
Mitochondrial Reactive Oxygen Species Formation Determines ACSL4/LPCAT2-Mediated Ferroptosis
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture, Compounds, and Treatments
2.2. RT-(q)PCR
2.3. Protein Analysis and Western Blot
2.4. Cell Viability Measurements
2.5. Flow Cytometry
2.6. ATP Bioluminescent Assay and Seahorse Measurement
2.7. Glutathione Assay
2.8. DPPH Assay
2.9. Statistical Analysis
3. Results
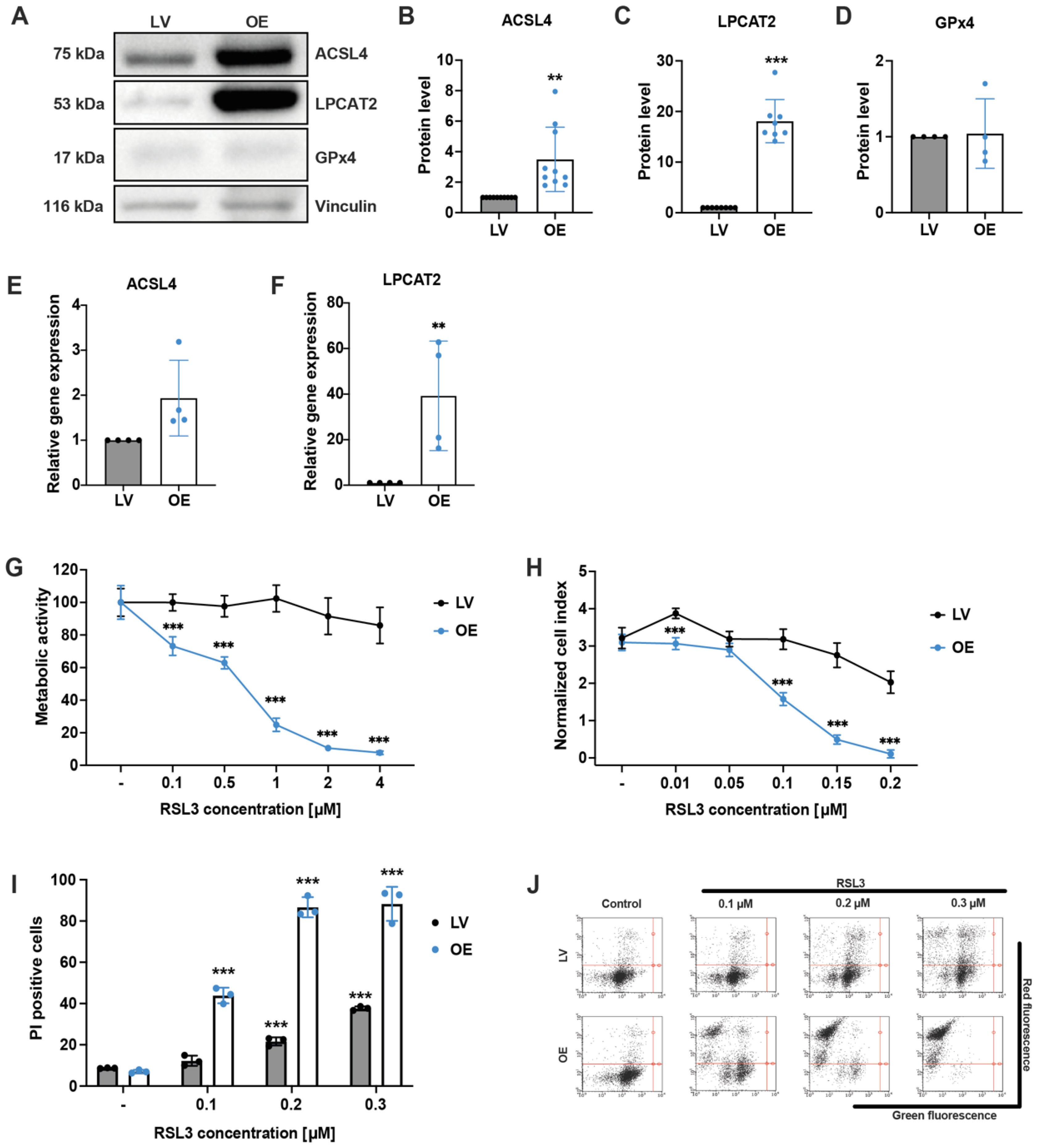
3.1. ACSL4 and LPCAT2 Overexpression Increases the Responsiveness of HEK293T Cells to Ferroptosis
3.2. Ferroptosis Inhibitors Deferoxamine and Ferrostatin-1 Protect against RSL3-Mediated Cell Death
3.3. Pharmacological ACSL4 Inhibition by Thiazolidinediones Prevented Ferroptosis in ACSL4/LPCAT2 Overexpressing Cells
3.4. ACSL4 and LPCAT2 Overexpression Causes Massive Mitochondrial Impairment
3.5. Mitochondrial ROS Scavenger Mitoquinone Prevents RSL3-Mediated Ferroptosis in ACSL4/LPCAT2 OE Cells
3.6. Inability to Protect ACSL4/LPCAT2-Driven Ferroptosis via Metabolic Intervention by Metformin
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and Function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Kroemer, G. Ferroptosis. Curr. Biol. 2020, 30, R1292–R1297. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, Biology and Role in Disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Yan, H.; Zou, T.; Tuo, Q.; Xu, S.; Li, H.; Belaidi, A.A.; Lei, P. Ferroptosis: Mechanisms and Links with Diseases. Signal Transduct. Target. Ther. 2021, 6, 1–16. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the Ferroptosis Regulator Gpx4 Triggers Acute Renal Failure in Mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Jelinek, A.; Heyder, L.; Daude, M.; Plessner, M.; Krippner, S.; Grosse, R.; Diederich, W.E.; Culmsee, C. Mitochondrial Rescue Prevents Glutathione Peroxidase-Dependent Ferroptosis. Free Radic. Biol. Med. 2018, 117, 45–57. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The Molecular Machinery of Regulated Cell Death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [Green Version]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 Dictates Ferroptosis Sensitivity by Shaping Cellular Lipid Composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized Arachidonic and Adrenic PEs Navigate Cells to Ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Soupene, E.; Kuypers, F.A. Mammalian Long-Chain Acyl-CoA Synthetases. Exp. Biol. Med. 2008, 233, 507–521. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. Identification of ACSL4 as a Biomarker and Contributor of Ferroptosis. Biochem. Biophys. Res. Commun. 2016, 478, 1338–1343. [Google Scholar] [CrossRef] [PubMed]
- Shindou, H.; Shimizu, T. Acyl-CoA:Lysophospholipid Acyltransferases *. J. Biol. Chem. 2009, 284, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, S.J.; Winter, G.E.; Musavi, L.S.; Lee, E.D.; Snijder, B.; Rebsamen, M.; Superti-Furga, G.; Stockwell, B.R. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem. Biol. 2015, 10, 1604–1609. [Google Scholar] [CrossRef] [Green Version]
- Seiler, A.; Schneider, M.; Förster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Rådmark, O.; Wurst, W.; et al. Glutathione Peroxidase 4 Senses and Translates Oxidative Stress into 12/15-Lipoxygenase Dependent- and AIF-Mediated Cell Death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Shintoku, R.; Takigawa, Y.; Yamada, K.; Kubota, C.; Yoshimoto, Y.; Takeuchi, T.; Koshiishi, I.; Torii, S. Lipoxygenase-Mediated Generation of Lipid Peroxides Enhances Ferroptosis Induced by Erastin and RSL3. Cancer Sci. 2017, 108, 2187–2194. [Google Scholar] [CrossRef]
- Sha, W.; Hu, F.; Xi, Y.; Chu, Y.; Bu, S. Mechanism of Ferroptosis and Its Role in Type 2 Diabetes Mellitus. J. Diabetes Res. 2021, 2021, e9999612. [Google Scholar] [CrossRef]
- Culmsee, C.; Landshamer, S. Molecular Insights into Mechanisms of the Cell Death Program:Role in the Progression of Neurodegenerative Disorders. Curr. Alzheimer Res. 2006, 3, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Landshamer, S.; Hoehn, M.; Barth, N.; Duvezin-Caubet, S.; Schwake, G.; Tobaben, S.; Kazhdan, I.; Becattini, B.; Zahler, S.; Vollmar, A.; et al. Bid-Induced Release of AIF from Mitochondria Causes Immediate Neuronal Cell Death. Cell Death Differ. 2008, 15, 1553–1563. [Google Scholar] [CrossRef]
- Mattson, M.P.; Gleichmann, M.; Cheng, A. Mitochondria in Neuroplasticity and Neurological Disorders. Neuron 2008, 60, 748–766. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–363.e3. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, L.; Waclawczyk, M.S.; Tang, S.; Hanschmann, E.-M.; Gellert, M.; Rust, M.B.; Culmsee, C. Cofilin1 Oxidation Links Oxidative Distress to Mitochondrial Demise and Neuronal Cell Death. Cell Death Dis. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Neitemeier, S.; Jelinek, A.; Laino, V.; Hoffmann, L.; Eisenbach, I.; Eying, R.; Ganjam, G.K.; Dolga, A.M.; Oppermann, S.; Culmsee, C. BID Links Ferroptosis to Mitochondrial Cell Death Pathways. Redox Biol. 2017, 12, 558–570. [Google Scholar] [CrossRef]
- Battaglia, A.M.; Chirillo, R.; Aversa, I.; Sacco, A.; Costanzo, F.; Biamonte, F. Ferroptosis and Cancer: Mitochondria Meet the “Iron Maiden” Cell Death. Cells 2020, 9, 1505. [Google Scholar] [CrossRef]
- Dan Dunn, J.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive Oxygen Species and Mitochondria: A Nexus of Cellular Homeostasis. Redox Biol. 2015, 6, 472–485. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Tobaben, S.; Grohm, J.; Seiler, A.; Conrad, M.; Plesnila, N.; Culmsee, C. Bid-Mediated Mitochondrial Damage Is a Key Mechanism in Glutamate-Induced Oxidative Stress and AIF-Dependent Cell Death in Immortalized HT-22 Hippocampal Neurons. Cell Death Differ. 2011, 18, 282–292. [Google Scholar] [CrossRef] [Green Version]
- Kowarz, E.; Löscher, D.; Marschalek, R. Optimized Sleeping Beauty Transposons Rapidly Generate Stable Transgenic Cell Lines. Biotechnol. J. 2015, 10, 647–653. [Google Scholar] [CrossRef]
- Zilka, O.; Shah, R.; Li, B.; Friedmann Angeli, J.P.; Griesser, M.; Conrad, M.; Pratt, D.A. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent. Sci. 2017, 3, 232–243. [Google Scholar] [CrossRef]
- Kretschmer, S.B.M.; Woltersdorf, S.; Rödl, C.B.; Vogt, D.; Häfner, A.K.; Steinhilber, D.; Stark, H.; Hofmann, B. Development of Novel Aminothiazole-Comprising 5-LO Inhibitors. Future Med. Chem. 2016, 8, 149–164. [Google Scholar] [CrossRef]
- Kretschmer, S.B.; Woltersdorf, S.; Vogt, D.; Lillich, F.F.; Rühl, M.; Karas, M.; Maucher, I.V.; Roos, J.; Häfner, A.K.; Kaiser, A.; et al. Characterization of the Molecular Mechanism of 5-Lipoxygenase Inhibition by 2-Aminothiazoles. Biochem. Pharmacol. 2017, 123, 52–62. [Google Scholar] [CrossRef]
- Bridges, H.R.; Sirviö, V.A.; Agip, A.-N.A.; Hirst, J. Molecular Features of Biguanides Required for Targeting of Mitochondrial Respiratory Complex I and Activation of AMP-Kinase. BMC Biol. 2016, 14, 65. [Google Scholar] [CrossRef] [Green Version]
- Cordes, T.; Lucas, A.; Divakaruni, A.S.; Murphy, A.N.; Cabrales, P.; Metallo, C.M. Itaconate Modulates Tricarboxylic Acid and Redox Metabolism to Mitigate Reperfusion Injury. Mol. Metab. 2020, 32, 122–135. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Liu, Y.; Peterson, D.A.; Kimura, H.; Schubert, D. Mechanism of Cellular 3-(4,5-Dimethylthiazol-2-Yl)-2,5-Diphenyltetrazolium Bromide (MTT) Reduction. J. Neurochem. 1997, 69, 581–593. [Google Scholar] [CrossRef]
- Diemert, S.; Dolga, A.M.; Tobaben, S.; Grohm, J.; Pfeifer, S.; Oexler, E.; Culmsee, C. Impedance Measurement for Real Time Detection of Neuronal Cell Death. J. Neurosci. Methods 2012, 203, 69–77. [Google Scholar] [CrossRef]
- Kim, J.-H.; Lewin, T.M.; Coleman, R.A. Expression and Characterization of Recombinant Rat Acyl-CoA Synthetases 1, 4, and 5: Selective Inhibition by Triacsin C and Thiazolidinediones. J. Biol. Chem. 2001, 276, 24667–24673. [Google Scholar] [CrossRef] [Green Version]
- Gale, E.A. Lessons from the Glitazones: A Story of Drug Development. Lancet 2001, 357, 1870–1875. [Google Scholar] [CrossRef]
- Lea, M.A.; Sura, M.; Desbordes, C. Inhibition of Cell Proliferation by Potential Peroxisome Proliferator-Activated Receptor (PPAR) Gamma Agonists and Antagonists. Anticancer Res. 2004, 24, 2765–2772. [Google Scholar]
- Kelso, G.F.; Porteous, C.M.; Coulter, C.V.; Hughes, G.; Porteous, W.K.; Ledgerwood, E.C.; Smith, R.A.J.; Murphy, M.P. Selective Targeting of a Redox-Active Ubiquinone to Mitochondria within Cells: Antioxidant and Antiapoptotic Properties. J. Biol. Chem. 2001, 276, 4588–4596. [Google Scholar] [CrossRef] [Green Version]
- Kelso, G.F.; Porteous, C.M.; Hughes, G.; Ledgerwood, E.C.; Gane, A.M.; Smith, R.A.J.; Murphy, M.P. Prevention of Mitochondrial Oxidative Damage Using Targeted Antioxidants. Ann. N. Y. Acad. Sci. 2002, 959, 263–274. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, R.L. Iron Accumulation, Glutathione Depletion, and Lipid Peroxidation Must Occur Simultaneously during Ferroptosis and Are Mutually Amplifying Events. Med. Hypotheses 2017, 101, 69–74. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Hu, F.; Feng, H.; Linkermann, A.; Min, W.; Stockwell, B.R. Determination of the Subcellular Localization and Mechanism of Action of Ferrostatins in Suppressing Ferroptosis. ACS Chem. Biol. 2018, 13, 1013–1020. [Google Scholar] [CrossRef]
- Lei, P.; Bai, T.; Sun, Y. Mechanisms of Ferroptosis and Relations With Regulated Cell Death: A Review. Front. Physiol. 2019, 10, 139. [Google Scholar] [CrossRef] [Green Version]
- Grohm, J.; Plesnila, N.; Culmsee, C. Bid Mediates Fission, Membrane Permeabilization and Peri-Nuclear Accumulation of Mitochondria as a Prerequisite for Oxidative Neuronal Cell Death. Brain. Behav. Immun. 2010, 24, 831–838. [Google Scholar] [CrossRef]
- Grohm, J.; Kim, S.-W.; Mamrak, U.; Tobaben, S.; Cassidy-Stone, A.; Nunnari, J.; Plesnila, N.; Culmsee, C. Inhibition of Drp1 Provides Neuroprotection in Vitro and in Vivo. Cell Death Differ. 2012, 19, 1446–1458. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.; Schubert, D.; Maher, P. Oxytosis: A Novel form of Programmed Cell Death. Curr. Top. Med. Chem. 2001, 1, 497–506. [Google Scholar] [CrossRef]
- Culmsee, C.; Zhu, C.; Landshamer, S.; Becattini, B.; Wagner, E.; Pellecchia, M.; Blomgren, K.; Plesnila, N. Apoptosis-Inducing Factor Triggered by Poly(ADP-Ribose) Polymerase and Bid Mediates Neuronal Cell Death after Oxygen-Glucose Deprivation and Focal Cerebral Ischemia. J. Neurosci. 2005, 25, 10262–10272. [Google Scholar] [CrossRef] [Green Version]
- Barho, M.T.; Oppermann, S.; Schrader, F.C.; Degenhardt, I.; Elsässer, K.; Wegscheid-Gerlach, C.; Culmsee, C.; Schlitzer, M. N-Acyl Derivatives of 4-Phenoxyaniline as Neuroprotective Agents. ChemMedChem 2014, 9, 2260–2273. [Google Scholar] [CrossRef]
- Rehklau, K.; Hoffmann, L.; Gurniak, C.B.; Ott, M.; Witke, W.; Scorrano, L.; Culmsee, C.; Rust, M.B. Cofilin1-Dependent Actin Dynamics Control DRP1-Mediated Mitochondrial Fission. Cell Death Dis. 2017, 8, e3063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, Z.; Tian, W.; Ye, Y.; Gu, W.; Bao, Z.; Xu, L.; Sun, G.; Li, C.; Tu, Y.; Chao, H.; et al. Hsp90 Induces Acsl4-Dependent Glioma Ferroptosis via Dephosphorylating Ser637 at Drp1. Cell Death Dis. 2022, 13, 1–14. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merkel, M.; Goebel, B.; Boll, M.; Adhikari, A.; Maurer, V.; Steinhilber, D.; Culmsee, C. Mitochondrial Reactive Oxygen Species Formation Determines ACSL4/LPCAT2-Mediated Ferroptosis. Antioxidants 2023, 12, 1590. https://doi.org/10.3390/antiox12081590
Merkel M, Goebel B, Boll M, Adhikari A, Maurer V, Steinhilber D, Culmsee C. Mitochondrial Reactive Oxygen Species Formation Determines ACSL4/LPCAT2-Mediated Ferroptosis. Antioxidants. 2023; 12(8):1590. https://doi.org/10.3390/antiox12081590
Chicago/Turabian StyleMerkel, Melanie, Bjarne Goebel, Moritz Boll, Aasha Adhikari, Viktoria Maurer, Dieter Steinhilber, and Carsten Culmsee. 2023. "Mitochondrial Reactive Oxygen Species Formation Determines ACSL4/LPCAT2-Mediated Ferroptosis" Antioxidants 12, no. 8: 1590. https://doi.org/10.3390/antiox12081590