, R.; Taschuk, M.; Mann, J.; Passos, J.F. Telomeres Are Favoured Targets of a Persistent DNA Damage Response in Ageing and Stress-Induced Senescence. Nat. Commun. 2012, 3, 708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fumagalli, M.; Rossiello, F.; Mondello, C.; d’Adda di Fagagna, F. Stable Cellular Senescence Is Associated with Persistent DDR Activation. PLoS ONE 2014, 9, e110969. [Google Scholar] [CrossRef] [PubMed]
- Jacome Burbano, M.S.; Cherfils-Vicini, J.; Gilson, E. Neutrophils: Mediating TelOxidation and Senescence. EMBO J. 2021, 40, e108164. [Google Scholar] [CrossRef]
- Oikawa, S.; Kawanishi, S. Site-Specific DNA Damage at GGG Sequence by Oxidative Stress May Accelerate Telomere Shortening. FEBS Lett. 1999, 453, 365–368. [Google Scholar] [CrossRef] [Green Version]
- Fouquerel, E.; Barnes, R.P.; Uttam, S.; Watkins, S.C.; Bruchez, M.P.; Opresko, P.L. Targeted and Persistent 8-Oxoguanine Base Damage at Telomeres Promotes Telomere Loss and Crisis. Mol. Cell 2019, 75, 117–130.e6. [Google Scholar] [CrossRef]
- Sekiguchi, M.; Tsuzuki, T. Oxidative Nucleotide Damage: Consequences and Prevention. Oncogene 2002, 21, 8895–8904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouquerel, E.; Lormand, J.; Bose, A.; Lee, H.-T.; Kim, G.S.; Li, J.; Sobol, R.W.; Freudenthal, B.D.; Myong, S.; Opresko, P.L. Oxidative Guanine Base Damage Regulates Human Telomerase Activity. Nat. Struct. Mol. Biol. 2016, 23, 1092–1100. [Google Scholar] [CrossRef] [Green Version]
- Aeby, E.; Ahmed, W.; Redon, S.; Simanis, V.; Lingner, J. Peroxiredoxin 1 Protects Telomeres from Oxidative Damage and Preserves Telomeric DNA for Extension by Telomerase. Cell Rep. 2016, 17, 3107–3114. [Google Scholar] [CrossRef] [Green Version]
- Smith, S. Telomerase Can’t Handle the Stress. Genes Dev. 2018, 32, 597–599. [Google Scholar] [CrossRef]
- Opresko, P.L.; Fan, J.; Danzy, S.; Wilson, D.M.; Bohr, V.A. Oxidative Damage in Telomeric DNA Disrupts Recognition by TRF1 and TRF2. Nucleic Acids Res. 2005, 33, 1230–1239. [Google Scholar] [CrossRef]
- Barbouti, A.; Doulias, P.T.; Zhu, B.Z.; Frei, B.; Galaris, D. Intracellular Iron, but Not Copper, Plays a Critical Role in Hydrogen Peroxide-Induced DNA Damage. Free Radic. Biol. Med. 2001, 31, 490–498. [Google Scholar] [CrossRef]
- Barbouti, A.; Amorgianiotis, C.; Kolettas, E.; Kanavaros, P.; Galaris, D. Hydrogen Peroxide Inhibits Caspase-Dependent Apoptosis by Inactivating Procaspase-9 in an Iron-Dependent Manner. Free Radic. Biol. Med. 2007, 43, 1377–1387. [Google Scholar] [CrossRef]
- Doulias, P.-T.; Christoforidis, S.; Brunk, U.T.; Galaris, D. Endosomal and Lysosomal Effects of Desferrioxamine: Protection of HeLa Cells from Hydrogen Peroxide-Induced DNA Damage and Induction of Cell-Cycle Arrest. Free Radic. Biol. Med. 2003, 35, 719–728. [Google Scholar] [CrossRef]
- Tenopoulou, M.; Doulias, P.-T.; Barbouti, A.; Brunk, U.; Galaris, D. Role of Compartmentalized Redox-Active Iron in Hydrogen Peroxide-Induced DNA Damage and Apoptosis. Biochem. J. 2005, 387, 703–710. [Google Scholar] [CrossRef]
- Mantelou, A.G.; Barbouti, A.; Goussia, A.; Zacharioudaki, A.; Papoudou-Bai, A.; Vlachou, C.; Kokkoris, S.; Papalois, A.; Galaris, D.; Glantzounis, G.K. Combined Administration of Membrane-Permeable and Impermeable Iron-Chelating Drugs Attenuates Ischemia/Reperfusion-Induced Hepatic Injury. Free Radic. Biol. Med. 2022, 193, 227–237. [Google Scholar] [CrossRef]
- Sfera, A.; Bullock, K.; Price, A.; Inderias, L.; Osorio, C. Ferrosenescence: The Iron Age of Neurodegeneration? Mech. Ageing Dev. 2018, 174, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Fracanzani, A.L.; Cairo, G.; Megazzini, C.P.; Gatti, S.; Rametta, R.; Fargion, S.; Valenti, L. Iron-Dependent Regulation of MDM2 Influences P53 Activity and Hepatic Carcinogenesis. Am. J. Pathol. 2010, 176, 1006–1017. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Cuervo, A.M. Proteostasis and Aging. Nat. Med. 2015, 21, 1406–1415. [Google Scholar] [CrossRef]
- Kehm, R.; Baldensperger, T.; Raupbach, J.; Höhn, A. Protein Oxidation—Formation Mechanisms, Detection and Relevance as Biomarkers in Human Diseases. Redox Biol. 2021, 42, 101901. [Google Scholar] [CrossRef]
- Castro, J.P.; Jung, T.; Grune, T.; Siems, W. 4-Hydroxynonenal (HNE) Modified Proteins in Metabolic Diseases. Free Radic. Biol. Med. 2017, 111, 309–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grune, T. Oxidized Protein Aggregates: Formation and Biological Effects. Free Radic. Biol. Med. 2020, 150, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Labbadia, J.; Morimoto, R.I. The Biology of Proteostasis in Aging and Disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of Cellular Proteostasis in Aging and Disease. J. Cell Biol. 2018, 217, 51–63. [Google Scholar] [CrossRef]
- Cavinato, M.; Madreiter-Sokolowski, C.T.; Büttner, S.; Schosserer, M.; Zwerschke, W.; Wedel, S.; Grillari, J.; Graier, W.F.; Jansen-Dürr, P. Targeting Cellular Senescence Based on Interorganelle Communication, Multilevel Proteostasis, and Metabolic Control. FEBS J. 2021, 288, 3834–3854. [Google Scholar] [CrossRef]
- Wiley, C.D.; Campisi, J. The Metabolic Roots of Senescence: Mechanisms and Opportunities for Intervention. Nat. Metab. 2021, 3, 1290–1301. [Google Scholar] [CrossRef]
- Ademowo, O.S.; Dias, H.K.I.; Burton, D.G.A.; Griffiths, H.R. Lipid (per) Oxidation in Mitochondria: An Emerging Target in the Ageing Process? Biogerontology 2017, 18, 859–879. [Google Scholar] [CrossRef] [Green Version]
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria Are Required for Pro-ageing Features of the Senescent Phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef] [PubMed]
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular Senescence Drives Age-Dependent Hepatic Steatosis. Nat. Commun. 2017, 8, 15691. [Google Scholar] [CrossRef] [Green Version]
- Kurz, T.; Terman, A.; Brunk, U.T. Autophagy, Ageing and Apoptosis: The Role of Oxidative Stress and Lysosomal Iron. Arch. Biochem. Biophys. 2007, 462, 220–230. [Google Scholar] [CrossRef]
- Terman, A.; Brunk, U.T. Oxidative Stress, Accumulation of Biological “Garbage”, and Aging. Antioxid. Redox Signal. 2006, 8, 197–204. [Google Scholar] [CrossRef]
- Salmonowicz, H.; Passos, J.F. Detecting Senescence: A New Method for an Old Pigment. Aging Cell 2017, 16, 432–434. [Google Scholar] [CrossRef]
- Evangelou, K.; Lougiakis, N.; Rizou, S.V.; Kotsinas, A.; Kletsas, D.; Muñoz-Espín, D.; Kastrinakis, N.G.; Pouli, N.; Marakos, P.; Townsend, P.; et al. Robust, Universal Biomarker Assay to Detect Senescent Cells in Biological Specimens. Aging Cell 2017, 16, 192–197. [Google Scholar] [CrossRef]
- Sitte, N.; Merker, K.; Grune, T.; von Zglinicki, T. Lipofuscin Accumulation in Proliferating Fibroblasts in Vitro: An Indicator of Oxidative Stress. Exp. Gerontol. 2001, 36, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.; Bader, N.; Grune, T. Lipofuscin: Formation, Distribution, and Metabolic Consequences. Ann. N. Y. Acad. Sci. 2007, 1119, 97–111. [Google Scholar] [CrossRef] [PubMed]
- König, J.; Ott, C.; Hugo, M.; Jung, T.; Bulteau, A.-L.; Grune, T.; Höhn, A. Mitochondrial Contribution to Lipofuscin Formation. Redox Biol. 2017, 11, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Von Zglinicki, T.; Nilsson, E.; Döcke, W.D.; Brunk, U.T. Lipofuscin Accumulation and Ageing of Fibroblasts. Gerontology 1995, 41, 95–108. [Google Scholar] [CrossRef]
- Terman, A.; Brunk, U.T. Lipofuscin. Int. J. Biochem. Cell Biol. 2004, 36, 1400–1404. [Google Scholar] [CrossRef]
- Moreno-García, A.; Kun, A.; Calero, O.; Medina, M.; Calero, M. An Overview of the Role of Lipofuscin in Age-Related Neurodegeneration. Front. Neurosci. 2018, 12, 464. [Google Scholar] [CrossRef] [Green Version]
- Yim, W.W.-Y.; Mizushima, N. Lysosome Biology in Autophagy. Cell Discov. 2020, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- Kurz, T.; Terman, A.; Gustafsson, B.; Brunk, U.T. Lysosomes in Iron Metabolism, Ageing and Apoptosis. Histochem. Cell Biol. 2008, 129, 389–406. [Google Scholar] [CrossRef] [Green Version]
- Marzabadi, M.R.; Sohal, R.S.; Brunk, U.T. Effect of Ferric Iron and Desferrioxamine on Lipofuscin Accumulation in Cultured Rat Heart Myocytes. Mech. Ageing Dev. 1988, 46, 145–157. [Google Scholar] [CrossRef]
- Martini, H.; Passos, J.F. Cellular Senescence: All Roads Lead to Mitochondria. FEBS J. 2022, 290, 1186–1202. [Google Scholar] [CrossRef] [PubMed]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between P21 and Reactive Oxygen Production Is Necessary for Cell Senescence. Mol. Syst. Biol. 2010, 6, 347. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.; Fielder, E.; Passos, J.F. Mitochondrial Dysfunction and Cell Senescence: Deciphering a Complex Relationship. FEBS Lett. 2019, 593, 1566–1579. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-C.; Yin, P.-H.; Chi, C.-W.; Wei, Y.-H. Increase in Mitochondrial Mass in Human Fibroblasts under Oxidative Stress and during Replicative Cell Senescence. J. Biomed. Sci. 2002, 9, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Tai, H.; Wang, Z.; Gong, H.; Han, X.; Zhou, J.; Wang, X.; Wei, X.; Ding, Y.; Huang, N.; Qin, J.; et al. Autophagy Impairment with Lysosomal and Mitochondrial Dysfunction Is an Important Characteristic of Oxidative Stress-Induced Senescence. Autophagy 2017, 13, 99–113. [Google Scholar] [CrossRef] [Green Version]
- Hutter, E.; Renner, K.; Pfister, G.; Stöckl, P.; Jansen-Dürr, P.; Gnaiger, E. Senescence-Associated Changes in Respiration and Oxidative Phosphorylation in Primary Human Fibroblasts. Biochem. J. 2004, 380, 919–928. [Google Scholar] [CrossRef] [Green Version]
- Moiseeva, O.; Bourdeau, V.; Roux, A.; Deschênes-Simard, X.; Ferbeyre, G. Mitochondrial Dysfunction Contributes to Oncogene-Induced Senescence. Mol. Cell. Biol. 2009, 29, 4495–4507. [Google Scholar] [CrossRef] [Green Version]
- Stöckl, P.; Hütter, E.; Zwerschke, W.; Jansen-Dürr, P. Sustained Inhibition of Oxidative Phosphorylation Impairs Cell Proliferation and Induces Premature Senescence in Human Fibroblasts. Exp. Gerontol. 2006, 41, 674–682. [Google Scholar] [CrossRef]
- Korolchuk, V.I.; Miwa, S.; Carroll, B.; von Zglinicki, T. Mitochondria in Cell Senescence: Is Mitophagy the Weakest Link? eBioMedicine 2017, 21, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Hamon, M.; Ahmed, E.K.; Baraibar, M.A.; Friguet, B. Proteome Oxidative Modifications and Impairment of Specific Metabolic Pathways During Cellular Senescence and Aging. Proteomics 2020, 20, 1800421. [Google Scholar] [CrossRef]
- Nelson, G.; Wordsworth, J.; Wang, C.; Jurk, D.; Lawless, C.; Martin-Ruiz, C.; von Zglinicki, T. A Senescent Cell Bystander Effect: Senescence-induced Senescence. Aging Cell 2012, 11, 345–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Victorelli, S.; Lagnado, A.; Halim, J.; Moore, W.; Talbot, D.; Barrett, K.; Chapman, J.; Birch, J.; Ogrodnik, M.; Meves, A.; et al. Senescent Human Melanocytes Drive Skin Ageing via Paracrine Telomere Dysfunction. EMBO J. 2019, 38, e101982. [Google Scholar] [CrossRef] [PubMed]
- Ward, D.M.; Cloonan, S.M. Mitochondrial Iron in Human Health and Disease. Annu. Rev. Physiol. 2019, 81, 453–482. [Google Scholar] [CrossRef]
- Seo, A.Y.; Xu, J.; Servais, S.; Hofer, T.; Marzetti, E.; Wohlgemuth, S.E.; Knutson, M.D.; Chung, H.Y.; Leeuwenburgh, C. Mitochondrial Iron Accumulation with Age and Functional Consequences. Aging Cell 2008, 7, 706–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolinches-AmorÃ3s, A.; Mollá, B.; Pla-MartÃn, D.; Palau, F.; González-Cabo, P. Mitochondrial Dysfunction Induced by Frataxin Deficiency Is Associated with Cellular Senescence and Abnormal Calcium Metabolism. Front. Cell. Neurosci. 2014, 8, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Issitt, T.; Bosseboeuf, E.; De Winter, N.; Dufton, N.; Gestri, G.; Senatore, V.; Chikh, A.; Randi, A.M.; Raimondi, C. Neuropilin-1 Controls Endothelial Homeostasis by Regulating Mitochondrial Function and Iron-Dependent Oxidative Stress. iScience 2019, 11, 205–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Abbate, V.; Hider, R.C. Iron-Sensitive Fluorescent Probes: Monitoring Intracellular Iron Pools. Met. Integr. Biometal Sci. 2015, 7, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Naguro, I.; Ichijo, H. Iron Homeostasis and Iron-Regulated ROS in Cell Death, Senescence and Human Diseases. Biochim. Biophys. Acta BBA—Gen. Subj. 2019, 1863, 1398–1409. [Google Scholar] [CrossRef]
- Shatrova, A.; Burova, E.; Kharchenko, M.; Smirnova, I.; Lyublinskaya, O.; Nikolsky, N.; Borodkina, A. Outcomes of Deferoxamine Action on H2O2-Induced Growth Inhibition and Senescence Progression of Human Endometrial Stem Cells. Int. J. Mol. Sci. 2021, 22, 6035. [Google Scholar] [CrossRef]
- Masaldan, S.; Clatworthy, S.A.S.; Gamell, C.; Meggyesy, P.M.; Rigopoulos, A.-T.; Haupt, S.; Haupt, Y.; Denoyer, D.; Adlard, P.A.; Bush, A.I.; et al. Iron Accumulation in Senescent Cells Is Coupled with Impaired Ferritinophagy and Inhibition of Ferroptosis. Redox Biol. 2018, 14, 100–115. [Google Scholar] [CrossRef]
- Killilea, D.W.; Wong, S.L.; Cahaya, H.S.; Atamna, H.; Ames, B.N. Iron Accumulation during Cellular Senescence. Ann. N. Y. Acad. Sci. 2004, 1019, 365–367. [Google Scholar] [CrossRef]
- Devos, D.; Moreau, C.; Devedjian, J.C.; Kluza, J.; Petrault, M.; Laloux, C.; Jonneaux, A.; Ryckewaert, G.; Garçon, G.; Rouaix, N.; et al. Targeting Chelatable Iron as a Therapeutic Modality in Parkinson’s Disease. Antioxid. Redox Signal. 2014, 21, 195–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mclachlan, D. Intramuscular Desferrioxamine in Patients with Alzheimer’s Disease. Lancet 1991, 337, 1304–1308. [Google Scholar] [CrossRef] [PubMed]
- Belaidi, A.A.; Bush, A.I. Iron Neurochemistry in Alzheimer’s Disease and Parkinson’s Disease: Targets for Therapeutics. J. Neurochem. 2016, 139, 179–197. [Google Scholar] [CrossRef] [Green Version]
- Hašková, P.; Applová, L.; Jansová, H.; Homola, P.; Franz, K.J.; Vávrová, K.; Roh, J.; Šimůnek, T. Examination of Diverse Iron-Chelating Agents for the Protection of Differentiated PC12 Cells against Oxidative Injury Induced by 6-Hydroxydopamine and Dopamine. Sci. Rep. 2022, 12, 9765. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Wu, R.; Shang, M.; Sato, T.; Chen, C.; Shapiro, J.S.; Liu, T.; Thakur, A.; Sawicki, K.T.; Prasad, S.V.; et al. Reduction in Mitochondrial Iron Alleviates Cardiac Damage during Injury. EMBO Mol. Med. 2016, 8, 247–267. [Google Scholar] [CrossRef] [PubMed]
- Koskenkorva-Frank, T.S.; Weiss, G.; Koppenol, W.H.; Burckhardt, S. The Complex Interplay of Iron Metabolism, Reactive Oxygen Species, and Reactive Nitrogen Species: Insights into the Potential of Various Iron Therapies to Induce Oxidative and Nitrosative Stress. Free Radic. Biol. Med. 2013, 65, 1174–1194. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nousis, L.; Kanavaros, P.; Barbouti, A. Oxidative Stress-Induced Cellular Senescence: Is Labile Iron the Connecting Link? Antioxidants 2023, 12, 1250. https://doi.org/10.3390/antiox12061250
Nousis L, Kanavaros P, Barbouti A. Oxidative Stress-Induced Cellular Senescence: Is Labile Iron the Connecting Link? Antioxidants. 2023; 12(6):1250. https://doi.org/10.3390/antiox12061250
Chicago/Turabian StyleNousis, Lambros, Panagiotis Kanavaros, and Alexandra Barbouti. 2023. "Oxidative Stress-Induced Cellular Senescence: Is Labile Iron the Connecting Link?" Antioxidants 12, no. 6: 1250. https://doi.org/10.3390/antiox12061250