
Role of Hydrogen Sulfide in Inflammatory Bowel Disease

 , , and
, , and
Abstract
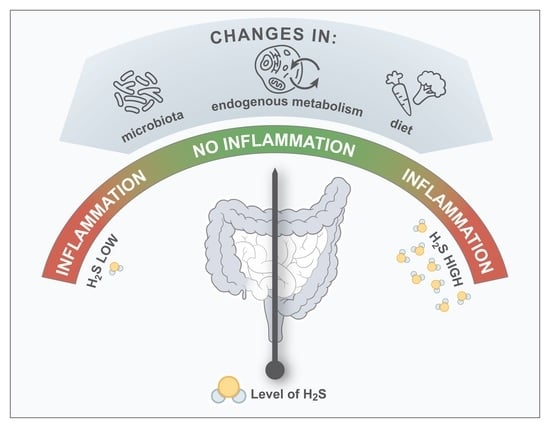
:
1. Introduction
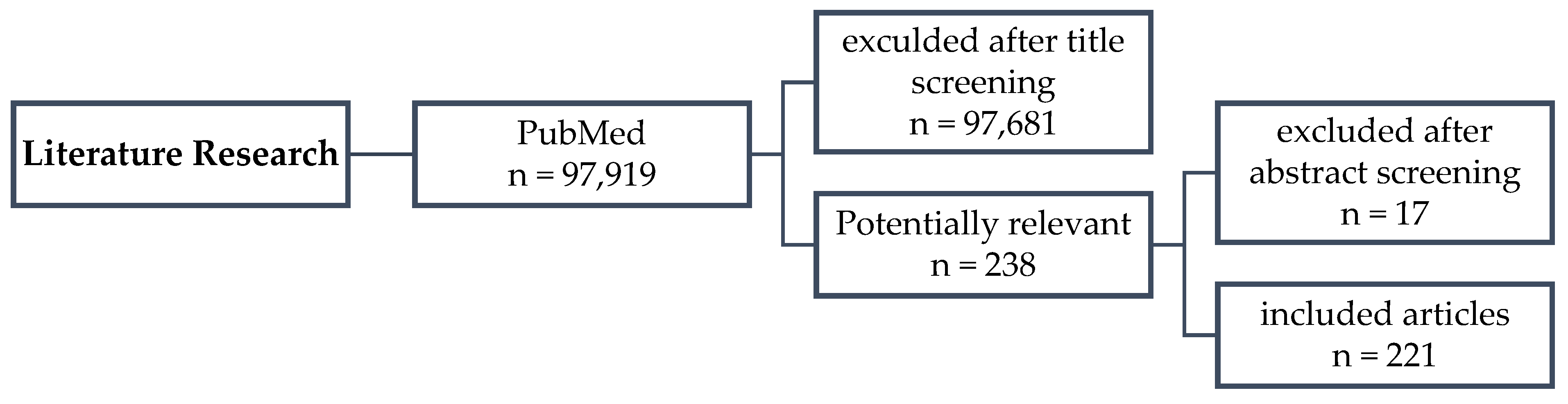
2. Materials and Methods
2.1. Data Source/Search Strategy
2.2. Study Selection
3. The Connection between H2S and the Pathogenesis of IBD
3.1. Microbial H2S
3.2. Non-Microbial and Endogenous H2S
3.3. Dietary H2S
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Evans, C.L. The toxicity of hydrogen sulphide and other sulphides. Q. J. Exp. Physiol. Cogn. Med. Sci. 1967, 52, 231–248. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Kimura, H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 1996, 16, 1066–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R. The gasotransmitter role of hydrogen sulfide. Antioxid. Redox Signal 2003, 5, 493–501. [Google Scholar] [CrossRef]
- Yang, G.; Wu, L.; Jiang, B.; Yang, W.; Qi, J.; Cao, K.; Meng, Q.; Mustafa, A.K.; Mu, W.; Zhang, S.; et al. H2S as a physiologic vasorelaxant: Hypertension in mice with deletion of cystathionine gamma-lyase. Science 2008, 322, 587–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, B.D.; Sbodio, J.I.; Xu, R.; Vandiver, M.S.; Cha, J.Y.; Snowman, A.M.; Snyder, S.H. Cystathionine γ-lyase deficiency mediates neurodegeneration in Huntington’s disease. Nature 2014, 509, 96–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hine, C.; Mitchell, J.R. Calorie restriction and methionine restriction in control of endogenous hydrogen sulfide production by the transsulfuration pathway. Exp. Gerontol. 2015, 68, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Wang, R. Two’s company, three’s a crowd: Can H2S be the third endogenous gaseous transmitter? FASEB J. 2002, 16, 1792–1798. [Google Scholar] [CrossRef] [Green Version]
- Wang, R. Gasotransmitters: Growing pains and joys. Trends Biochem. Sci. 2014, 39, 227–232. [Google Scholar] [CrossRef]
- Blachier, F.; Beaumont, M.; Kim, E. Cysteine-derived hydrogen sulfide and gut health: A matter of endogenous or bacterial origin. Curr. Opin. Clin. Nutr. Metab. Care 2019, 22, 68–75. [Google Scholar] [CrossRef]
- Kabil, O.; Banerjee, R. Redox biochemistry of hydrogen sulfide. J. Biol. Chem. 2010, 285, 21903–21907. [Google Scholar] [CrossRef] [Green Version]
- Kimura, H. Signaling molecules: Hydrogen sulfide and polysulfide. Antioxid. Redox Signal 2015, 22, 362–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiku, T.; Padovani, D.; Zhu, W.; Singh, S.; Vitvitsky, V.; Banerjee, R. H2S biogenesis by human cystathionine gamma-lyase leads to the novel sulfur metabolites lanthionine and homolanthionine and is responsive to the grade of hyperhomocysteinemia. J. Biol. Chem. 2009, 284, 11601–11612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Padovani, D.; Leslie, R.A.; Chiku, T.; Banerjee, R. Relative contributions of cystathionine beta-synthase and gamma-cystathionase to H2S biogenesis via alternative trans-sulfuration reactions. J. Biol. Chem. 2009, 284, 22457–22466. [Google Scholar] [CrossRef] [Green Version]
- Wallace, J.L.; Vong, L.; McKnight, W.; Dicay, M.; Martin, G.R. Endogenous and exogenous hydrogen sulfide promotes resolution of colitis in rats. Gastroenterology 2009, 137, 569–578, 578.e1. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.F.; Yu, T.C.; Hong, J.; Fang, J.Y. Emerging Roles of Hydrogen Sulfide in Inflammatory and Neoplastic Colonic Diseases. Front. Physiol. 2016, 7, 156. [Google Scholar] [CrossRef] [Green Version]
- De Cicco, P.; Sanders, T.; Cirino, G.; Maloy, K.J.; Ianaro, A. Hydrogen Sulfide Reduces Myeloid-Derived Suppressor Cell-Mediated Inflammatory Response in a Model of Helicobacter hepaticus-Induced Colitis. Front. Immunol. 2018, 9, 499. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, E.; Matsunami, M.; Kimura, T.; Yonezawa, D.; Ishiki, T.; Sekiguchi, F.; Nishikawa, H.; Maeda, Y.; Ishikura, H.; Kawabata, A. Rhodanese, but not cystathionine-gamma-lyase, is associated with dextran sulfate sodium-evoked colitis in mice: A sign of impaired colonic sulfide detoxification? Toxicology 2009, 264, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Linden, D.R.; Sha, L.; Mazzone, A.; Stoltz, G.J.; Bernard, C.E.; Furne, J.K.; Levitt, M.D.; Farrugia, G.; Szurszewski, J.H. Production of the gaseous signal molecule hydrogen sulfide in mouse tissues. J. Neurochem. 2008, 106, 1577–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flannigan, K.L.; Ferraz, J.G.; Wang, R.; Wallace, J.L. Enhanced synthesis and diminished degradation of hydrogen sulfide in experimental colitis: A site-specific, pro-resolution mechanism. PLoS ONE 2013, 8, e71962. [Google Scholar] [CrossRef] [Green Version]
- Blachier, F.; Davila, A.M.; Mimoun, S.; Benetti, P.H.; Atanasiu, C.; Andriamihaja, M.; Benamouzig, R.; Bouillaud, F.; Tomé, D. Luminal sulfide and large intestine mucosa: Friend or foe? Amino Acids 2010, 39, 335–347. [Google Scholar] [CrossRef]
- Barton, L.L.; Ritz, N.L.; Fauque, G.D.; Lin, H.C. Sulfur Cycling and the Intestinal Microbiome. Dig. Dis. Sci. 2017, 62, 2241–2257. [Google Scholar] [CrossRef]
- Hale, V.L.; Jeraldo, P.; Mundy, M.; Yao, J.; Keeney, G.; Scott, N.; Cheek, E.H.; Davidson, J.; Greene, M.; Martinez, C.; et al. Synthesis of multi-omic data and community metabolic models reveals insights into the role of hydrogen sulfide in colon cancer. Methods 2018, 149, 59–68. [Google Scholar] [CrossRef]
- Tomasova, L.; Konopelski, P.; Ufnal, M. Gut Bacteria and Hydrogen Sulfide: The New Old Players in Circulatory System Homeostasis. Molecules 2016, 21, 1558. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Stams, A.J.M.; de Vos, W.M.; Sánchez-Andrea, I. Enrichment of sulfidogenic bacteria from the human intestinal tract. FEMS Microbiol. Lett. 2017, 364, fnx028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X.; Ding, L.; Xie, Z.Z.; Yang, Y.; Whiteman, M.; Moore, P.K.; Bian, J.S. A Review of Hydrogen Sulfide Synthesis, Metabolism, and Measurement: Is Modulation of Hydrogen Sulfide a Novel Therapeutic for Cancer? Antioxid. Redox Signal 2019, 31, 1–38. [Google Scholar] [CrossRef] [Green Version]
- Augustyn, K.D.; Jackson, M.R.; Jorns, M.S. Use of Tissue Metabolite Analysis and Enzyme Kinetics to Discriminate between Alternate Pathways for Hydrogen Sulfide Metabolism. Biochemistry 2017, 56, 986–996. [Google Scholar] [CrossRef]
- Libiad, M.; Sriraman, A.; Banerjee, R. Polymorphic Variants of Human Rhodanese Exhibit Differences in Thermal Stability and Sulfur Transfer Kinetics. J. Biol. Chem. 2015, 290, 23579–23588. [Google Scholar] [CrossRef] [Green Version]
- Jackson, M.R.; Melideo, S.L.; Jorns, M.S. Human sulfide: Quinone oxidoreductase catalyzes the first step in hydrogen sulfide metabolism and produces a sulfane sulfur metabolite. Biochemistry 2012, 51, 6804–6815. [Google Scholar] [CrossRef]
- Jackson, M.R.; Melideo, S.L.; Jorns, M.S. Role of human sulfide: Quinone oxidoreductase in H2S metabolism. Methods Enzymol. 2015, 554, 255–270. [Google Scholar] [CrossRef]
- Kabil, O.; Motl, N.; Strack, M.; Seravalli, J.; Metzler-Nolte, N.; Banerjee, R. Mechanism-based inhibition of human persulfide dioxygenase by γ-glutamyl-homocysteinyl-glycine. J. Biol. Chem. 2018, 293, 12429–12439. [Google Scholar] [CrossRef] [Green Version]
- Kabil, O.; Banerjee, R. Characterization of patient mutations in human persulfide dioxygenase (ETHE1) involved in H2S catabolism. J. Biol. Chem. 2012, 287, 44561–44567. [Google Scholar] [CrossRef] [Green Version]
- Pettinati, I.; Brem, J.; McDonough, M.A.; Schofield, C.J. Crystal structure of human persulfide dioxygenase: Structural basis of ethylmalonic encephalopathy. Hum. Mol. Genet. 2015, 24, 2458–2469. [Google Scholar] [CrossRef] [Green Version]
- Zhen, Y.; Pan, W.; Hu, F.; Wu, H.; Feng, J.; Zhang, Y.; Chen, J. Exogenous hydrogen sulfide exerts proliferation/anti-apoptosis/angiogenesis/migration effects via amplifying the activation of NF-κB pathway in PLC/PRF/5 hepatoma cells. Int. J. Oncol. 2015, 46, 2194–2204. [Google Scholar] [CrossRef] [Green Version]
- Zhen, Y.; Zhang, W.; Liu, C.; He, J.; Lu, Y.; Guo, R.; Feng, J.; Zhang, Y.; Chen, J. Exogenous hydrogen sulfide promotes C6 glioma cell growth through activation of the p38 MAPK/ERK1/2-COX-2 pathways. Oncol. Rep. 2015, 34, 2413–2422. [Google Scholar] [CrossRef] [Green Version]
- Lei, Y.; Zhen, Y.; Zhang, W.; Sun, X.; Lin, X.; Feng, J.; Luo, H.; Chen, Z.; Su, C.; Zeng, B.; et al. Exogenous hydrogen sulfide exerts proliferation, anti-apoptosis, angiopoiesis and migration effects via activating HSP90 pathway in EC109 cells. Oncol. Rep. 2016, 35, 3714–3720. [Google Scholar] [CrossRef] [Green Version]
- Yang, N.; Liu, Y.; Li, T.; Tuo, Q. Role of Hydrogen Sulfide in Chronic Diseases. DNA Cell Biol. 2020, 39, 187–196. [Google Scholar] [CrossRef]
- Blachier, F.; Andriamihaja, M.; Larraufie, P.; Ahn, E.; Lan, A.; Kim, E. Production of hydrogen sulfide by the intestinal microbiota and epithelial cells and consequences for the colonic and rectal mucosa. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 320, G125–G135. [Google Scholar] [CrossRef]
- Medani, M.; Collins, D.; Docherty, N.G.; Baird, A.W.; O’Connell, P.R.; Winter, D.C. Emerging role of hydrogen sulfide in colonic physiology and pathophysiology. Inflamm. Bowel Dis. 2011, 17, 1620–1625. [Google Scholar] [CrossRef]
- Mottawea, W.; Chiang, C.K.; Mühlbauer, M.; Starr, A.E.; Butcher, J.; Abujamel, T.; Deeke, S.A.; Brandel, A.; Zhou, H.; Shokralla, S.; et al. Altered intestinal microbiota-host mitochondria crosstalk in new onset Crohn’s disease. Nat. Commun. 2016, 7, 13419. [Google Scholar] [CrossRef] [Green Version]
- Motta, J.P.; Flannigan, K.L.; Agbor, T.A.; Beatty, J.K.; Blackler, R.W.; Workentine, M.L.; Da Silva, G.J.; Wang, R.; Buret, A.G.; Wallace, J.L. Hydrogen sulfide protects from colitis and restores intestinal microbiota biofilm and mucus production. Inflamm. Bowel Dis. 2015, 21, 1006–1017. [Google Scholar] [CrossRef]
- Podolsky, D.K. Inflammatory bowel disease. N. Engl. J. Med. 2002, 347, 417–429. [Google Scholar] [CrossRef]
- Geboes, K.; Van Eyken, P. Inflammatory bowel disease unclassified and indeterminate colitis: The role of the pathologist. J. Clin. Pathol. 2009, 62, 201–205. [Google Scholar] [CrossRef]
- Tremaine, W.J. Diagnosis and treatment of indeterminate colitis. Gastroenterol. Hepatol. 2011, 7, 826–828. [Google Scholar]
- Baumgart, D.C.; Sandborn, W.J. Crohn’s disease. Lancet 2012, 380, 1590–1605. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.C.; Stappenbeck, T.S. Genetics and Pathogenesis of Inflammatory Bowel Disease. Annu. Rev. Pathol. 2016, 11, 127–148. [Google Scholar] [CrossRef] [Green Version]
- Torres, J.; Mehandru, S.; Colombel, J.F.; Peyrin-Biroulet, L. Crohn’s disease. Lancet 2017, 389, 1741–1755. [Google Scholar] [CrossRef]
- Ordás, I.; Eckmann, L.; Talamini, M.; Baumgart, D.C.; Sandborn, W.J. Ulcerative colitis. Lancet 2012, 380, 1606–1619. [Google Scholar] [CrossRef] [Green Version]
- Sairenji, T.; Collins, K.L.; Evans, D.V. An Update on Inflammatory Bowel Disease. Prim. Care 2017, 44, 673–692. [Google Scholar] [CrossRef]
- Molodecky, N.A.; Soon, I.S.; Rabi, D.M.; Ghali, W.A.; Ferris, M.; Chernoff, G.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Barkema, H.W.; et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012, 142, 46–54.e42; quiz e30. [Google Scholar] [CrossRef] [Green Version]
- Chaparro, M.; Garre, A.; Núñez Ortiz, A.; Diz-Lois Palomares, M.T.; Rodríguez, C.; Riestra, S.; Vela, M.; Benítez, J.M.; Fernández Salgado, E.; Sánchez Rodríguez, E.; et al. Incidence, Clinical Characteristics and Management of Inflammatory Bowel Disease in Spain: Large-Scale Epidemiological Study. J. Clin. Med. 2021, 10, 2885. [Google Scholar] [CrossRef]
- Sauer, C.G.; Kugathasan, S. Pediatric inflammatory bowel disease: Highlighting pediatric differences in IBD. Med. Clin. N. Am. 2010, 94, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Van Limbergen, J.; Russell, R.K.; Drummond, H.E.; Aldhous, M.C.; Round, N.K.; Nimmo, E.R.; Smith, L.; Gillett, P.M.; McGrogan, P.; Weaver, L.T.; et al. Definition of phenotypic characteristics of childhood-onset inflammatory bowel disease. Gastroenterology 2008, 135, 1114–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benchimol, E.I.; Fortinsky, K.J.; Gozdyra, P.; Van den Heuvel, M.; Van Limbergen, J.; Griffiths, A.M. Epidemiology of pediatric inflammatory bowel disease: A systematic review of international trends. Inflamm. Bowel Dis. 2011, 17, 423–439. [Google Scholar] [CrossRef]
- Sýkora, J.; Pomahačová, R.; Kreslová, M.; Cvalínová, D.; Štych, P.; Schwarz, J. Current global trends in the incidence of pediatric-onset inflammatory bowel disease. World J. Gastroenterol. 2018, 24, 2741–2763. [Google Scholar] [CrossRef]
- Ghione, S.; Sarter, H.; Fumery, M.; Armengol-Debeir, L.; Savoye, G.; Ley, D.; Spyckerelle, C.; Pariente, B.; Peyrin-Biroulet, L.; Turck, D.; et al. Dramatic Increase in Incidence of Ulcerative Colitis and Crohn’s Disease (1988–2011): A Population-Based Study of French Adolescents. Am. J. Gastroenterol. 2018, 113, 265–272. [Google Scholar] [CrossRef]
- Jakobsen, C.; Paerregaard, A.; Munkholm, P.; Faerk, J.; Lange, A.; Andersen, J.; Jakobsen, M.; Kramer, I.; Czernia-Mazurkiewicz, J.; Wewer, V. Pediatric inflammatory bowel disease: Increasing incidence, decreasing surgery rate, and compromised nutritional status: A prospective population-based cohort study 2007–2009. Inflamm. Bowel Dis. 2011, 17, 2541–2550. [Google Scholar] [CrossRef]
- Vatn, M.H.; Sandvik, A.K. Inflammatory bowel disease. Scand. J. Gastroenterol. 2015, 50, 748–762. [Google Scholar] [CrossRef]
- Guan, Q. A Comprehensive Review and Update on the Pathogenesis of Inflammatory Bowel Disease. J. Immunol. Res. 2019, 2019, 7247238. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.Z.; Li, Y.Y. Inflammatory bowel disease: Pathogenesis. World J. Gastroenterol. 2014, 20, 91–99. [Google Scholar] [CrossRef]
- Dixon, L.J.; Kabi, A.; Nickerson, K.P.; McDonald, C. Combinatorial effects of diet and genetics on inflammatory bowel disease pathogenesis. Inflamm. Bowel Dis. 2015, 21, 912–922. [Google Scholar] [CrossRef]
- Leone, V.; Chang, E.B.; Devkota, S. Diet, microbes, and host genetics: The perfect storm in inflammatory bowel diseases. J. Gastroenterol. 2013, 48, 315–321. [Google Scholar] [CrossRef] [Green Version]
- Zuk, O.; Hechter, E.; Sunyaev, S.R.; Lander, E.S. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc. Natl. Acad. Sci. USA 2012, 109, 1193–1198. [Google Scholar] [CrossRef] [PubMed]
- Loftus, E.V. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology 2004, 126, 1504–1517. [Google Scholar] [CrossRef] [PubMed]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the human intestinal microbial flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, S.J.; Musfeldt, M.; Wenderoth, D.F.; Hampe, J.; Brant, O.; Fölsch, U.R.; Timmis, K.N.; Schreiber, S. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut 2004, 53, 685–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, M.E.; Phillipson, M.; Petersson, J.; Velcich, A.; Holm, L.; Hansson, G.C. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc. Natl. Acad. Sci. USA 2008, 105, 15064–15069. [Google Scholar] [CrossRef] [PubMed]
- Liberati, A.; Altman, D.G.; Tetzlaff, J.; Mulrow, C.; Gotzsche, P.C.; Ioannidis, J.P.; Clarke, M.; Devereaux, P.J.; Kleijnen, J.; Moher, D. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate healthcare interventions: Explanation and elaboration. BMJ 2009, 339, b2700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dordević, D.; Jančíková, S.; Vítězová, M.; Kushkevych, I. Hydrogen sulfide toxicity in the gut environment: Meta-analysis of sulfate-reducing and lactic acid bacteria in inflammatory processes. J. Adv. Res. 2021, 27, 55–69. [Google Scholar] [CrossRef]
- Tremaroli, V.; Bäckhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, D.R.; Garge, N.; Zhang, X.; Sun, W.; O’Connell, T.M.; Bunger, M.K.; Bultman, S.J. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011, 13, 517–526. [Google Scholar] [CrossRef] [Green Version]
- Awano, N.; Wada, M.; Mori, H.; Nakamori, S.; Takagi, H. Identification and functional analysis of Escherichia coli cysteine desulfhydrases. Appl. Environ. Microbiol. 2005, 71, 4149–4152. [Google Scholar] [CrossRef] [Green Version]
- Rolfe, R.D. The role of probiotic cultures in the control of gastrointestinal health. J. Nutr. 2000, 130, 396S–402S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Yang, B.; Ma, D.; Wang, L.; Duan, W. Hydrogen Sulfide, Adipose Tissue and Diabetes Mellitus. Diabetes Metab. Syndr. Obes. 2020, 13, 1873–1886. [Google Scholar] [CrossRef] [PubMed]
- Elson, C.O.; Sartor, R.B.; Tennyson, G.S.; Riddell, R.H. Experimental models of inflammatory bowel disease. Gastroenterology 1995, 109, 1344–1367. [Google Scholar] [CrossRef]
- Smith, E.A.; Macfarlane, G.T. Dissimilatory amino Acid metabolism in human colonic bacteria. Anaerobe 1997, 3, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Florin, T.; Neale, G.; Gibson, G.R.; Christl, S.U.; Cummings, J.H. Metabolism of dietary sulphate: Absorption and excretion in humans. Gut 1991, 32, 766–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, G.R.; Cummings, J.H.; Macfarlane, G.T. Competition for hydrogen between sulphate-reducing bacteria and methanogenic bacteria from the human large intestine. J. Appl. Bacteriol. 1988, 65, 241–247. [Google Scholar] [CrossRef]
- Liau, Y.H.; Horowitz, M.I. The importance of PAPS in determining sulfation in gastrointestinal mucosa. Digestion 1976, 14, 372–375. [Google Scholar] [CrossRef]
- Sartor, R.B. Microbial influences in inflammatory bowel diseases. Gastroenterology 2008, 134, 577–594. [Google Scholar] [CrossRef]
- Levine, J.; Ellis, C.J.; Furne, J.K.; Springfield, J.; Levitt, M.D. Fecal hydrogen sulfide production in ulcerative colitis. Am. J. Gastroenterol. 1998, 93, 83–87. [Google Scholar] [CrossRef]
- Marchesi, J.R.; Holmes, E.; Khan, F.; Kochhar, S.; Scanlan, P.; Shanahan, F.; Wilson, I.D.; Wang, Y. Rapid and noninvasive metabonomic characterization of inflammatory bowel disease. J. Proteome Res. 2007, 6, 546–551. [Google Scholar] [CrossRef]
- Pitcher, M.C.; Beatty, E.R.; Cummings, J.H. The contribution of sulphate reducing bacteria and 5-aminosalicylic acid to faecal sulphide in patients with ulcerative colitis. Gut 2000, 46, 64–72. [Google Scholar] [CrossRef] [Green Version]
- Kushkevych, I.; Dordević, D.; Vítězová, M. Possible synergy effect of hydrogen sulfide and acetate produced by sulfate-reducing bacteria on inflammatory bowel disease development. J. Adv. Res. 2021, 27, 71–78. [Google Scholar] [CrossRef]
- Rowan, F.E.; Docherty, N.G.; Coffey, J.C.; O’Connell, P.R. Sulphate-reducing bacteria and hydrogen sulphide in the aetiology of ulcerative colitis. Br. J. Surg. 2009, 96, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.P.; Schippa, S.; Zamboni, I.; Penta, M.; Chiarini, F.; Seganti, L.; Osborn, J.; Falconieri, P.; Borrelli, O.; Cucchiara, S. Gut-associated bacterial microbiota in paediatric patients with inflammatory bowel disease. Gut 2006, 55, 1760–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwiertz, A.; Jacobi, M.; Frick, J.S.; Richter, M.; Rusch, K.; Köhler, H. Microbiota in pediatric inflammatory bowel disease. J. Pediatr. 2010, 157, 240–244.e241. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermúdez-Humarán, L.G.; Gratadoux, J.J.; Blugeon, S.; Bridonneau, C.; Furet, J.P.; Corthier, G.; et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef]
- Sultan, S.; El-Mowafy, M.; Elgaml, A.; Ahmed, T.A.E.; Hassan, H.; Mottawea, W. Metabolic Influences of Gut Microbiota Dysbiosis on Inflammatory Bowel Disease. Front. Physiol. 2021, 12, 715506. [Google Scholar] [CrossRef]
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vázquez-Baeza, Y.; Van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M.; et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 2014, 15, 382–392. [Google Scholar] [CrossRef] [Green Version]
- Finnie, I.A.; Dwarakanath, A.D.; Taylor, B.A.; Rhodes, J.M. Colonic mucin synthesis is increased by sodium butyrate. Gut 1995, 36, 93–99. [Google Scholar] [CrossRef] [Green Version]
- Ramasamy, S.; Singh, S.; Taniere, P.; Langman, M.J.; Eggo, M.C. Sulfide-detoxifying enzymes in the human colon are decreased in cancer and upregulated in differentiation. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291, G288–G296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byndloss, M.X.; Olsan, E.E.; Rivera-Chávez, F.; Tiffany, C.R.; Cevallos, S.A.; Lokken, K.L.; Torres, T.P.; Byndloss, A.J.; Faber, F.; Gao, Y.; et al. Microbiota-activated PPAR-γ signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 2017, 357, 570–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litvak, Y.; Byndloss, M.X.; Tsolis, R.M.; Bäumler, A.J. Dysbiotic Proteobacteria expansion: A microbial signature of epithelial dysfunction. Curr. Opin. Microbiol. 2017, 39, 1–6. [Google Scholar] [CrossRef]
- Babidge, W.; Millard, S.; Roediger, W. Sulfides impair short chain fatty acid beta-oxidation at acyl-CoA dehydrogenase level in colonocytes: Implications for ulcerative colitis. Mol. Cell. Biochem. 1998, 181, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Teigen, L.M.; Geng, Z.; Sadowsky, M.J.; Vaughn, B.P.; Hamilton, M.J.; Khoruts, A. Dietary Factors in Sulfur Metabolism and Pathogenesis of Ulcerative Colitis. Nutrients 2019, 11, 931. [Google Scholar] [CrossRef] [Green Version]
- Ungaro, R.; Bernstein, C.N.; Gearry, R.; Hviid, A.; Kolho, K.L.; Kronman, M.P.; Shaw, S.; Van Kruiningen, H.; Colombel, J.F.; Atreja, A. Antibiotics associated with increased risk of new-onset Crohn’s disease but not ulcerative colitis: A meta-analysis. Am. J. Gastroenterol. 2014, 109, 1728–1738. [Google Scholar] [CrossRef]
- Ohge, H.; Furne, J.K.; Springfield, J.; Sueda, T.; Madoff, R.D.; Levitt, M.D. The effect of antibiotics and bismuth on fecal hydrogen sulfide and sulfate-reducing bacteria in the rat. FEMS Microbiol. Lett. 2003, 228, 137–142. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.B.; Lin, H.C. Hydrogen Sulfide in Physiology and Diseases of the Digestive Tract. Microorganisms 2015, 3, 866–889. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Chan, A.; Ali, S.; Saha, A.; Haushalter, K.J.; Lam, W.L.; Glasheen, M.; Parker, J.; Brenner, M.; Mahon, S.B.; et al. Hydrogen Sulfide—Mechanisms of Toxicity and Development of an Antidote. Sci. Rep. 2016, 6, 20831. [Google Scholar] [CrossRef]
- De Preter, V.; Arijs, I.; Windey, K.; Vanhove, W.; Vermeire, S.; Schuit, F.; Rutgeerts, P.; Verbeke, K. Decreased mucosal sulfide detoxification is related to an impaired butyrate oxidation in ulcerative colitis. Inflamm. Bowel Dis. 2012, 18, 2371–2380. [Google Scholar] [CrossRef]
- Wolf, P.G.; Biswas, A.; Morales, S.E.; Greening, C.; Gaskins, H.R. H2 metabolism is widespread and diverse among human colonic microbes. Gut Microbes 2016, 7, 235–245. [Google Scholar] [CrossRef] [Green Version]
- Carbonero, F.; Benefiel, A.C.; Gaskins, H.R. Contributions of the microbial hydrogen economy to colonic homeostasis. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 504–518. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, S.; Macfarlane, G.T. Regulation of short-chain fatty acid production. Proc. Nutr. Soc. 2003, 62, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Duboc, H.; Rajca, S.; Rainteau, D.; Benarous, D.; Maubert, M.A.; Quervain, E.; Thomas, G.; Barbu, V.; Humbert, L.; Despras, G.; et al. Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut 2013, 62, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.; Schmitt-Kopplin, P. The role of fecal sulfur metabolome in inflammatory bowel diseases. Int. J. Med. Microbiol. 2021, 311, 151513. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Xavier, R.J.; Gevers, D. The microbiome in inflammatory bowel disease: Current status and the future ahead. Gastroenterology 2014, 146, 1489–1499. [Google Scholar] [CrossRef] [Green Version]
- Khodakivskyi, P.V.; Lauber, C.L.; Yevtodiyenko, A.; Bazhin, A.A.; Bruce, S.; Ringel-Kulka, T.; Ringel, Y.; Bétrisey, B.; Torres, J.; Hu, J.; et al. Noninvasive imaging and quantification of bile salt hydrolase activity: From bacteria to humans. Sci. Adv. 2021, 7, eaaz9857. [Google Scholar] [CrossRef]
- Jones, B.V.; Begley, M.; Hill, C.; Gahan, C.G.; Marchesi, J.R. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc. Natl. Acad. Sci. USA 2008, 105, 13580–13585. [Google Scholar] [CrossRef]
- Lopez-Siles, M.; Khan, T.M.; Duncan, S.H.; Harmsen, H.J.; Garcia-Gil, L.J.; Flint, H.J. Cultured representatives of two major phylogroups of human colonic Faecalibacterium prausnitzii can utilize pectin, uronic acids, and host-derived substrates for growth. Appl. Environ. Microbiol. 2012, 78, 420–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Z.; Cai, Y.; Lao, X.; Wang, X.; Lin, X.; Cui, Y.; Kalavagunta, P.K.; Liao, J.; Jin, L.; Shang, J.; et al. Taxonomic profiling and populational patterns of bacterial bile salt hydrolase (BSH) genes based on worldwide human gut microbiome. Microbiome 2019, 7, 9. [Google Scholar] [CrossRef] [Green Version]
- Cummings, J.H.; Macfarlane, G.T.; Macfarlane, S. Intestinal bacteria and ulcerative colitis. Curr. Issues Intest. Microbiol. 2003, 4, 9–20. [Google Scholar] [PubMed]
- Kushkevych, I.; Kotrsová, V.; Dordević, D.; Buňková, L.; Vítězová, M.; Amedei, A. Hydrogen Sulfide Effects on the Survival of Lactobacilli with Emphasis on the Development of Inflammatory Bowel Diseases. Biomolecules 2019, 9, 752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, P.; Marcišauskas, S.; Ji, B.; Nielsen, J. Metagenomic analysis of bile salt biotransformation in the human gut microbiome. BMC Genom. 2019, 20, 517. [Google Scholar] [CrossRef] [Green Version]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef]
- Laue, H.; Denger, K.; Cook, A.M. Taurine reduction in anaerobic respiration of Bilophila wadsworthia RZATAU. Appl. Environ. Microbiol. 1997, 63, 2016–2021. [Google Scholar] [CrossRef] [PubMed]
- Edmond, L.M.; Hopkins, M.J.; Magee, E.A.; Cummings, J.H. The effect of 5-aminosalicylic acid-containing drugs on sulfide production by sulfate-reducing and amino acid-fermenting bacteria. Inflamm. Bowel Dis. 2003, 9, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Dubuquoy, L.; Rousseaux, C.; Thuru, X.; Peyrin-Biroulet, L.; Romano, O.; Chavatte, P.; Chamaillard, M.; Desreumaux, P. PPARgamma as a new therapeutic target in inflammatory bowel diseases. Gut 2006, 55, 1341–1349. [Google Scholar] [CrossRef] [Green Version]
- Alex, S.; Lange, K.; Amolo, T.; Grinstead, J.S.; Haakonsson, A.K.; Szalowska, E.; Koppen, A.; Mudde, K.; Haenen, D.; Al-Lahham, S.; et al. Short-chain fatty acids stimulate angiopoietin-like 4 synthesis in human colon adenocarcinoma cells by activating peroxisome proliferator-activated receptor γ. Mol. Cell. Biol. 2013, 33, 1303–1316. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Chen, N.; Wu, Z.; Song, Y.; Zhang, Y.; Wu, N.; Zhang, F.; Ren, X.; Liu, Y. 5-Aminosalicylic Acid Alters the Gut Bacterial Microbiota in Patients with Ulcerative Colitis. Front. Microbiol. 2018, 9, 1274. [Google Scholar] [CrossRef]
- Fiorucci, S.; Orlandi, S.; Mencarelli, A.; Caliendo, G.; Santagada, V.; Distrutti, E.; Santucci, L.; Cirino, G.; Wallace, J.L. Enhanced activity of a hydrogen sulphide-releasing derivative of mesalamine (ATB-429) in a mouse model of colitis. Br. J. Pharmacol. 2007, 150, 996–1002. [Google Scholar] [CrossRef] [Green Version]
- Roediger, W.E. Decreased sulphur aminoacid intake in ulcerative colitis. Lancet 1998, 351, 1555. [Google Scholar] [CrossRef]
- Ijssennagger, N.; van der Meer, R.; van Mil, S.W.C. Sulfide as a Mucus Barrier-Breaker in Inflammatory Bowel Disease? Trends Mol. Med. 2016, 22, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Langendijk, P.S.; Hanssen, J.T.; Van der Hoeven, J.S. Sulfate-reducing bacteria in association with human periodontitis. J. Clin. Periodontol. 2000, 27, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Ohge, H.; Furne, J.K.; Springfield, J.; Rothenberger, D.A.; Madoff, R.D.; Levitt, M.D. Association between fecal hydrogen sulfide production and pouchitis. Dis. Colon. Rectum 2005, 48, 469–475. [Google Scholar] [CrossRef]
- Sahami, S.; Kooij, I.A.; Meijer, S.L.; Van den Brink, G.R.; Buskens, C.J.; Te Velde, A.A. The Link between the Appendix and Ulcerative Colitis: Clinical Relevance and Potential Immunological Mechanisms. Am. J. Gastroenterol. 2016, 111, 163–169. [Google Scholar] [CrossRef]
- Rogers, M.B.; Brower-Sinning, R.; Firek, B.; Zhong, D.; Morowitz, M.J. Acute Appendicitis in Children Is Associated with a Local Expansion of Fusobacteria. Clin. Infect. Dis. 2016, 63, 71–78. [Google Scholar] [CrossRef] [Green Version]
- Basic, A.; Blomqvist, S.; Carlén, A.; Dahlén, G. Estimation of bacterial hydrogen sulfide production in vitro. J. Oral. Microbiol. 2015, 7, 28166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, D.; Brower-Sinning, R.; Firek, B.; Morowitz, M.J. Acute appendicitis in children is associated with an abundance of bacteria from the phylum Fusobacteria. J. Pediatr. Surg. 2014, 49, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.; Babidge, W.; Millard, S.; Roediger, W. Colonic luminal hydrogen sulfide is not elevated in ulcerative colitis. Dig. Dis. Sci. 1998, 43, 162–165. [Google Scholar] [CrossRef]
- Jørgensen, J.; Mortensen, P.B. Hydrogen sulfide and colonic epithelial metabolism: Implications for ulcerative colitis. Dig. Dis. Sci. 2001, 46, 1722–1732. [Google Scholar] [CrossRef]
- Levitt, M.D.; Springfield, J.; Furne, J.; Koenig, T.; Suarez, F.L. Physiology of sulfide in the rat colon: Use of bismuth to assess colonic sulfide production. J. Appl. Physiol. 2002, 92, 1655–1660. [Google Scholar] [CrossRef] [Green Version]
- Macfarlane, S.; McBain, A.J.; Macfarlane, G.T. Consequences of biofilm and sessile growth in the large intestine. Adv. Dent. Res. 1997, 11, 59–68. [Google Scholar] [CrossRef]
- Swidsinski, A.; Weber, J.; Loening-Baucke, V.; Hale, L.P.; Lochs, H. Spatial organization and composition of the mucosal flora in patients with inflammatory bowel disease. J. Clin. Microbiol. 2005, 43, 3380–3389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleessen, B.; Kroesen, A.J.; Buhr, H.J.; Blaut, M. Mucosal and invading bacteria in patients with inflammatory bowel disease compared with controls. Scand. J. Gastroenterol. 2002, 37, 1034–1041. [Google Scholar] [CrossRef]
- Swidsinski, A.; Ladhoff, A.; Pernthaler, A.; Swidsinski, S.; Loening-Baucke, V.; Ortner, M.; Weber, J.; Hoffmann, U.; Schreiber, S.; Dietel, M.; et al. Mucosal flora in inflammatory bowel disease. Gastroenterology 2002, 122, 44–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, M.E.; Gustafsson, J.K.; Holmén-Larsson, J.; Jabbar, K.S.; Xia, L.; Xu, H.; Ghishan, F.K.; Carvalho, F.A.; Gewirtz, A.T.; Sjövall, H.; et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut 2014, 63, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Swidsinski, A.; Loening-Baucke, V.; Theissig, F.; Engelhardt, H.; Bengmark, S.; Koch, S.; Lochs, H.; Dörffel, Y. Comparative study of the intestinal mucus barrier in normal and inflamed colon. Gut 2007, 56, 343–350. [Google Scholar] [CrossRef]
- Pullan, R.D.; Thomas, G.A.; Rhodes, M.; Newcombe, R.G.; Williams, G.T.; Allen, A.; Rhodes, J. Thickness of adherent mucus gel on colonic mucosa in humans and its relevance to colitis. Gut 1994, 35, 353–359. [Google Scholar] [CrossRef] [Green Version]
- Strugala, V.; Dettmar, P.W.; Pearson, J.P. Thickness and continuity of the adherent colonic mucus barrier in active and quiescent ulcerative colitis and Crohn’s disease. Int. J. Clin. Pract. 2008, 62, 762–769. [Google Scholar] [CrossRef]
- Rankin, B.J.; Srivastava, E.D.; Record, C.O.; Pearson, J.P.; Allen, A. Patients with ulcerative colitis have reduced mucin polymer content in the adherent colonic mucus gel. Biochem. Soc. Trans. 1995, 23, 104S. [Google Scholar] [CrossRef] [Green Version]
- Gardiner, K.R.; Halliday, M.I.; Barclay, G.R.; Milne, L.; Brown, D.; Stephens, S.; Maxwell, R.J.; Rowlands, B.J. Significance of systemic endotoxaemia in inflammatory bowel disease. Gut 1995, 36, 897–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ijssennagger, N.; Belzer, C.; Hooiveld, G.J.; Dekker, J.; van Mil, S.W.; Müller, M.; Kleerebezem, M.; van der Meer, R. Gut microbiota facilitates dietary heme-induced epithelial hyperproliferation by opening the mucus barrier in colon. Proc. Natl. Acad. Sci. USA 2015, 112, 10038–10043. [Google Scholar] [CrossRef] [PubMed]
- Blackler, R.W.; Motta, J.P.; Manko, A.; Workentine, M.; Bercik, P.; Surette, M.G.; Wallace, J.L. Hydrogen sulphide protects against NSAID-enteropathy through modulation of bile and the microbiota. Br. J. Pharmacol. 2015, 172, 992–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, J.L.; Ianaro, A.; de Nucci, G. Gaseous Mediators in Gastrointestinal Mucosal Defense and Injury. Dig. Dis. Sci. 2017, 62, 2223–2230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stummer, N.; Weghuber, D.; Feichtinger, R.G.; Huber, S.; Mayr, J.A.; Kofler, B.; Neureiter, D.; Klieser, E.; Hochmann, S.; Lauth, W.; et al. Hydrogen Sulfide Metabolizing Enzymes in the Intestinal Mucosa in Pediatric and Adult Inflammatory Bowel Disease. Antioxidants 2022, 11, 2235. [Google Scholar] [CrossRef] [PubMed]
- Eichele, D.D.; Kharbanda, K.K. Dextran sodium sulfate colitis murine model: An indispensable tool for advancing our understanding of inflammatory bowel diseases pathogenesis. World J. Gastroenterol. 2017, 23, 6016–6029. [Google Scholar] [CrossRef]
- Beaumont, M.; Andriamihaja, M.; Lan, A.; Khodorova, N.; Audebert, M.; Blouin, J.M.; Grauso, M.; Lancha, L.; Benetti, P.H.; Benamouzig, R.; et al. Detrimental effects for colonocytes of an increased exposure to luminal hydrogen sulfide: The adaptive response. Free Radic. Biol. Med. 2016, 93, 155–164. [Google Scholar] [CrossRef]
- Cooper, C.E.; Brown, G.C. The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: Chemical mechanism and physiological significance. J. Bioenerg. Biomembr. 2008, 40, 533–539. [Google Scholar] [CrossRef]
- Andriamihaja, M.; Lan, A.; Beaumont, M.; Grauso, M.; Gotteland, M.; Pastene, E.; Cires, M.J.; Carrasco-Pozo, C.; Tomé, D.; Blachier, F. Proanthocyanidin-containing polyphenol extracts from fruits prevent the inhibitory effect of hydrogen sulfide on human colonocyte oxygen consumption. Amino Acids 2018, 50, 755–763. [Google Scholar] [CrossRef]
- Mimoun, S.; Andriamihaja, M.; Chaumontet, C.; Atanasiu, C.; Benamouzig, R.; Blouin, J.M.; Tomé, D.; Bouillaud, F.; Blachier, F. Detoxification of H2S by differentiated colonic epithelial cells: Implication of the sulfide oxidizing unit and of the cell respiratory capacity. Antioxid. Redox Signal 2012, 17, 1–10. [Google Scholar] [CrossRef]
- Hirata, I.; Naito, Y.; Takagi, T.; Mizushima, K.; Suzuki, T.; Omatsu, T.; Handa, O.; Ichikawa, H.; Ueda, H.; Yoshikawa, T. Endogenous hydrogen sulfide is an anti-inflammatory molecule in dextran sodium sulfate-induced colitis in mice. Dig. Dis. Sci. 2011, 56, 1379–1386. [Google Scholar] [CrossRef]
- Picton, R.; Eggo, M.C.; Langman, M.J.; Singh, S. Impaired detoxication of hydrogen sulfide in ulcerative colitis? Dig. Dis. Sci. 2007, 52, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cen, L.; Zhang, X.; Tang, C.; Chen, Y.; Zhang, Y.; Yu, M.; Lu, C.; Li, M.; Li, S.; et al. MPST deficiency promotes intestinal epithelial cell apoptosis and aggravates inflammatory bowel disease via AKT. Redox Biol. 2022, 56, 102469. [Google Scholar] [CrossRef]
- Arijs, I.; Vanhove, W.; Rutgeerts, P.; Schuit, F.; Verbeke, K.; De Preter, V. Decreased mucosal sulfide detoxification capacity in patients with Crohn’s disease. Inflamm. Bowel Dis. 2013, 19, E70–E72. [Google Scholar] [CrossRef] [PubMed]
- Furne, J.; Springfield, J.; Koenig, T.; DeMaster, E.; Levitt, M.D. Oxidation of hydrogen sulfide and methanethiol to thiosulfate by rat tissues: A specialized function of the colonic mucosa. Biochem. Pharmacol. 2001, 62, 255–259. [Google Scholar] [CrossRef]
- Flannigan, K.L.; Agbor, T.A.; Blackler, R.W.; Kim, J.J.; Khan, W.I.; Verdu, E.F.; Ferraz, J.G.; Wallace, J.L. Impaired hydrogen sulfide synthesis and IL-10 signaling underlie hyperhomocysteinemia-associated exacerbation of colitis. Proc. Natl. Acad. Sci. USA 2014, 111, 13559–13564. [Google Scholar] [CrossRef] [PubMed]
- Vagianos, K.; Bector, S.; McConnell, J.; Bernstein, C.N. Nutrition assessment of patients with inflammatory bowel disease. JPEN J. Parenter. Enteral Nutr. 2007, 31, 311–319. [Google Scholar] [CrossRef]
- Goubern, M.; Andriamihaja, M.; Nübel, T.; Blachier, F.; Bouillaud, F. Sulfide, the first inorganic substrate for human cells. FASEB J. 2007, 21, 1699–1706. [Google Scholar] [CrossRef] [PubMed]
- Zanardo, R.C.; Brancaleone, V.; Distrutti, E.; Fiorucci, S.; Cirino, G.; Wallace, J.L. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. 2006, 20, 2118–2120. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.L.; Caliendo, G.; Santagada, V.; Cirino, G.; Fiorucci, S. Gastrointestinal safety and anti-inflammatory effects of a hydrogen sulfide-releasing diclofenac derivative in the rat. Gastroenterology 2007, 132, 261–271. [Google Scholar] [CrossRef]
- Distrutti, E.; Sediari, L.; Mencarelli, A.; Renga, B.; Orlandi, S.; Antonelli, E.; Roviezzo, F.; Morelli, A.; Cirino, G.; Wallace, J.L.; et al. Evidence that hydrogen sulfide exerts antinociceptive effects in the gastrointestinal tract by activating KATP channels. J. Pharmacol. Exp. Ther. 2006, 316, 325–335. [Google Scholar] [CrossRef] [Green Version]
- Wallace, J.L.; Ferraz, J.G.; Muscara, M.N. Hydrogen sulfide: An endogenous mediator of resolution of inflammation and injury. Antioxid. Redox Signal 2012, 17, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Lee, H.G.; Choi, K.S.; Surh, Y.J.; Na, H.K. Diallyl trisulfide suppresses dextran sodium sulfate-induced mouse colitis: NF-κB and STAT3 as potential targets. Biochem. Biophys. Res. Commun. 2013, 437, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Peake, B.F.; Nicholson, C.K.; Lambert, J.P.; Hood, R.L.; Amin, H.; Amin, S.; Calvert, J.W. Hydrogen sulfide preconditions the db/db diabetic mouse heart against ischemia-reperfusion injury by activating Nrf2 signaling in an Erk-dependent manner. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1215–H1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Zhao, K.; Ju, Y.; Mani, S.; Cao, Q.; Puukila, S.; Khaper, N.; Wu, L.; Wang, R. Hydrogen sulfide protects against cellular senescence via S-sulfhydration of Keap1 and activation of Nrf2. Antioxid. Redox Signal 2013, 18, 1906–1919. [Google Scholar] [CrossRef]
- Elrod, J.W.; Calvert, J.W.; Morrison, J.; Doeller, J.E.; Kraus, D.W.; Tao, L.; Jiao, X.; Scalia, R.; Kiss, L.; Szabo, C.; et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc. Natl. Acad. Sci. USA 2007, 104, 15560–15565. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.V.; Wallace, J.L. Hydrogen sulfide-based therapeutics and gastrointestinal diseases: Translating physiology to treatments. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G467–G473. [Google Scholar] [CrossRef] [Green Version]
- Oh, G.S.; Pae, H.O.; Lee, B.S.; Kim, B.N.; Kim, J.M.; Kim, H.R.; Jeon, S.B.; Jeon, W.K.; Chae, H.J.; Chung, H.T. Hydrogen sulfide inhibits nitric oxide production and nuclear factor-kappaB via heme oxygenase-1 expression in RAW264.7 macrophages stimulated with lipopolysaccharide. Free Radic. Biol. Med. 2006, 41, 106–119. [Google Scholar] [CrossRef]
- Wallace, J.L.; Dicay, M.; McKnight, W.; Martin, G.R. Hydrogen sulfide enhances ulcer healing in rats. FASEB J. 2007, 21, 4070–4076. [Google Scholar] [CrossRef]
- Zhao, H.; Yan, R.; Zhou, X.; Ji, F.; Zhang, B. Hydrogen sulfide improves colonic barrier integrity in DSS-induced inflammation in Caco-2 cells and mice. Int. Immunopharmacol. 2016, 39, 121–127. [Google Scholar] [CrossRef]
- Li, T.; Zhao, B.; Wang, C.; Wang, H.; Liu, Z.; Li, W.; Jin, H.; Tang, C.; Du, J. Regulatory effects of hydrogen sulfide on IL-6, IL-8 and IL-10 levels in the plasma and pulmonary tissue of rats with acute lung injury. Exp. Biol. Med. 2008, 233, 1081–1087. [Google Scholar] [CrossRef]
- Wu, J.; Wei, J.; You, X.; Chen, X.; Zhu, H.; Zhu, X.; Liu, Y.; Xu, M. Inhibition of hydrogen sulfide generation contributes to lung injury after experimental orthotopic lung transplantation. J. Surg. Res. 2013, 182, e25–e33. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Lin, X.; Fan, H.; Li, C. Hydrogen sulfide attenuates the inflammatory response in a mouse burn injury model. Mol. Med. Rep. 2013, 8, 1204–1208. [Google Scholar] [CrossRef] [Green Version]
- Tokuda, K.; Kida, K.; Marutani, E.; Crimi, E.; Bougaki, M.; Khatri, A.; Kimura, H.; Ichinose, F. Inhaled hydrogen sulfide prevents endotoxin-induced systemic inflammation and improves survival by altering sulfide metabolism in mice. Antioxid. Redox Signal 2012, 17, 11–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noguchi, E.; Homma, Y.; Kang, X.; Netea, M.G.; Ma, X. A Crohn’s disease-associated NOD2 mutation suppresses transcription of human IL10 by inhibiting activity of the nuclear ribonucleoprotein hnRNP-A1. Nat. Immunol. 2009, 10, 471–479. [Google Scholar] [CrossRef] [Green Version]
- van der Linde, K.; Boor, P.P.; Sandkuijl, L.A.; Meijssen, M.A.; Savelkoul, H.F.; Wilson, J.H.; de Rooij, F.W. A Gly15Arg mutation in the interleukin-10 gene reduces secretion of interleukin-10 in Crohn disease. Scand. J. Gastroenterol. 2003, 38, 611–617. [Google Scholar] [PubMed]
- Franke, A.; Balschun, T.; Karlsen, T.H.; Sventoraityte, J.; Nikolaus, S.; Mayr, G.; Domingues, F.S.; Albrecht, M.; Nothnagel, M.; Ellinghaus, D.; et al. Sequence variants in IL10, ARPC2 and multiple other loci contribute to ulcerative colitis susceptibility. Nat. Genet. 2008, 40, 1319–1323. [Google Scholar] [CrossRef]
- Shouval, D.S.; Biswas, A.; Goettel, J.A.; McCann, K.; Conaway, E.; Redhu, N.S.; Mascanfroni, I.D.; Al Adham, Z.; Lavoie, S.; Ibourk, M.; et al. Interleukin-10 receptor signaling in innate immune cells regulates mucosal immune tolerance and anti-inflammatory macrophage function. Immunity 2014, 40, 706–719. [Google Scholar] [CrossRef]
- Fuss, I.J.; Boirivant, M.; Lacy, B.; Strober, W. The interrelated roles of TGF-beta and IL-10 in the regulation of experimental colitis. J. Immunol. 2002, 168, 900–908. [Google Scholar] [CrossRef]
- Lesage, S.; Zouali, H.; Cézard, J.P.; Colombel, J.F.; Belaiche, J.; Almer, S.; Tysk, C.; O’Morain, C.; Gassull, M.; Binder, V.; et al. CARD15/NOD2 mutational analysis and genotype-phenotype correlation in 612 patients with inflammatory bowel disease. Am. J. Hum. Genet. 2002, 70, 845–857. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.X.; Liu, S.J.; Tang, X.L.; Duan, G.L.; Ni, X.; Zhu, X.Y.; Liu, Y.J.; Wang, C.N. H2S Attenuates LPS-Induced Acute Lung Injury by Reducing Oxidative/Nitrative Stress and Inflammation. Cell. Physiol. Biochem. 2016, 40, 1603–1612. [Google Scholar] [CrossRef] [PubMed]
- Younis, N.N.; Shaheen, M.A.; Mahmoud, M.F. Silymarin preconditioning protected insulin resistant rats from liver ischemia-reperfusion injury: Role of endogenous H2S. J. Surg. Res. 2016, 204, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Qu, H.; Xie, J.; Long, J.; Bai, Q.; Chen, Y.; Mao, H. H2S-mediated aerobic exercise antagonizes the hippocampal inflammatory response in CUMS-depressed mice. J. Affect. Disord. 2021, 283, 410–419. [Google Scholar] [CrossRef]
- Martin, G.R.; Wallace, J.L. Gastrointestinal inflammation: A central component of mucosal defense and repair. Exp. Biol. Med. 2006, 231, 130–137. [Google Scholar] [CrossRef]
- Fiorucci, S.; Antonelli, E.; Distrutti, E.; Rizzo, G.; Mencarelli, A.; Orlandi, S.; Zanardo, R.; Renga, B.; Di Sante, M.; Morelli, A.; et al. Inhibition of hydrogen sulfide generation contributes to gastric injury caused by anti-inflammatory nonsteroidal drugs. Gastroenterology 2005, 129, 1210–1224. [Google Scholar] [CrossRef] [Green Version]
- Martin, G.R.; McKnight, G.W.; Dicay, M.S.; Coffin, C.S.; Ferraz, J.G.; Wallace, J.L. Hydrogen sulphide synthesis in the rat and mouse gastrointestinal tract. Dig. Liver Dis. 2010, 42, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Gaudichon, C.; Bos, C.; Morens, C.; Petzke, K.J.; Mariotti, F.; Everwand, J.; Benamouzig, R.; Daré, S.; Tomé, D.; Metges, C.C. Ileal losses of nitrogen and amino acids in humans and their importance to the assessment of amino acid requirements. Gastroenterology 2002, 123, 50–59. [Google Scholar] [CrossRef]
- Florin, T.H. Hydrogen sulphide and total acid-volatile sulphide in faeces, determined with a direct spectrophotometric method. Clin. Chim. Acta 1991, 196, 127–134. [Google Scholar] [CrossRef]
- Buffière, C.; Gaudichon, C.; Hafnaoui, N.; Migné, C.; Scislowsky, V.; Khodorova, N.; Mosoni, L.; Blot, A.; Boirie, Y.; Dardevet, D.; et al. In the elderly, meat protein assimilation from rare meat is lower than that from meat that is well done. Am. J. Clin. Nutr. 2017, 106, 1257–1266. [Google Scholar] [CrossRef] [Green Version]
- Mills, D.J.; Tuohy, K.M.; Booth, J.; Buck, M.; Crabbe, M.J.; Gibson, G.R.; Ames, J.M. Dietary glycated protein modulates the colonic microbiota towards a more detrimental composition in ulcerative colitis patients and non-ulcerative colitis subjects. J. Appl. Microbiol. 2008, 105, 706–714. [Google Scholar] [CrossRef]
- Rémond, D.; Machebeuf, M.; Yven, C.; Buffière, C.; Mioche, L.; Mosoni, L.; Patureau Mirand, P. Postprandial whole-body protein metabolism after a meat meal is influenced by chewing efficiency in elderly subjects. Am. J. Clin. Nutr. 2007, 85, 1286–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, S.; Cochrane, S. Alteration of sulfate and hydrogen metabolism in the human colon by changing intestinal transit rate. Am. J. Gastroenterol. 2007, 102, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Flynn, S.; Eisenstein, S. Inflammatory Bowel Disease Presentation and Diagnosis. Surg. Clin. N. Am. 2019, 99, 1051–1062. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, P.C.; Reigstad, C.S.; Loftus, E.V. Role of diet and gut microbiota in management of inflammatory bowel disease in an Asian migrant. J. Allergy Clin. Immunol. 2013, 132, 250–250.e5. [Google Scholar] [CrossRef]
- Chiba, M.; Tsuda, S.; Komatsu, M.; Tozawa, H.; Takayama, Y. Onset of Ulcerative Colitis during a Low-Carbohydrate Weight-Loss Diet and Treatment with a Plant-Based Diet: A Case Report. Perm. J. 2016, 20, 80–84. [Google Scholar] [CrossRef] [Green Version]
- Chiba, M.; Sugawara, T.; Komatsu, M.; Tozawa, H. Onset of Ulcerative Colitis in the Second Trimester after Emesis Gravidarum: Treatment with Plant-based Diet. Inflamm. Bowel Dis. 2018, 24, e8–e9. [Google Scholar] [CrossRef]
- Chiba, M.; Nakane, K.; Tsuji, T.; Tsuda, S.; Ishii, H.; Ohno, H.; Watanabe, K.; Ito, M.; Komatsu, M.; Yamada, K.; et al. Relapse Prevention in Ulcerative Colitis by Plant-Based Diet Through Educational Hospitalization: A Single-Group Trial. Perm. J. 2018, 22, 17–167. [Google Scholar] [CrossRef] [Green Version]
- Roediger, W.E.; Moore, J.; Babidge, W. Colonic sulfide in pathogenesis and treatment of ulcerative colitis. Dig. Dis. Sci. 1997, 42, 1571–1579. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [Green Version]
- Mu, C.; Yang, Y.; Luo, Z.; Guan, L.; Zhu, W. The Colonic Microbiome and Epithelial Transcriptome Are Altered in Rats Fed a High-Protein Diet Compared with a Normal-Protein Diet. J. Nutr. 2016, 146, 474–483. [Google Scholar] [CrossRef] [Green Version]
- Fuentes, S.; Rossen, N.G.; van der Spek, M.J.; Hartman, J.H.; Huuskonen, L.; Korpela, K.; Salojärvi, J.; Aalvink, S.; de Vos, W.M.; D’Haens, G.R.; et al. Microbial shifts and signatures of long-term remission in ulcerative colitis after faecal microbiota transplantation. ISME J. 2017, 11, 1877–1889. [Google Scholar] [CrossRef] [Green Version]
- Bajer, L.; Kverka, M.; Kostovcik, M.; Macinga, P.; Dvorak, J.; Stehlikova, Z.; Brezina, J.; Wohl, P.; Spicak, J.; Drastich, P. Distinct gut microbiota profiles in patients with primary sclerosing cholangitis and ulcerative colitis. World J. Gastroenterol. 2017, 23, 4548–4558. [Google Scholar] [CrossRef]
- Vermeire, S.; Joossens, M.; Verbeke, K.; Wang, J.; Machiels, K.; Sabino, J.; Ferrante, M.; Van Assche, G.; Rutgeerts, P.; Raes, J. Donor Species Richness Determines Faecal Microbiota Transplantation Success in Inflammatory Bowel Disease. J. Crohns Colitis 2016, 10, 387–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macfarlane, G.T.; Gibson, G.R.; Cummings, J.H. Comparison of fermentation reactions in different regions of the human colon. J. Appl. Bacteriol. 1992, 72, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.; Kuo, B.; McCallum, R.W.; Chey, W.D.; DiBaise, J.K.; Hasler, W.L.; Koch, K.L.; Lackner, J.M.; Miller, C.; Saad, R.; et al. Investigation of colonic and whole-gut transit with wireless motility capsule and radiopaque markers in constipation. Clin. Gastroenterol. Hepatol. 2009, 7, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.W.; Duncan, S.H.; McWilliam Leitch, E.C.; Child, M.W.; Flint, H.J. pH and peptide supply can radically alter bacterial populations and short-chain fatty acid ratios within microbial communities from the human colon. Appl. Environ. Microbiol. 2005, 71, 3692–3700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, G.R.; Cummings, J.H.; Macfarlane, G.T.; Allison, C.; Segal, I.; Vorster, H.H.; Walker, A.R. Alternative pathways for hydrogen disposal during fermentation in the human colon. Gut 1990, 31, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, J.; Lange, B.; Frick, J.S.; Sauer, H.; Zimmermann, K.; Schwiertz, A.; Rusch, K.; Klosterhalfen, S.; Enck, P. A vegan or vegetarian diet substantially alters the human colonic faecal microbiota. Eur. J. Clin. Nutr. 2012, 66, 53–60. [Google Scholar] [CrossRef]
- Zhu, Q.C.; Gao, R.Y.; Wu, W.; Guo, B.M.; Peng, J.Y.; Qin, H.L. Effect of a high-fat diet in development of colonic adenoma in an animal model. World J. Gastroenterol. 2014, 20, 8119–8129. [Google Scholar] [CrossRef]
- Devkota, S.; Wang, Y.; Musch, M.W.; Leone, V.; Fehlner-Peach, H.; Nadimpalli, A.; Antonopoulos, D.A.; Jabri, B.; Chang, E.B. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/- mice. Nature 2012, 487, 104–108. [Google Scholar] [CrossRef] [Green Version]
- de Vries, J.H.M.; Dijkhuizen, M.; Tap, P.; Witteman, B.J.M. Patient’s Dietary Beliefs and Behaviours in Inflammatory Bowel Disease. Dig. Dis. 2019, 37, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Ruemmele, F.M.; Veres, G.; Kolho, K.L.; Griffiths, A.; Levine, A.; Escher, J.C.; Amil Dias, J.; Barabino, A.; Braegger, C.P.; Bronsky, J.; et al. Consensus guidelines of ECCO/ESPGHAN on the medical management of pediatric Crohn’s disease. J. Crohns Colitis 2014, 8, 1179–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, A.S.; Whitten, K.E.; Sidler, M.; Lemberg, D.A. Systematic review: Nutritional therapy in paediatric Crohn’s disease. Aliment. Pharmacol. Ther. 2008, 27, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Otley, A.R.; Russell, R.K.; Day, A.S. Nutritional therapy for the treatment of pediatric Crohn’s disease. Expert. Rev. Clin. Immunol. 2010, 6, 667–676. [Google Scholar] [CrossRef]
- Levine, A.; Turner, D.; Pfeffer Gik, T.; Amil Dias, J.; Veres, G.; Shaoul, R.; Staiano, A.; Escher, J.; Kolho, K.L.; Paerregaard, A.; et al. Comparison of outcomes parameters for induction of remission in new onset pediatric Crohn’s disease: Evaluation of the porto IBD group “growth relapse and outcomes with therapy” (GROWTH CD) study. Inflamm. Bowel Dis. 2014, 20, 278–285. [Google Scholar] [CrossRef]
- MacLellan, A.; Moore-Connors, J.; Grant, S.; Cahill, L.; Langille, M.G.I.; Van Limbergen, J. The Impact of Exclusive Enteral Nutrition (EEN) on the Gut Microbiome in Crohn’s Disease: A Review. Nutrients 2017, 9, 447. [Google Scholar] [CrossRef]
- Levine, A.; Wine, E. Effects of enteral nutrition on Crohn’s disease: Clues to the impact of diet on disease pathogenesis. Inflamm. Bowel Dis. 2013, 19, 1322–1329. [Google Scholar] [CrossRef] [PubMed]
- Day, A.S.; Lopez, R.N. Exclusive enteral nutrition in children with Crohn’s disease. World J. Gastroenterol. 2015, 21, 6809–6816. [Google Scholar] [CrossRef]
- Quince, C.; Ijaz, U.Z.; Loman, N.; Eren, A.M.; Saulnier, D.; Russell, J.; Haig, S.J.; Calus, S.T.; Quick, J.; Barclay, A.; et al. Extensive Modulation of the Fecal Metagenome in Children With Crohn’s Disease During Exclusive Enteral Nutrition. Am. J. Gastroenterol. 2015, 110, 1718–1729; quiz 1730. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [Green Version]
- Gerasimidis, K.; Bertz, M.; Hanske, L.; Junick, J.; Biskou, O.; Aguilera, M.; Garrick, V.; Russell, R.K.; Blaut, M.; McGrogan, P.; et al. Decline in presumptively protective gut bacterial species and metabolites are paradoxically associated with disease improvement in pediatric Crohn’s disease during enteral nutrition. Inflamm. Bowel Dis. 2014, 20, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Colombel, J.F.; Mahadevan, U. Inflammatory Bowel Disease 2017: Innovations and Changing Paradigms. Gastroenterology 2017, 152, 309–312. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Enzyme | Method | Difference in Enzyme Expression in Comparison to the Control Group | Species | Reference | Year |
|---|---|---|---|---|---|
| CBS | mRNA measurement | increased | Mouse (DSS-induced colitis) | Hirata et al. [151] | 2011 |
| CBS | mRNA and protein expression | decreased | Mouse (Helicobacter hepaticus induced colitis in mice without adaptive immune system | De Cicco et al. [16] | 2018 |
| CBS | Protein expression (Western blot) | first lower (days 3–7 after induction), then increased | Rat (trinitrobenzene sulfonic acid induced colitis) | Wallace et al. [14] | 2009 |
| CBS | Immunohistochemical staining | increased | Rat (trinitrobenzene sulfonic acid induced colitis) | Wallace et al. [14] | 2009 |
| CSE | mRNA measurement | increased | Mouse (DSS-induced colitis) | Hirata et al. [151] | 2011 |
| CSE | Protein expression (Western blot) | decreased | Rat (trinitrobenzene sulfonic acid induced colitis) | Wallace et al. [14] | 2009 |
| CSE | Immunohistochemical staining | Unstained epithelial cells, while increased staining of mucosa and submucosa | Rat (trinitrobenzene sulfonic acid induced colitis) | Wallace et al. [14] | 2009 |
| CSE | mRNA and protein expression (Western blot) | decreased protein levels, decreased mRNA | Rat and mouse (DSS-induced colitis) | Taniguchi et al. [17] | 2009 |
| CSE | Immunohistochemical staining | decreased | Human (CD + UC) | Stummer et al. [145] | 2022 |
| 3-MST | Protein levels (Western blot) | decreased | Human (CD + UC) | Zhang et al. [153] | 2022 |
| 3-MST | Immunohistochemical staining | decreased | Human (CD + UC) | Zhang et al. [153] | 2022 |
| 3-MST | mRNA and protein expression | decreased | Mouse (DSS induced colitis) | Zhang et al. [153] | 2022 |
| 3-MST | Immunohistochemical staining | decreased | Human (CD + UC) | Stummer et al. [145] | 2022 |
| ETHE1 | mRNA | decreased | Human (CD) | Mottawea et al. [39] | 2016 |
| ETHE1 | Immunohistochemical staining | decreased, except for the terminal ileum in pediatric patients | Human (CD + UC) | Stummer et al. [145] | 2022 |
| SQOR | mRNA | decreased | Human (CD) | Mottawea et al. [39] | 2016 |
| SQOR | Immunohistochemical staining | decreased, except for the terminal ileum | Human (CD + UC) | Stummer et al. [145] | 2022 |
| TST | mRNA and protein expression (Western blot) | decreased | Rat and mouse (DSS induced colitis) | Taniguchi et al. [17] | 2009 |
| TST | mRNA | decreased | Human (UC) | De Preter et al. [100] | 2012 |
| TST | mRNA | decreased | Human (CD) | Mottawea et al. [39] | 2016 |
| TST | Immunohistochemical staining | decreased | Human (CD + UC) | Stummer et al. [145] | 2022 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stummer, N.; Feichtinger, R.G.; Weghuber, D.; Kofler, B.; Schneider, A.M. Role of Hydrogen Sulfide in Inflammatory Bowel Disease. Antioxidants 2023, 12, 1570. https://doi.org/10.3390/antiox12081570
Stummer N, Feichtinger RG, Weghuber D, Kofler B, Schneider AM. Role of Hydrogen Sulfide in Inflammatory Bowel Disease. Antioxidants. 2023; 12(8):1570. https://doi.org/10.3390/antiox12081570
Chicago/Turabian StyleStummer, Nathalie, René G. Feichtinger, Daniel Weghuber, Barbara Kofler, and Anna M. Schneider. 2023. "Role of Hydrogen Sulfide in Inflammatory Bowel Disease" Antioxidants 12, no. 8: 1570. https://doi.org/10.3390/antiox12081570
APA StyleStummer, N., Feichtinger, R. G., Weghuber, D., Kofler, B., & Schneider, A. M. (2023). Role of Hydrogen Sulfide in Inflammatory Bowel Disease. Antioxidants, 12(8), 1570. https://doi.org/10.3390/antiox12081570