

Bradykinin B1 Receptor Affects Tumor-Associated Macrophage Activity and Glioblastoma Progression

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Cell Transfection
2.4. Real-Time PCR (qPCR)
2.5. Western Blotting
2.6. Cell Migration (Wound Healing) Assay
2.7. Flow Cytometry Analysis
2.8. Monocyte Binding Assay
2.9. ROS Production Analysis
2.10. Co-Culture of GBM Cells and Macrophages
2.11. Animals
2.12. Intracranial Tumor Models
2.13. Statistical Analysis
3. Results
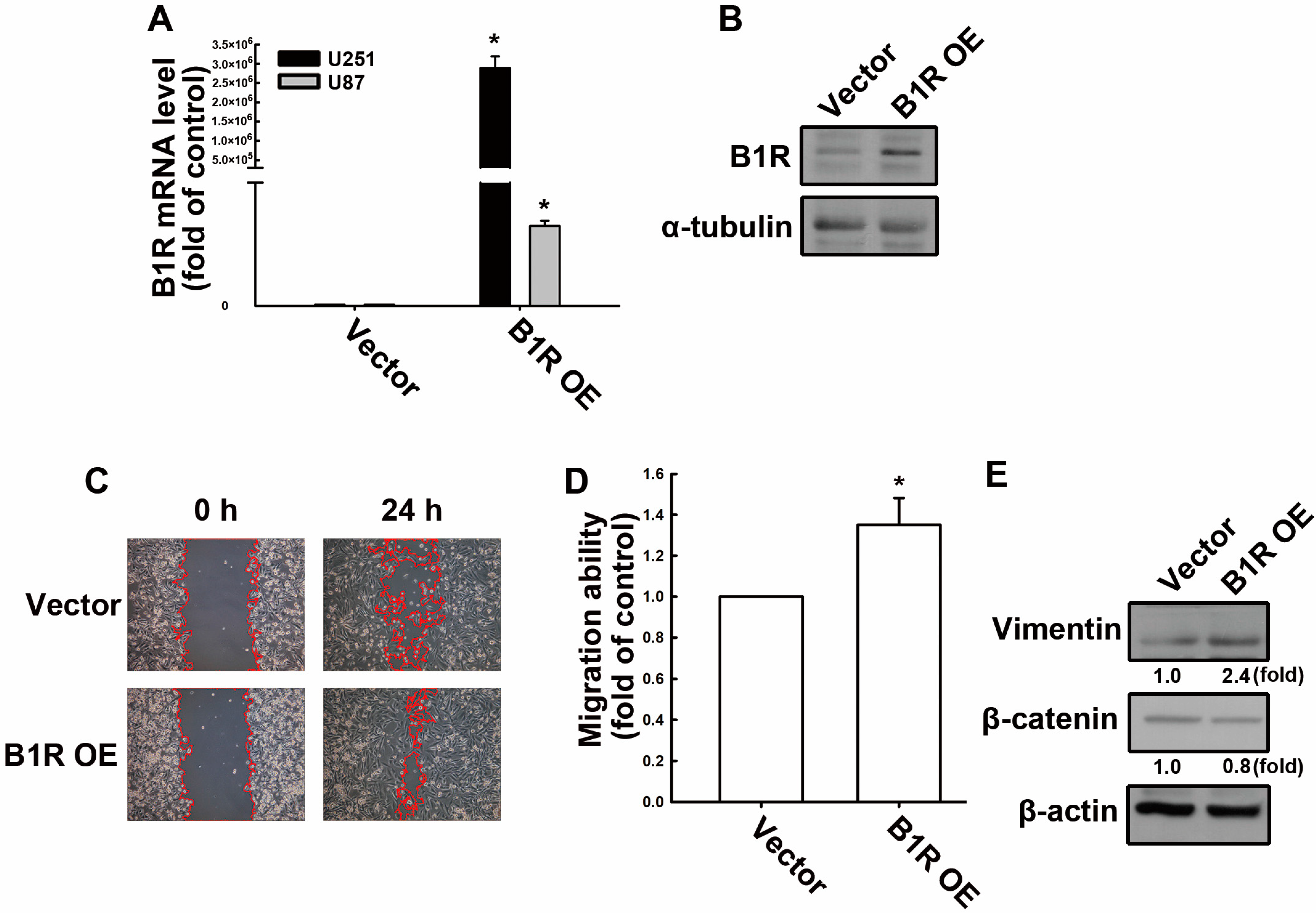
3.1. B1R OE Promotes GBM Cell Migration
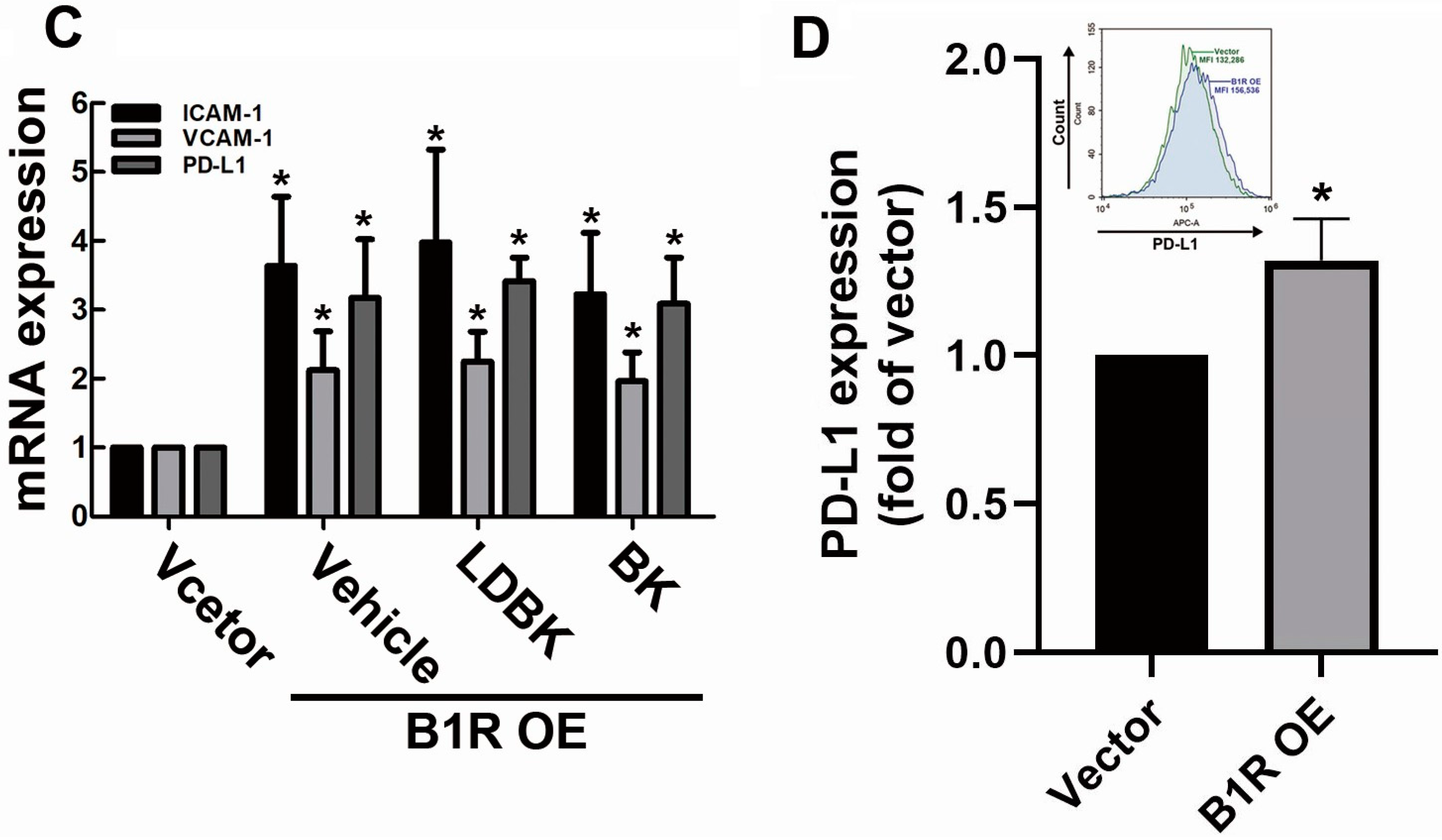
3.2. B1R OE in GBM Cells Induces the Expression of Adhesion Molecules and Immune Checkpoints
3.3. B1R OE Induces Monocyte Binding to GBM Cells
3.4. B1R OE Inhibits ROS Production in GBM Cells
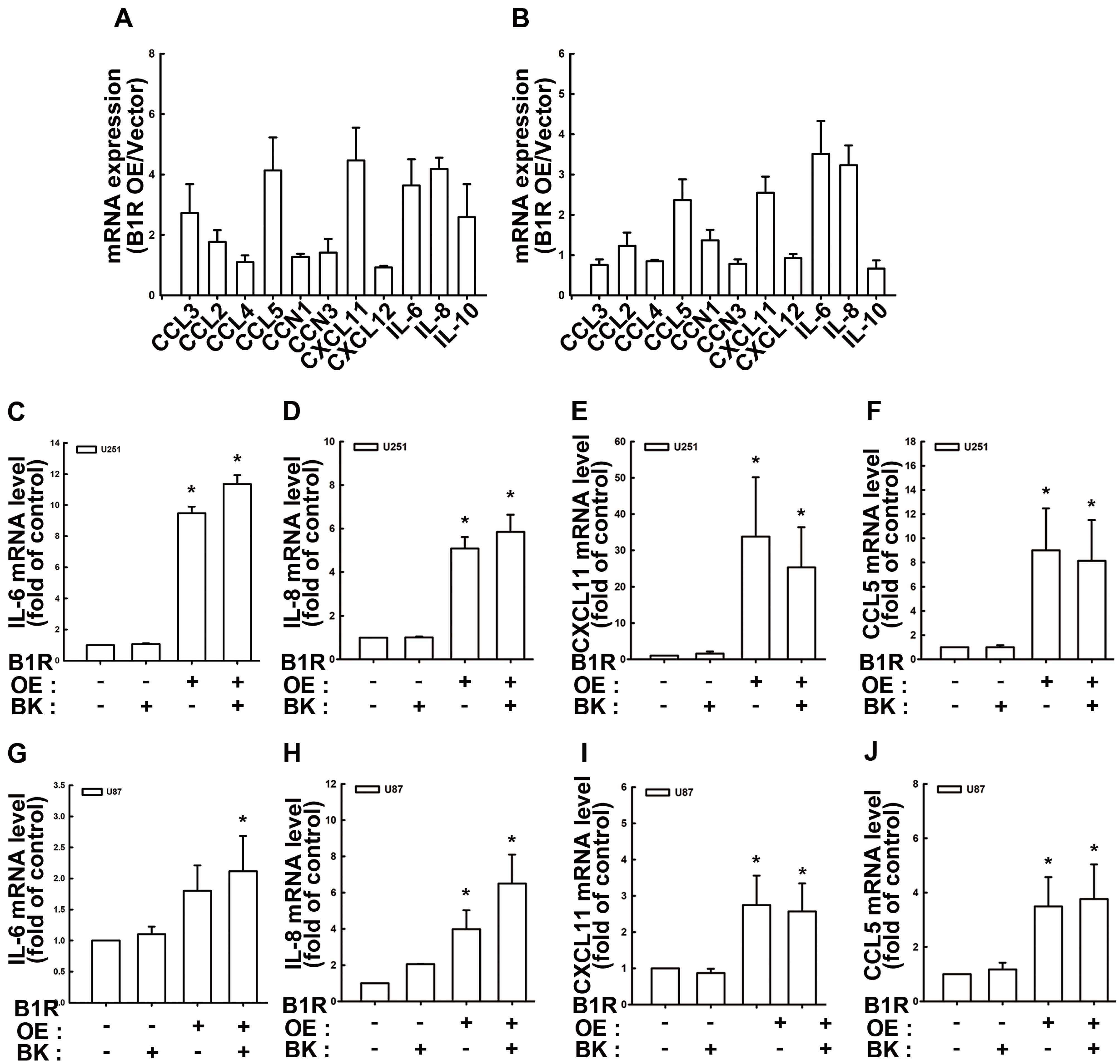
3.5. B1R OE Induces Expression of Protumorigenic Cytokines and Chemokines in GBM Cells
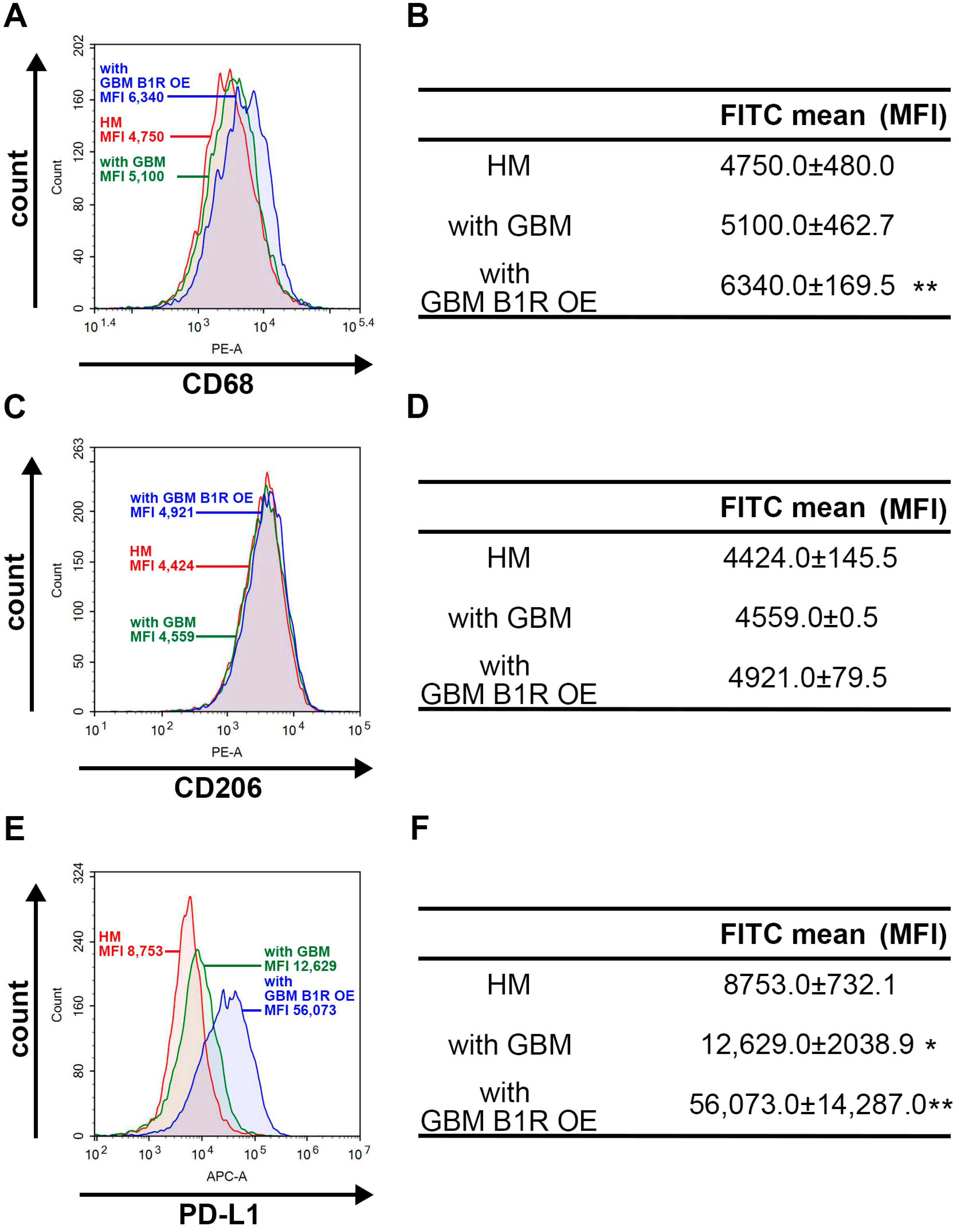
3.6. B1R OE in GBM Cells Affects Macrophage Polarization and PD-L1 Expression in Macrophages
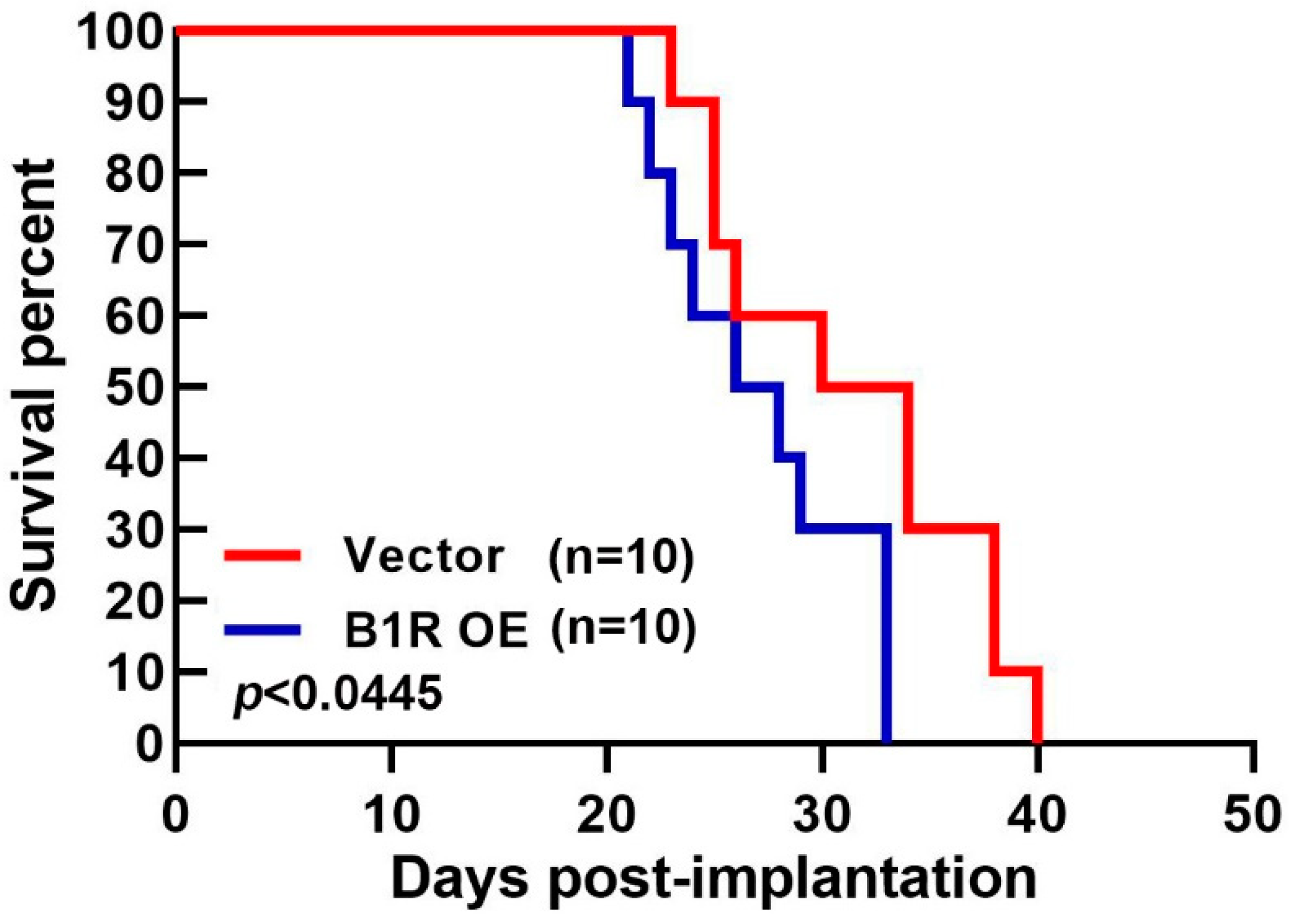
3.7. B1R OE in GBM Reduces the Survival Rate of an Intracranial Xenograft Mouse Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alves, J.M.; Martins, A.H.; Lameu, C.; Glaser, T.; Boukli, N.M.; Bassaneze, V.; Dariolli, R.; Nascimento, I.C.; Martins, P.C.M.; de Souza, H.D.N.; et al. Kinin-B2 receptor activity in skeletal muscle regeneration and myoblast differentiation. Stem Cell Rev. Rep. 2019, 15, 48–58. [Google Scholar] [CrossRef]
- Abbott, N.J. Inflammatory mediators and modulation of blood-brain barrier permeability. Cell Mol. Neurobiol. 2000, 20, 131–147. [Google Scholar] [CrossRef]
- Lehmberg, J.; Beck, J.; Baethmann, A.; Uhl, E. Bradykinin antagonists reduce leukocyte-endothelium interactions after global cerebral ischemia. J. Cereb. Blood Flow. Metab. 2003, 23, 441–448. [Google Scholar] [CrossRef] [Green Version]
- Dutra, R.C.; Bento, A.F.; Leite, D.F.; Manjavachi, M.N.; Marcon, R.; Bicca, M.A.; Pesquero, J.B.; Calixto, J.B. The role of kinin b1 and b2 receptors in the persistent pain induced by experimental autoimmune encephalomyelitis (EAE) in mice: Evidence for the involvement of astrocytes. Neurobiol. Dis. 2013, 54, 82–93. [Google Scholar] [CrossRef]
- Gobel, K.; Pankratz, S.; Schneider-Hohendorf, T.; Bittner, S.; Schuhmann, M.K.; Langer, H.F.; Stoll, G.; Wiendl, H.; Kleinschnitz, C.; Meuth, S.G. Blockade of the kinin receptor B1 protects from autoimmune CNS disease by reducing leukocyte trafficking. J. Autoimmun. 2011, 36, 106–114. [Google Scholar] [CrossRef]
- Alexander-Curtis, M.; Pauls, R.; Chao, J.; Volpi, J.J.; Bath, P.M.; Verdoorn, T.A. Human tissue kallikrein in the treatment of acute ischemic stroke. Ther. Adv. Neurol. Disord. 2019, 12, 1756286418821918. [Google Scholar] [CrossRef] [Green Version]
- Regoli, D.; Barabe, J.; Park, W.K. Receptors for bradykinin in rabbit aortae. Can. J. Physiol. Pharmacol. 1977, 55, 855–867. [Google Scholar] [CrossRef]
- Roberts, R.A. Bradykinin receptors: Characterization, distribution and mechanisms of signal transduction. Prog. Growth Factor. Res. 1989, 1, 237–252. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, W.; Wei, R.; Jiang, G.; Li, F.; Chen, X.; Wang, X.; Long, S.; Ma, D.; Xi, L. Serum bradykinin levels as a diagnostic marker in cervical cancer with a potential mechanism to promote VEGF expression via BDKRB2. Int. J. Oncol. 2019, 55, 131–141. [Google Scholar] [CrossRef] [Green Version]
- da Costa, P.L.N.; Wynne, D.; Fifis, T.; Nguyen, L.; Perini, M.; Christophi, C. The kallikrein-kinin system modulates the progression of colorectal liver metastases in a mouse model. BMC Cancer 2018, 18, 382. [Google Scholar] [CrossRef] [Green Version]
- da Costa, P.L.N.; Sirois, P.; Tannock, I.F.; Chammas, R. The role of kinin receptors in cancer and therapeutic opportunities. Cancer Lett. 2014, 345, 27–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawant, S.; Snyman, C.; Bhoola, K. Comparison of tissue kallikrein and kinin receptor expression in gastric ulcers and neoplasms. Int. Immunopharmacol. 2001, 1, 2063–2080. [Google Scholar] [CrossRef] [PubMed]
- Molina, L.; Matus, C.E.; Astroza, A.; Pavicic, F.; Tapia, E.; Toledo, C.; Perez, J.A.; Nualart, F.; Gonzalez, C.B.; Burgos, R.A.; et al. Stimulation of the bradykinin B1 receptor induces the proliferation of estrogen-sensitive breast cancer cells and activates the ERK1/2 signaling pathway. Breast Cancer Res. Treat. 2009, 118, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Barki-Harrington, L.; Daaka, Y. Bradykinin induced mitogenesis of androgen independent prostate cancer cells. J. Urol. 2001, 165, 2121–2125. [Google Scholar] [CrossRef] [PubMed]
- Chee, J.; Naran, A.; Misso, N.L.; Thompson, P.J.; Bhoola, K.D. Expression of tissue and plasma kallikreins and kinin B1 and B2 receptors in lung cancer. Biol. Chem. 2008, 389, 1225–1233. [Google Scholar] [CrossRef] [PubMed]
- Cote, J.; Bovenzi, V.; Savard, M.; Dubuc, C.; Fortier, A.; Neugebauer, W.; Tremblay, L.; Muller-Esterl, W.; Tsanaclis, A.M.; Lepage, M.; et al. Induction of selective blood-tumor barrier permeability and macromolecular transport by a biostable kinin B1 receptor agonist in a glioma rat model. PLoS ONE 2012, 7, e37485. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, M.N.; Pillat, M.M.; Motaln, H.; Ulrich, H.; Lah, T.T. Kinin-B1 receptor stimulation promotes invasion and is involved in cell-cell interaction of co-cultured glioblastoma and mesenchymal stem cells. Sci. Rep. 2018, 8, 1299. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Xue, Y.; Liu, Y.; Fu, W.; Jiang, N.; An, P.; Wang, P.; Yang, Z.; Wang, Y. Study of correlation between expression of bradykinin B2 receptor and pathological grade in human gliomas. Br. J. Neurosurg. 2005, 19, 322–326. [Google Scholar] [CrossRef]
- Huse, J.T.; Holland, E.C. Targeting brain cancer: Advances in the molecular pathology of malignant glioma and medulloblastoma. Nat. Rev. Cancer 2010, 10, 319–331. [Google Scholar] [CrossRef]
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef] [Green Version]
- Zinnhardt, B.; Pigeon, H.; Theze, B.; Viel, T.; Wachsmuth, L.; Fricke, I.B.; Schelhaas, S.; Honold, L.; Schwegmann, K.; Wagner, S.; et al. Combined pet imaging of the inflammatory tumor microenvironment identifies margins of unique radiotracer uptake. Cancer Res. 2017, 77, 1831–1841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Aaroe, A.; Liang, J.; Puduvalli, V.K. Tumor microenvironment in glioblastoma: Current and emerging concepts. Neuro-Oncol. Adv. 2023, 5, vdad009. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Simon, D.I. Integrin signals, transcription factors, and monocyte differentiation. Trends Cardiovasc. Med. 2006, 16, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [Green Version]
- Irvin, D.M.; McNeill, R.S.; Bash, R.E.; Miller, C.R. Intrinsic astrocyte heterogeneity influences tumor growth in glioma mouse models. Brain Pathol. 2017, 27, 36–50. [Google Scholar] [CrossRef]
- Mestas, J.; Ley, K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc. Med. 2008, 18, 228–232. [Google Scholar] [CrossRef] [Green Version]
- Imhof, B.A.; Aurrand-Lions, M. Adhesion mechanisms regulating the migration of monocytes. Nat. Rev. Immunol. 2004, 4, 432–444. [Google Scholar] [CrossRef]
- Huo, Y.; Ley, K. Adhesion molecules and atherogenesis. Acta Physiol. Scand. 2001, 173, 35–43. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Yang, W.; Aldape, K.; He, J.; Lu, Z. Epidermal growth factor (EGF)-enhanced vascular cell adhesion molecule-1 (vcam-1) expression promotes macrophage and glioblastoma cell interaction and tumor cell invasion. J. Biol. Chem. 2013, 288, 31488–31495. [Google Scholar] [CrossRef] [Green Version]
- Khan, B.V.; Parthasarathy, S.S.; Alexander, R.W.; Medford, R.M. Modified low density lipoprotein and its constituents augment cytokine-activated vascular cell adhesion molecule-1 gene expression in human vascular endothelial cells. J. Clin. Investig. 1995, 95, 1262–1270. [Google Scholar] [CrossRef] [Green Version]
- Kong, D.-H.; Kim, Y.K.; Kim, M.R.; Jang, J.H.; Lee, S. Emerging roles of vascular cell adhesion molecule-1 (VCAM-1) in immunological disorders and cancer. Int. J. Mol. Sci. 2018, 19, 1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, S.-W.; Liu, Y.-S.; Lu, D.-Y.; Tsai, C.-F. Melatonin modulates the microenvironment of glioblastoma multiforme by targeting sirtuin 1. Nutrients 2019, 11, 1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Movahedi, K.; Laoui, D.; Gysemans, C.; Baeten, M.; Stange, G.; Van den Bossche, J.; Mack, M.; Pipeleers, D.; In’t Veld, P.; De Baetselier, P.; et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010, 70, 5728–5739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.H.; Huang, B.R.; Lai, S.W.; Lin, C.; Lin, H.Y.; Yang, L.Y.; Lu, D.Y. Sirt1 activation by minocycline on regulation of microglial polarization homeostasis. Aging 2020, 12, 17990–18007. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.R.; Chen, T.S.; Bau, D.T.; Chuang, I.C.; Tsai, C.F.; Chang, P.C.; Lu, D.Y. EGFR is a pivotal regulator of thrombin-mediated inflammation in primary human nucleus pulposus culture. Sci. Rep. 2017, 7, 8578. [Google Scholar] [CrossRef] [Green Version]
- Son, H.; Moon, A. Epithelial-mesenchymal transition and cell invasion. Toxicol. Res. 2010, 26, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Turaga, S.M.; Lathia, J.D. Adhering towards tumorigenicity: Altered adhesion mechanisms in glioblastoma cancer stem cells. CNS Oncol. 2016, 5, 251–259. [Google Scholar] [CrossRef]
- Shen, C.-K.; Huang, B.-R.; Yeh, W.-L.; Chen, C.-W.; Liu, Y.-S.; Lai, S.-W.; Tseng, W.-P.; Lu, D.-Y.; Tsai, C.-F. Regulatory effects of IL-1β in the interaction of GBM and tumor-associated monocyte through VCAM-1 and ICAM-1. Eur. J. Pharmacol. 2021, 905, 174216. [Google Scholar] [CrossRef]
- Lee, H.W.; Choi, H.J.; Ha, S.J.; Lee, K.T.; Kwon, Y.G. Recruitment of monocytes/macrophages in different tumor microenvironments. Biochim. Biophys. Acta 2013, 1835, 170–179. [Google Scholar] [CrossRef]
- Hourani, T.; Holden, J.A.; Li, W.; Lenzo, J.C.; Hadjigol, S.; O’Brien-Simpson, N.M. Tumor associated macrophages: Origin, recruitment, phenotypic diversity, and targeting. Front. Oncol. 2021, 11, 788365. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, M.; Caffo, M.; Minutoli, L.; Marini, H.; Abbritti, R.V.; Squadrito, F.; Trichilo, V.; Valenti, A.; Barresi, V.; Altavilla, D.; et al. Ros and brain gliomas: An overview of potential and innovative therapeutic strategies. Int. J. Mol. Sci. 2016, 17, 984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, E.C.F.; Brown, M.P.; Gargett, T.; Ebert, L.M. The role of cytokines and chemokines in shaping the immune microenvironment of glioblastoma: Implications for immunotherapy. Cells 2021, 10, 607. [Google Scholar] [CrossRef]
- de Groot, J.; Penas-Prado, M.; Alfaro-Munoz, K.; Hunter, K.; Pei, B.L.; O’Brien, B.; Weathers, S.P.; Loghin, M.; Kamiya Matsouka, C.; Yung, W.K.A.; et al. Window-of-opportunity clinical trial of pembrolizumab in patients with recurrent glioblastoma reveals predominance of immune-suppressive macrophages. Neuro Oncol. 2020, 22, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Storz, P. Reactive oxygen species in tumor progression. Front. Biosci. 2005, 10, 1881–1896. [Google Scholar] [CrossRef] [Green Version]
- Vurusaner, B.; Poli, G.; Basaga, H. Tumor suppressor genes and ROS: Complex networks of interactions. Free Radic. Biol. Med. 2012, 52, 7–18. [Google Scholar] [CrossRef]
- Festa, M.; Capasso, A.; D’Acunto, C.W.; Masullo, M.; Rossi, A.G.; Pizza, C.; Piacente, S. Xanthohumol induces apoptosis in human malignant glioblastoma cells by increasing reactive oxygen species and activating MAPK pathways. J. Nat. Prod. 2011, 74, 2505–2513. [Google Scholar] [CrossRef]
- Chen, S.; Ma, J.; Yang, L.; Teng, M.; Lai, Z.-Q.; Chen, X.; He, J. Anti-glioblastoma activity of kaempferol via programmed cell death induction: Involvement of autophagy and pyroptosis. Front. Bioeng. Biotechnol. 2020, 8, 614419. [Google Scholar] [CrossRef]
- Eller-Borges, R.; Rodrigues, E.G.; Teodoro, A.C.S.; Moraes, M.S.; Arruda, D.C.; Paschoalin, T.; Curcio, M.F.; da Costa, P.E.; Do Nascimento, I.R.; Calixto, L.A.; et al. Bradykinin promotes murine melanoma cell migration and invasion through endogenous production of superoxide and nitric oxide. Nitric Oxide 2023, 132, 15–26. [Google Scholar] [CrossRef]
- Luu Hoang, K.N.; Anstee, J.E.; Arnold, J.N. The diverse roles of heme oxygenase-1 in tumor progression. Front. Immunol. 2021, 12, 658315. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Jozkowicz, A.; Dulak, J. HO-1/CO system in tumor growth, angiogenesis and metabolism—Targeting Ho-1 as an anti-tumor therapy. Vasc. Pharmacol. 2015, 74, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Deng, R.; Wang, S.M.; Yin, T.; Ye, T.H.; Shen, G.B.; Li, L.; Zhao, J.Y.; Sang, Y.X.; Duan, X.G.; Wei, Y.Q. Inhibition of tumor growth and alteration of associated macrophage cell type by an HO-1 inhibitor in breast carcinoma-bearing mice. Oncol. Res. 2013, 20, 473–482. [Google Scholar] [CrossRef]
- Gozzelino, R.; Jeney, V.; Soares, M.P. Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 323–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castruccio Castracani, C.; Longhitano, L.; Distefano, A.; Di Rosa, M.; Pittala, V.; Lupo, G.; Caruso, M.; Corona, D.; Tibullo, D.; Li Volti, G. Heme oxygenase-1 and carbon monoxide regulate growth and progression in glioblastoma cells. Mol. Neurobiol. 2020, 57, 2436–2446. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.Y.; Yeh, W.L.; Huang, S.M.; Tang, C.H.; Lin, H.Y.; Chou, S.J. Osteopontin increases heme oxygenase-1 expression and subsequently induces cell migration and invasion in glioma cells. Neuro Oncol. 2012, 14, 1367–1378. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.-M.; Hsieh, H.-L.; Lin, C.-C.; Shih, R.-H.; Chi, P.-L.; Cheng, S.-E.; Hsiao, L.-D. Multiple factors from bradykinin-challenged astrocytes contribute to the neuronal apoptosis: Involvement of astroglial ROS, MMP-9, and HO-1/CO system. Mol. Neurobiol. 2013, 47, 1020–1033. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Dong, P.; Xiong, Y.; Yue, J.; Hanley, S.J.B.; Watari, H. Tumor-intrinsic PD-L1 signaling in cancer initiation, development and treatment: Beyond immune evasion. Front. Oncol. 2018, 8, 386. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Li, S.; Xue, J.; Qi, M.; Liu, X.; Huang, Y.; Hu, J.; Dong, H.; Ling, K. PD-L1 tumor-intrinsic signaling and its therapeutic implication in triple-negative breast cancer. JCI Insight 2021, 6, e131458. [Google Scholar] [CrossRef]
- Bloch, O.; Crane, C.A.; Kaur, R.; Safaee, M.; Rutkowski, M.J.; Parsa, A.T. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin. Cancer Res. 2013, 19, 3165–3175. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Carlsson, R.; Ambjorn, M.; Hasan, M.; Badn, W.; Darabi, A.; Siesjo, P.; Issazadeh-Navikas, S. PD-L1 expression by neurons nearby tumors indicates better prognosis in glioblastoma patients. J. Neurosci. 2013, 33, 14231–14245. [Google Scholar] [CrossRef]
- Wang, S.; Yao, F.; Lu, X.; Li, Q.; Su, Z.; Lee, J.H.; Wang, C.; Du, L. Temozolomide promotes immune escape of GBM cells via upregulating PD-L1. Am. J. Cancer Res. 2019, 9, 1161–1171. [Google Scholar] [PubMed]
- Xue, S.; Song, G.; Yu, J. The prognostic significance of Pd-L1 expression in patients with glioma: A meta-analysis. Sci. Rep. 2017, 7, 4231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, S.; Hu, M.; Li, P.; Ma, J.; Xie, L.; Teng, F.; Zhu, Y.; Fan, B.; Mu, D.; Yu, J. Relationship between expression of PD-L1 and tumor angiogenesis, proliferation, and invasion in glioma. Oncotarget 2017, 8, 49702–49712. [Google Scholar] [CrossRef] [Green Version]
- Berghoff, A.S.; Kiesel, B.; Widhalm, G.; Rajky, O.; Ricken, G.; Wohrer, A.; Dieckmann, K.; Filipits, M.; Brandstetter, A.; Weller, M.; et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro Oncol. 2015, 17, 1064–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nduom, E.K.; Wei, J.; Yaghi, N.K.; Huang, N.; Kong, L.Y.; Gabrusiewicz, K.; Ling, X.; Zhou, S.; Ivan, C.; Chen, J.Q.; et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro Oncol. 2016, 18, 195–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Zhang, C.; Liu, X.; Wang, Z.; Sun, L.; Li, G.; Liang, J.; Hu, H.; Liu, Y.; Zhang, W.; et al. Molecular and clinical characterization of PD-L1 expression at transcriptional level via 976 samples of brain glioma. Oncoimmunology 2016, 5, e1196310. [Google Scholar] [CrossRef] [Green Version]
- Zhai, L.; Ladomersky, E.; Dostal, C.R.; Lauing, K.L.; Swoap, K.; Billingham, L.K.; Gritsina, G.; Wu, M.; McCusker, R.H.; Binder, D.C.; et al. Non-tumor cell IDO1 predominantly contributes to enzyme activity and response to CTLA-4/PD-L1 inhibition in mouse glioblastoma. Brain Behav. Immun. 2017, 62, 24–29. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.S.; Huang, B.R.; Lin, C.J.; Shen, C.K.; Lai, S.W.; Chen, C.W.; Lin, H.J.; Lin, C.H.; Hsieh, Y.C.; Lu, D.Y. Paliperidone inhibits glioblastoma growth in mouse brain tumor model and reduces PD-L1 expression. Cancers 2021, 13, 4357. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Zhang, H.; Chen, B.; Liu, X.; Zhang, S.; Zong, Z.; Gao, M. Pd-l1-mediated immunosuppression in glioblastoma is associated with the infiltration and M2-polarization of tumor-associated macrophages. Front. Immunol. 2020, 11, 588552. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Aldo, P.; You, Y.; Ding, J.; Kaislasuo, J.; Petersen, J.F.; Lokkegaard, E.; Peng, G.; Paidas, M.J.; Simpson, S.; et al. Trophoblast-secreted soluble-PD-L1 modulates macrophage polarization and function. J. Leukoc. Biol. 2020, 108, 983–998. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Mittman, S.; Rodriguez, R.; Moskalenko, M.; Pacheco-Sanchez, P.; Yang, Y.; Nickles, D.; Cubas, R. Anti-PD-L1 treatment results in functional remodeling of the macrophage compartment. Cancer Res. 2019, 79, 1493–1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueroa, C.D.; Matus, C.E.; Pavicic, F.; Sarmiento, J.; Hidalgo, M.A.; Burgos, R.A.; Gonzalez, C.B.; Bhoola, K.D.; Ehrenfeld, P. Kinin B1 receptor regulates interactions between neutrophils and endothelial cells by modulating the levels of Mac-1, LFA-1 and intercellular adhesion molecule-1. Innate Immun. 2015, 21, 289–304. [Google Scholar] [CrossRef] [PubMed]
- Maria, A.G.; Dillemburg-Pilla, P.; Durand, M.T.; Floriano, E.M.; Manfiolli, A.O.; Ramos, S.G.; Pesquero, J.B.; Nahmias, C.; Costa-Neto, C.M. Activation of the kinin B1 receptor by its agonist reduces melanoma metastasis by playing a dual effect on tumor cells and host immune response. Front. Pharmacol. 2019, 10, 1106. [Google Scholar] [CrossRef]
- Liu, Y.S.; Hsu, J.W.; Lin, H.Y.; Lai, S.W.; Huang, B.R.; Tsai, C.F.; Lu, D.Y. Bradykinin b1 receptor contributes to interleukin-8 production and glioblastoma migration through interaction of STAT3 and SP-1. Neuropharmacology 2019, 144, 143–154. [Google Scholar] [CrossRef]
- Lu, D.Y.; Leung, Y.M.; Huang, S.M.; Wong, K.L. Bradykinin-induced cell migration and COX-2 production mediated by the bradykinin B1 receptor in glioma cells. J. Cell Biochem. 2010, 110, 141–150. [Google Scholar] [CrossRef]
- Ohtani, Y.; Minami, M.; Kawaguchi, N.; Nishiyori, A.; Yamamoto, J.; Takami, S.; Satoh, M. Expression of stromal cell-derived factor-1 and CXCR4 chemokine receptor mRNAs in cultured rat glial and neuronal cells. Neurosci. Lett. 1998, 249, 163–166. [Google Scholar] [CrossRef] [PubMed]
- Chow, M.T.; Luster, A.D. Chemokines in cancer. Cancer Immunol. Res. 2014, 2, 1125–1131. [Google Scholar] [CrossRef] [Green Version]
- Seo, W.; Shimizu, K.; Kojo, S.; Okeke, A.; Kohwi-Shigematsu, T.; Fujii, S.I.; Taniuchi, I. Runx-mediated regulation of CCL5 via antagonizing two enhancers influences immune cell function and anti-tumor immunity. Nat. Commun. 2020, 11, 1562. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.Y.; Fong, Y.C.; Lee, C.Y.; Chen, M.Y.; Tsai, H.C.; Hsu, H.C.; Tang, C.H. CCL5 increases lung cancer migration via PI3K, Akt and NF-κB pathways. Biochem. Pharmacol. 2009, 77, 794–803. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.Y.; Lin, Y.C.; Mahalingam, J.; Huang, C.T.; Chen, T.W.; Kang, C.W.; Peng, H.M.; Chu, Y.Y.; Chiang, J.M.; Dutta, A.; et al. Tumor-derived chemokine CCL5 enhances TGF-β-mediated killing of CD8+ T cells in colon cancer by T-regulatory cells. Cancer Res. 2012, 72, 1092–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moogooei, M.; Shamaei, M.; Khorramdelazad, H.; Fattahpour, S.; Seyedmehdi, S.M.; Moogooei, M.; Hassanshahi, G.; Kalantari Khandani, B. The intricate expression of cc chemokines in glial tumors: Evidence for involvement of CCL2 and CCL5 but not CCL11. Acta Med. Iran. 2015, 53, 770–777. [Google Scholar]
- Li, Y.; Han, S.; Wu, B.; Zhong, C.; Shi, Y.; Lv, C.; Fu, L.; Zhang, Y.; Lang, Q.; Liang, Z.; et al. CXCL11 correlates with immune infiltration and impacts patient immunotherapy efficacy: A pan-cancer analysis. Front. Immunol. 2022, 13, 951247. [Google Scholar] [CrossRef]
- Ge, W.L.; Chen, Q.; Meng, L.D.; Huang, X.M.; Shi, G.D.; Zong, Q.Q.; Shen, P.; Lu, Y.C.; Zhang, Y.H.; Miao, Y.; et al. The YY1/miR-548t-5p/CXCL11 signaling axis regulates cell proliferation and metastasis in human pancreatic cancer. Cell Death Dis. 2020, 11, 294. [Google Scholar] [CrossRef]
- Sun, X.; Mao, Y.; Wang, J.; Zu, L.; Hao, M.; Cheng, G.; Qu, Q.; Cui, D.; Keller, E.T.; Chen, X.; et al. IL-6 secreted by cancer-associated fibroblasts induces tamoxifen resistance in luminal breast cancer. Oncogene 2014, 33, 4450. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Ye, Y.; Zhang, X.; Song, J. Bradykinin stimulates IL-6 production and cell invasion in colorectal cancer cells. Oncol. Rep. 2014, 32, 1709–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobrzycka, B.; Mackowiak-Matejczyk, B.; Terlikowska, K.M.; Kulesza-Bronczyk, B.; Kinalski, M.; Terlikowski, S.J. Serum levels of IL-6, IL-8 and CRP as prognostic factors in epithelial ovarian cancer. Eur. Cytokine Netw. 2013, 24, 106–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFarland, B.C.; Hong, S.W.; Rajbhandari, R.; Twitty, G.B., Jr.; Gray, G.K.; Yu, H.; Benveniste, E.N.; Nozell, S.E. NF-κB-induced IL-6 ensures STAT3 activation and tumor aggressiveness in glioblastoma. PLoS ONE 2013, 8, e78728. [Google Scholar] [CrossRef]
- Kesanakurti, D.; Chetty, C.; Dinh, D.H.; Gujrati, M.; Rao, J.S. Role of mmp-2 in the regulation of IL-6/STAT3 survival signaling via interaction with α5β1 integrin in glioma. Oncogene 2013, 32, 327–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domanska, U.M.; Kruizinga, R.C.; den Dunnen, W.F.; Timmer-Bosscha, H.; de Vries, E.G.; Walenkamp, A.M. The chemokine network, a newly discovered target in high grade gliomas. Crit. Rev. Oncol. Hematol. 2011, 79, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.; Gonzalez, J.; Decramer, S.; Bandin, F.; Neau, E.; Salant, D.J.; Heeringa, P.; Pesquero, J.B.; Schanstra, J.P.; Bascands, J.L. Blockade of the kinin B1 receptor ameloriates glomerulonephritis. J. Am. Soc. Nephrol. 2010, 21, 1157–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Zhong, M.; Wang, C.; Xu, Y.; Gao, W.Q.; Zhang, Y. Ccl5-deficiency enhances intratumoral infiltration of CD8+ T cells in colorectal cancer. Cell Death Dis. 2018, 9, 766. [Google Scholar] [CrossRef] [Green Version]
- Viola, A.; Sarukhan, A.; Bronte, V.; Molon, B. The pros and cons of chemokines in tumor immunology. Trends Immunol. 2012, 33, 496–504. [Google Scholar] [CrossRef]
- Biswas, S.K.; Allavena, P.; Mantovani, A. Tumor-associated macrophages: Functional diversity, clinical significance, and open questions. Semin. Immunopathol. 2013, 35, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Buranych, A.; Sarkar, D.; Fisher, P.B.; Wang, X.Y. The role of tumor-associated macrophages in tumor vascularization. Vasc. Cell 2013, 5, 20. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Ji, H.; Niu, X.; Yin, L.; Wang, Y.; Gu, Y.; Wang, J.; Zhou, X.; Zhang, H.; Zhang, Q. Tumor-associated macrophages secrete CC-chemokine ligand 2 and induce tamoxifen resistance by activating PI3K/Akt/mTOR in breast cancer. Cancer Sci. 2020, 111, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Hussain, S.F.; Yang, D.; Suki, D.; Aldape, K.; Grimm, E.; Heimberger, A.B. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol. 2006, 8, 261–279. [Google Scholar] [CrossRef] [Green Version]
- Boutilier, A.J.; Elsawa, S.F. Macrophage polarization states in the tumor microenvironment. Int. J. Mol. Sci. 2021, 22, 6995. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, C.-K.; Huang, B.-R.; Charoensaensuk, V.; Yang, L.-Y.; Tsai, C.-F.; Liu, Y.-S.; Lu, D.-Y.; Yeh, W.-L.; Lin, C. Bradykinin B1 Receptor Affects Tumor-Associated Macrophage Activity and Glioblastoma Progression. Antioxidants 2023, 12, 1533. https://doi.org/10.3390/antiox12081533
Shen C-K, Huang B-R, Charoensaensuk V, Yang L-Y, Tsai C-F, Liu Y-S, Lu D-Y, Yeh W-L, Lin C. Bradykinin B1 Receptor Affects Tumor-Associated Macrophage Activity and Glioblastoma Progression. Antioxidants. 2023; 12(8):1533. https://doi.org/10.3390/antiox12081533
Chicago/Turabian StyleShen, Ching-Kai, Bor-Ren Huang, Vichuda Charoensaensuk, Liang-Yo Yang, Cheng-Fang Tsai, Yu-Shu Liu, Dah-Yuu Lu, Wei-Lan Yeh, and Chingju Lin. 2023. "Bradykinin B1 Receptor Affects Tumor-Associated Macrophage Activity and Glioblastoma Progression" Antioxidants 12, no. 8: 1533. https://doi.org/10.3390/antiox12081533
APA StyleShen, C.-K., Huang, B.-R., Charoensaensuk, V., Yang, L.-Y., Tsai, C.-F., Liu, Y.-S., Lu, D.-Y., Yeh, W.-L., & Lin, C. (2023). Bradykinin B1 Receptor Affects Tumor-Associated Macrophage Activity and Glioblastoma Progression. Antioxidants, 12(8), 1533. https://doi.org/10.3390/antiox12081533