
Oxidative Stress in Liver Pathophysiology and Disease


 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
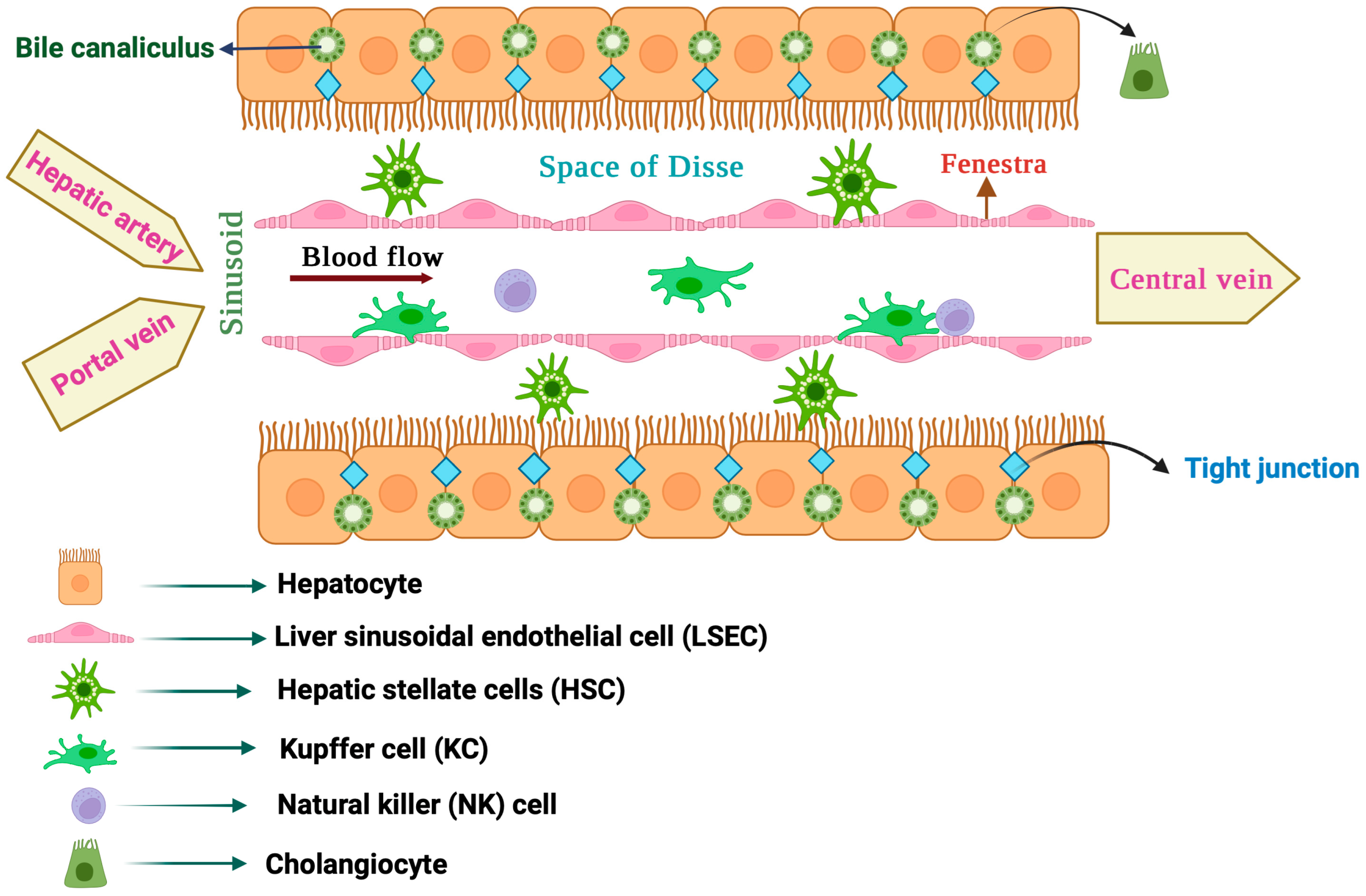
:1. Major Types of Liver Cells and Their Role in Liver Functions
1.1. Hepatocytes
1.2. Cholangiocytes
1.3. Liver Sinusoidal Endothelial Cells (LSECs)
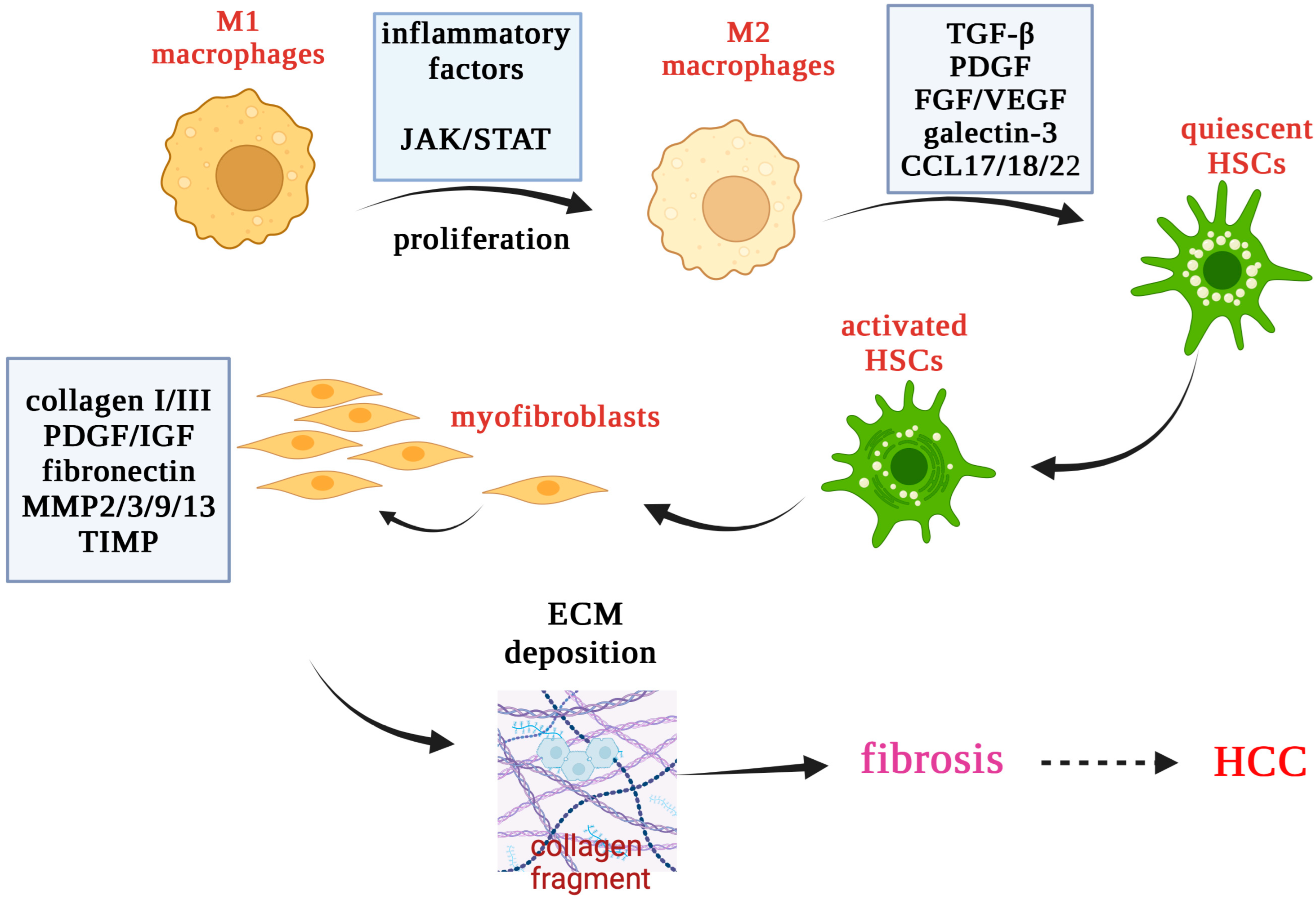
1.4. Hepatic Stellate Cells (HSCs)
1.5. Liver Macrophages
1.6. Liver Natural Killer (NK) Cells
2. Oxidative Stress in the Liver
3. Antioxidant Defense Mechanisms in the Liver
4. The Impact of Oxidative Stress on Liver Cells
5. Role of NOX Enzymes in Oxidative Damage to Liver Cells
6. Oxidative Stress in Drug-Induced Liver Injury
7. Oxidative Stress in Chronic Viral Hepatitis
8. Oxidative Stress in Fatty Liver Disease
9. Oxidative Stress in Genetic Liver Disorders
10. Oxidative Stress in Liver Fibrosis
11. Oxidative Stress in Hepatocellular Carcinoma
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sibulesky, L. Normal liver anatomy. Clin. Liver Dis. 2013, 2, S1–S3. [Google Scholar] [CrossRef] [PubMed]
- Vernon, H.; Wehrle, C.J.; Alia, V.S.K.; Kasi, A. Anatomy, Abdomen and Pelvis: Liver; StatPearls: Treasure Island, FL, USA, 2023. [Google Scholar]
- Werner, M.; Driftmann, S.; Kleinehr, K.; Kaiser, G.M.; Mathe, Z.; Treckmann, J.W.; Paul, A.; Skibbe, K.; Timm, J.; Canbay, A.; et al. All-In-One: Advanced preparation of Human Parenchymal and Non-Parenchymal Liver Cells. PLoS ONE 2015, 10, e0138655. [Google Scholar] [CrossRef]
- Seo, W.; Jeong, W.I. Hepatic non-parenchymal cells: Master regulators of alcoholic liver disease? World J. Gastroenterol. 2016, 22, 1348–1356. [Google Scholar] [CrossRef] [PubMed]
- Schulze, R.J.; Schott, M.B.; Casey, C.A.; Tuma, P.L.; McNiven, M.A. The cell biology of the hepatocyte: A membrane trafficking machine. J. Cell Biol. 2019, 218, 2096–2112. [Google Scholar] [CrossRef]
- Cocciolillo, S.; Sebastiani, G.; Blostein, M.; Pantopoulos, K. Liver Hormones. In Hormonal Signaling in Biology and Medicine; Litwack, G., Ed.; Academic Press: Cambridge, MA, USA, 2020; pp. 425–444. [Google Scholar]
- Boyer, J.L. Bile formation and secretion. Compr. Physiol. 2013, 3, 1035–1078. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Xu, M.J.; Gao, B. Hepatocytes: A key cell type for innate immunity. Cell Mol. Immunol. 2016, 13, 301–315. [Google Scholar] [CrossRef]
- Lorente, S.; Hautefeuille, M.; Sanchez-Cedillo, A. The liver, a functionalized vascular structure. Sci. Rep. 2020, 10, 16194. [Google Scholar] [CrossRef]
- Ehrlich, A.; Duche, D.; Ouedraogo, G.; Nahmias, Y. Challenges and Opportunities in the Design of Liver-on-Chip Microdevices. Annu. Rev. Biomed. Eng. 2019, 21, 219–239. [Google Scholar] [CrossRef]
- Banales, J.M.; Huebert, R.C.; Karlsen, T.; Strazzabosco, M.; LaRusso, N.F.; Gores, G.J. Cholangiocyte pathobiology. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 269–281. [Google Scholar] [CrossRef]
- Gracia-Sancho, J.; Caparros, E.; Fernandez-Iglesias, A.; Frances, R. Role of liver sinusoidal endothelial cells in liver diseases. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 411–431. [Google Scholar] [CrossRef]
- Poisson, J.; Lemoinne, S.; Boulanger, C.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P.E. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef] [PubMed]
- Shetty, S.; Lalor, P.F.; Adams, D.H. Liver sinusoidal endothelial cells—Gatekeepers of hepatic immunity. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, P.; Wang, S.; Friedman, S.L. The Power of Plasticity-Metabolic Regulation of Hepatic Stellate Cells. Cell Metab. 2021, 33, 242–257. [Google Scholar] [CrossRef] [PubMed]
- Kamm, D.R.; McCommis, K.S. Hepatic stellate cells in physiology and pathology. J. Physiol. 2022, 600, 1825–1837. [Google Scholar] [CrossRef] [PubMed]
- Kitto, L.J.; Henderson, N.C. Hepatic Stellate Cell Regulation of Liver Regeneration and Repair. Hepatol. Commun. 2021, 5, 358–370. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Woltman, A.M.; Boonstra, A.; Naito, M.; Leenen, P.J.M. Kupffer Cells in Health and Disease. In Macrophages: Biology and Role in the Pathology of Diseases; Biswas, S.K., Mantovani, A., Eds.; Springer: Cham, Switzerland, 2014; pp. 217–247. [Google Scholar]
- Korolnek, T.; Hamza, I. Macrophages and iron trafficking at the birth and death of red cells. Blood 2015, 125, 2893–2897. [Google Scholar] [CrossRef]
- Guilliams, M.; Bonnardel, J.; Haest, B.; Vanderborght, B.; Wagner, C.; Remmerie, A.; Bujko, A.; Martens, L.; Thone, T.; Browaeys, R.; et al. Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell 2022, 185, 379–396.e38. [Google Scholar] [CrossRef]
- Guilliams, M.; Scott, C.L. Liver macrophages in health and disease. Immunity 2022, 55, 1515–1529. [Google Scholar] [CrossRef]
- Liaskou, E.; Wilson, D.V.; Oo, Y.H. Innate immune cells in liver inflammation. Mediators Inflamm. 2012, 2012, 949157. [Google Scholar] [CrossRef]
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron homeostasis and oxidative stress: An intimate relationship. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118535. [Google Scholar] [CrossRef] [PubMed]
- Bayir, H.; Anthonymuthu, T.S.; Tyurina, Y.Y.; Patel, S.J.; Amoscato, A.A.; Lamade, A.M.; Yang, Q.; Vladimirov, G.K.; Philpott, C.C.; Kagan, V.E. Achieving Life through Death: Redox Biology of Lipid Peroxidation in Ferroptosis. Cell Chem. Biol. 2020, 27, 387–408. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative stress: Oxidants and antioxidants. Exp. Physiol. 1997, 82, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Martinez, M.C.; Andriantsitohaina, R. Reactive nitrogen species: Molecular mechanisms and potential significance in health and disease. Antioxid. Redox Signal. 2009, 11, 669–702. [Google Scholar] [CrossRef]
- Jaeschke, H.; Ramachandran, A. Reactive oxygen species in the normal and acutely injured liver. J. Hepatol. 2011, 55, 227–228. [Google Scholar] [CrossRef]
- Paik, Y.H.; Kim, J.; Aoyama, T.; De Minicis, S.; Bataller, R.; Brenner, D.A. Role of NADPH oxidases in liver fibrosis. Antioxid. Redox Signal. 2014, 20, 2854–2872. [Google Scholar] [CrossRef]
- Andueza, A.; Garde, N.; Garcia-Garzon, A.; Ansorena, E.; Lopez-Zabalza, M.J.; Iraburu, M.J.; Zalba, G.; Martinez-Irujo, J.J. NADPH oxidase 5 promotes proliferation and fibrosis in human hepatic stellate cells. Free Radic. Biol. Med. 2018, 126, 15–26. [Google Scholar] [CrossRef]
- Martyn, K.D.; Frederick, L.M.; von Loehneysen, K.; Dinauer, M.C.; Knaus, U.G. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell. Signal. 2006, 18, 69–82. [Google Scholar] [CrossRef]
- Halliwell, B. Antioxidant defence mechanisms: From the beginning to the end (of the beginning). Free Radic. Res. 1999, 31, 261–272. [Google Scholar] [CrossRef]
- Casas-Grajales, S.; Muriel, P. Antioxidants in liver health. World J. Gastrointest. Pharmacol. Ther. 2015, 6, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.M.; Lin, P.H.; Yao, Q.; Chen, C. Chemical and molecular mechanisms of antioxidants: Experimental approaches and model systems. J. Cell. Mol. Med. 2010, 14, 840–860. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Galicia-Moreno, M.; Lucano-Landeros, S.; Monroy-Ramirez, H.C.; Silva-Gomez, J.; Gutierrez-Cuevas, J.; Santos, A.; Armendariz-Borunda, J. Roles of Nrf2 in Liver Diseases: Molecular, Pharmacological, and Epigenetic Aspects. Antioxidants 2020, 9, 980. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Tan, H.Y.; Wang, N.; Zhang, Z.J.; Lao, L.; Wong, C.W.; Feng, Y. The Role of Oxidative Stress and Antioxidants in Liver Diseases. Int. J. Mol. Sci. 2015, 16, 26087–26124. [Google Scholar] [CrossRef]
- An, P.; Wei, L.L.; Zhao, S.; Sverdlov, D.Y.; Vaid, K.A.; Miyamoto, M.; Kuramitsu, K.; Lai, M.; Popov, Y.V. Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis. Nat. Commun. 2020, 11, 2362. [Google Scholar] [CrossRef]
- Leveille, M.; Estall, J.L. Mitochondrial Dysfunction in the Transition from NASH to HCC. Metabolites 2019, 9, 233. [Google Scholar] [CrossRef]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef]
- Zeng, X.; Yang, J.; Hu, O.; Huang, J.; Ran, L.; Chen, M.; Zhang, Y.; Zhou, X.; Zhu, J.; Zhang, Q.; et al. Dihydromyricetin Ameliorates Nonalcoholic Fatty Liver Disease by Improving Mitochondrial Respiratory Capacity and Redox Homeostasis Through Modulation of SIRT3 Signaling. Antioxid. Redox Signal. 2019, 30, 163–183. [Google Scholar] [CrossRef]
- Allameh, A.; Hüttmann, N.; Charlebois, E.; Katsarou, A.; Gu, W.; Gkouvatsos, K.; Pasini, E.; Bhat, M.; Minic, Z.; Berezovski, M.; et al. Hemojuvelin deficiency promotes liver mitochondrial dysfunction and predisposes mice to hepatocellular carcinoma. Commun. Biol. 2022, 5, 153. [Google Scholar] [CrossRef]
- Hara, Y.; Yanatori, I.; Tanaka, A.; Kishi, F.; Lemasters, J.J.; Nishina, S.; Sasaki, K.; Hino, K. Iron loss triggers mitophagy through induction of mitochondrial ferritin. EMBO Rep. 2020, 21, e50202. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Miyajima, A. Liver regeneration and fibrosis after inflammation. Inflamm. Regen. 2016, 36, 19. [Google Scholar] [CrossRef] [PubMed]
- Ermak, G.; Davies, K.J. Calcium and oxidative stress: From cell signaling to cell death. Mol. Immunol. 2002, 38, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Malhi, H.; Gores, G.J.; Lemasters, J.J. Apoptosis and necrosis in the liver: A tale of two deaths? Hepatology 2006, 43, S31–S44. [Google Scholar] [CrossRef]
- Basiglio, C.L.; Crocenzi, F.A.; Sanchez Pozzi, E.J.; Roma, M.G. Oxidative Stress and Localization Status of Hepatocellular Transporters: Impact on Bile Secretion and Role of Signaling Pathways. Antioxid. Redox Signal. 2021, 35, 808–831. [Google Scholar] [CrossRef]
- Gandhi, C.R. Oxidative Stress and Hepatic Stellate Cells: A Paradoxical Relationship. Trends Cell Mol. Biol. 2012, 7, 1–10. [Google Scholar]
- Ramos-Tovar, E.; Muriel, P. Molecular Mechanisms That Link Oxidative Stress, Inflammation, and Fibrosis in the Liver. Antioxidants 2020, 9, 1279. [Google Scholar] [CrossRef]
- Liang, S.; Kisseleva, T.; Brenner, D.A. The Role of NADPH Oxidases (NOXs) in Liver Fibrosis and the Activation of Myofibroblasts. Front. Physiol. 2016, 7, 17. [Google Scholar] [CrossRef]
- Kolios, G.; Valatas, V.; Kouroumalis, E. Role of Kupffer cells in the pathogenesis of liver disease. World J. Gastroenterol. 2006, 12, 7413–7420. [Google Scholar] [CrossRef]
- Luckey, S.W.; Petersen, D.R. Activation of Kupffer cells during the course of carbon tetrachloride-induced liver injury and fibrosis in rats. Exp. Mol. Pathol. 2001, 71, 226–240. [Google Scholar] [CrossRef]
- Arthur, M.J.; Bentley, I.S.; Tanner, A.R.; Saunders, P.K.; Millward-Sadler, G.H.; Wright, R. Oxygen-derived free radicals promote hepatic injury in the rat. Gastroenterology 1985, 89, 1114–1122. [Google Scholar] [CrossRef] [PubMed]
- Teufelhofer, O.; Parzefall, W.; Kainzbauer, E.; Ferk, F.; Freiler, C.; Knasmuller, S.; Elbling, L.; Thurman, R.; Schulte-Hermann, R. Superoxide generation from Kupffer cells contributes to hepatocarcinogenesis: Studies on NADPH oxidase knockout mice. Carcinogenesis 2005, 26, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Afford, S.C.; Randhawa, S.; Eliopoulos, A.G.; Hubscher, S.G.; Young, L.S.; Adams, D.H. CD40 activation induces apoptosis in cultured human hepatocytes via induction of cell surface fas ligand expression and amplifies fas-mediated hepatocyte death during allograft rejection. J. Exp. Med. 1999, 189, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Mandili, G.; Alchera, E.; Merlin, S.; Imarisio, C.; Chandrashekar, B.R.; Riganti, C.; Bianchi, A.; Novelli, F.; Follenzi, A.; Carini, R. Mouse hepatocytes and LSEC proteome reveal novel mechanisms of ischemia/reperfusion damage and protection by A2aR stimulation. J. Hepatol. 2015, 62, 573–580. [Google Scholar] [CrossRef]
- Hamer, I.; Wattiaux, R.; Wattiaux-De Coninck, S. Deleterious effects of xanthine oxidase on rat liver endothelial cells after ischemia/reperfusion. Biochim. Biophys. Acta 1995, 1269, 145–152. [Google Scholar] [CrossRef]
- Cogger, V.C.; Muller, M.; Fraser, R.; McLean, A.J.; Khan, J.; Le Couteur, D.G. The effects of oxidative stress on the liver sieve. J. Hepatol. 2004, 41, 370–376. [Google Scholar] [CrossRef]
- Zapotoczny, B.; Braet, F.; Kus, E.; Ginda-Makela, K.; Klejevskaja, B.; Campagna, R.; Chlopicki, S.; Szymonski, M. Actin-spectrin scaffold supports open fenestrae in liver sinusoidal endothelial cells. Traffic 2019, 20, 932–942. [Google Scholar] [CrossRef]
- Ni, Y.; Li, J.M.; Liu, M.K.; Zhang, T.T.; Wang, D.P.; Zhou, W.H.; Hu, L.Z.; Lv, W.L. Pathological process of liver sinusoidal endothelial cells in liver diseases. World J. Gastroenterol. 2017, 23, 7666–7677. [Google Scholar] [CrossRef]
- Ruart, M.; Chavarria, L.; Camprecios, G.; Suarez-Herrera, N.; Montironi, C.; Guixe-Muntet, S.; Bosch, J.; Friedman, S.L.; Garcia-Pagan, J.C.; Hernandez-Gea, V. Impaired endothelial autophagy promotes liver fibrosis by aggravating the oxidative stress response during acute liver injury. J. Hepatol. 2019, 70, 458–469. [Google Scholar] [CrossRef]
- Vairappan, B. Endothelial dysfunction in cirrhosis: Role of inflammation and oxidative stress. World J. Hepatol. 2015, 7, 443–459. [Google Scholar] [CrossRef]
- Nasiri-Ansari, N.; Androutsakos, T.; Flessa, C.M.; Kyrou, I.; Siasos, G.; Randeva, H.S.; Kassi, E.; Papavassiliou, A.G. Endothelial Cell Dysfunction and Nonalcoholic Fatty Liver Disease (NAFLD): A Concise Review. Cells 2022, 11, 2511. [Google Scholar] [CrossRef] [PubMed]
- Iwakiri, Y. Endothelial dysfunction in the regulation of cirrhosis and portal hypertension. Liver Int. 2012, 32, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Chao, X.; Williams, J.; Fulte, S.; Li, T.; Yang, L.; Ding, W.X. Autophagy in liver diseases: A review. Mol. Aspects Med. 2021, 82, 100973. [Google Scholar] [CrossRef] [PubMed]
- Hammoutene, A.; Biquard, L.; Lasselin, J.; Kheloufi, M.; Tanguy, M.; Vion, A.C.; Merian, J.; Colnot, N.; Loyer, X.; Tedgui, A.; et al. A defect in endothelial autophagy occurs in patients with non-alcoholic steatohepatitis and promotes inflammation and fibrosis. J. Hepatol. 2020, 72, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Meadows, V.; Baiocchi, L.; Kundu, D.; Sato, K.; Fuentes, Y.; Wu, C.; Chakraborty, S.; Glaser, S.; Alpini, G.; Kennedy, L.; et al. Biliary Epithelial Senescence in Liver Disease: There Will Be SASP. Front. Mol. Biosci. 2021, 8, 803098. [Google Scholar] [CrossRef]
- Ostrycharz, E.; Wasik, U.; Kempinska-Podhorodecka, A.; Banales, J.M.; Milkiewicz, P.; Milkiewicz, M. Melatonin Protects Cholangiocytes from Oxidative Stress-Induced Proapoptotic and Proinflammatory Stimuli via miR-132 and miR-34. Int. J. Mol. Sci. 2020, 21, 9667. [Google Scholar] [CrossRef]
- Nakamura, K.; Matsunaga, K. Susceptibility of natural killer (NK) cells to reactive oxygen species (ROS) and their restoration by the mimics of superoxide dismutase (SOD). Cancer Biother. Radiopharm. 1998, 13, 275–290. [Google Scholar] [CrossRef]
- Kotsafti, A.; Scarpa, M.; Castagliuolo, I.; Scarpa, M. Reactive Oxygen Species and Antitumor Immunity-From Surveillance to Evasion. Cancers 2020, 12, 1748. [Google Scholar] [CrossRef]
- Jiang, J.X.; Torok, N.J. NADPH Oxidases in Chronic Liver Diseases. Adv. Hepatol. 2014, 2014, 742931. [Google Scholar] [CrossRef]
- Paik, Y.H.; Iwaisako, K.; Seki, E.; Inokuchi, S.; Schnabl, B.; Osterreicher, C.H.; Kisseleva, T.; Brenner, D.A. The nicotinamide adenine dinucleotide phosphate oxidase (NOX) homologues NOX1 and NOX2/gp91(phox) mediate hepatic fibrosis in mice. Hepatology 2011, 53, 1730–1741. [Google Scholar] [CrossRef]
- Aoyama, T.; Paik, Y.H.; Watanabe, S.; Laleu, B.; Gaggini, F.; Fioraso-Cartier, L.; Molango, S.; Heitz, F.; Merlot, C.; Szyndralewiez, C.; et al. Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent. Hepatology 2012, 56, 2316–2327. [Google Scholar] [CrossRef]
- Lan, T.; Kisseleva, T.; Brenner, D.A. Deficiency of NOX1 or NOX4 Prevents Liver Inflammation and Fibrosis in Mice through Inhibition of Hepatic Stellate Cell Activation. PLoS ONE 2015, 10, e0129743. [Google Scholar] [CrossRef] [PubMed]
- Herranz-Iturbide, M.; Penuelas-Haro, I.; Espinosa-Sotelo, R.; Bertran, E.; Fabregat, I. The TGF-beta/NADPH Oxidases Axis in the Regulation of Liver Cell Biology in Health and Disease. Cells 2021, 10, 2312. [Google Scholar] [CrossRef]
- Fabregat, I.; Moreno-Caceres, J.; Sanchez, A.; Dooley, S.; Dewidar, B.; Giannelli, G.; Ten Dijke, P.; Consortium, I.-L. TGF-beta signalling and liver disease. FEBS J. 2016, 283, 2219–2232. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.M.; Desai, L.P. Reciprocal regulation of TGF-beta and reactive oxygen species: A perverse cycle for fibrosis. Redox Biol. 2015, 6, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Mehr, R.N.M.; Kheirollah, A.; Seif, F.; Dayati, P.M.; Babaahmadi-Rezaei, H. Reactive Oxygen Species and p38MAPK Have a Role in the Smad2 Linker Region Phosphorylation Induced by TGF-beta. Iran. J. Med. Sci. 2018, 43, 401–408. [Google Scholar]
- Carmona-Cuenca, I.; Roncero, C.; Sancho, P.; Caja, L.; Fausto, N.; Fernandez, M.; Fabregat, I. Upregulation of the NADPH oxidase NOX4 by TGF-beta in hepatocytes is required for its pro-apoptotic activity. J. Hepatol. 2008, 49, 965–976. [Google Scholar] [CrossRef]
- Jiang, J.X.; Chen, X.; Serizawa, N.; Szyndralewiez, C.; Page, P.; Schroder, K.; Brandes, R.P.; Devaraj, S.; Torok, N.J. Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831, a novel NOX4/NOX1 inhibitor in vivo. Free Radic. Biol. Med. 2012, 53, 289–296. [Google Scholar] [CrossRef]
- Bettaieb, A.; Jiang, J.X.; Sasaki, Y.; Chao, T.I.; Kiss, Z.; Chen, X.; Tian, J.; Katsuyama, M.; Yabe-Nishimura, C.; Xi, Y.; et al. Hepatocyte Nicotinamide Adenine Dinucleotide Phosphate Reduced Oxidase 4 Regulates Stress Signaling, Fibrosis, and Insulin Sensitivity During Development of Steatohepatitis in Mice. Gastroenterology 2015, 149, 468–480. [Google Scholar] [CrossRef]
- Sancho, P.; Mainez, J.; Crosas-Molist, E.; Roncero, C.; Fernandez-Rodriguez, C.M.; Pinedo, F.; Huber, H.; Eferl, R.; Mikulits, W.; Fabregat, I. NADPH oxidase NOX4 mediates stellate cell activation and hepatocyte cell death during liver fibrosis development. PLoS ONE 2012, 7, e45285. [Google Scholar] [CrossRef]
- Cui, W.; Matsuno, K.; Iwata, K.; Ibi, M.; Matsumoto, M.; Zhang, J.; Zhu, K.; Katsuyama, M.; Torok, N.J.; Yabe-Nishimura, C. NOX1/nicotinamide adenine dinucleotide phosphate, reduced form (NADPH) oxidase promotes proliferation of stellate cells and aggravates liver fibrosis induced by bile duct ligation. Hepatology 2011, 54, 949–958. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.X.; Venugopal, S.; Serizawa, N.; Chen, X.; Scott, F.; Li, Y.; Adamson, R.; Devaraj, S.; Shah, V.; Gershwin, M.E.; et al. Reduced nicotinamide adenine dinucleotide phosphate oxidase 2 plays a key role in stellate cell activation and liver fibrogenesis in vivo. Gastroenterology 2010, 139, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Lee, G.; Heo, S.Y.; Roh, Y.S. Oxidative Stress Is a Key Modulator in the Development of Nonalcoholic Fatty Liver Disease. Antioxidants 2021, 11, 91. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Nelson, L.J.; Gomez Del Moral, M.; Martinez-Naves, E.; Cubero, F.J. Dissecting the molecular pathophysiology of drug-induced liver injury. World J. Gastroenterol. 2018, 24, 1373–1385. [Google Scholar] [CrossRef]
- Villanueva-Paz, M.; Moran, L.; Lopez-Alcantara, N.; Freixo, C.; Andrade, R.J.; Lucena, M.I.; Cubero, F.J. Oxidative Stress in Drug-Induced Liver Injury (DILI): From Mechanisms to Biomarkers for Use in Clinical Practice. Antioxidants 2021, 10, 390. [Google Scholar] [CrossRef]
- Athersuch, T.J.; Antoine, D.J.; Boobis, A.R.; Coen, M.; Daly, A.K.; Possamai, L.; Nicholson, J.K.; Wilson, I.D. Paracetamol metabolism, hepatotoxicity, biomarkers and therapeutic interventions: A perspective. Toxicol. Res. 2018, 7, 347–357. [Google Scholar] [CrossRef]
- Mehta, P.; Reddivari, A.K.R. Hepatitis; StatPearls: Treasure Island, FL, USA, 2023. [Google Scholar]
- Castaneda, D.; Gonzalez, A.J.; Alomari, M.; Tandon, K.; Zervos, X.B. From hepatitis A to E: A critical review of viral hepatitis. World J. Gastroenterol. 2021, 27, 1691–1715. [Google Scholar] [CrossRef]
- Gupta, E.; Ballani, N.; Kumar, M.; Sarin, S.K. Role of non-hepatotropic viruses in acute sporadic viral hepatitis and acute-on-chronic liver failure in adults. Indian J. Gastroenterol. 2015, 34, 448–452. [Google Scholar] [CrossRef]
- Roehlen, N.; Crouchet, E.; Baumert, T.F. Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives. Cells 2020, 9, 875. [Google Scholar] [CrossRef]
- Zamor, P.J.; deLemos, A.S.; Russo, M.W. Viral hepatitis and hepatocellular carcinoma: Etiology and management. J. Gastrointest. Oncol. 2017, 8, 229–242. [Google Scholar] [CrossRef]
- Sebastiani, G. Oxidative stress in liver diseases. In Principles of Free Radical Biomedicine; Schipper, K.P.H.M., Ed.; Nova Science Publishers, Inc.: New York, NY, USA, 2012; Volume 3, pp. 65–105. [Google Scholar]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Tyurina, D.A.; Ivanova, O.N.; Kochetkov, S.N.; Bartosch, B.; Isaguliants, M.G. Oxidative stress, a trigger of hepatitis C and B virus-induced liver carcinogenesis. Oncotarget 2017, 8, 3895–3932. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.I.; Hwang, J.M.; Im, J.H.; Lee, Y.I.; Kim, N.S.; Kim, D.G.; Yu, D.Y.; Moon, H.B.; Park, S.K. Human hepatitis B virus-X protein alters mitochondrial function and physiology in human liver cells. J. Biol. Chem. 2004, 279, 15460–15471. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Liu, Y.; Feng, X.; Wang, X.; Wu, M.; Gong, L.; Shu, B.; Lu, Q.; Dong, J. HBx acts as an oncogene and promotes the invasion and metastasis of hepatocellular carcinoma both in vivo and vitro. Dig. Liver Dis. 2021, 53, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yan, Q.; Gong, L.; Xu, H.; Liu, B.; Fang, X.; Yu, D.; Li, L.; Wei, T.; Wang, Y.; et al. C-terminal truncated HBx initiates hepatocarcinogenesis by downregulating TXNIP and reprogramming glucose metabolism. Oncogene 2021, 40, 1147–1161. [Google Scholar] [CrossRef]
- Korenaga, M.; Wang, T.; Li, Y.; Showalter, L.A.; Chan, T.; Sun, J.; Weinman, S.A. Hepatitis C virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ROS) production. J. Biol. Chem. 2005, 280, 37481–37488. [Google Scholar] [CrossRef]
- Okuda, M.; Li, K.; Beard, M.R.; Showalter, L.A.; Scholle, F.; Lemon, S.M.; Weinman, S.A. Mitochondrial injury, oxidative stress, and antioxidant gene expression are induced by hepatitis C virus core protein. Gastroenterology 2002, 122, 366–375. [Google Scholar] [CrossRef]
- Moriya, K.; Fujie, H.; Shintani, Y.; Yotsuyanagi, H.; Tsutsumi, T.; Ishibashi, K.; Matsuura, Y.; Kimura, S.; Miyamura, T.; Koike, K. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med. 1998, 4, 1065–1067. [Google Scholar] [CrossRef]
- Moriya, K.; Nakagawa, K.; Santa, T.; Shintani, Y.; Fujie, H.; Miyoshi, H.; Tsutsumi, T.; Miyazawa, T.; Ishibashi, K.; Horie, T.; et al. Oxidative stress in the absence of inflammation in a mouse model for hepatitis C virus-associated hepatocarcinogenesis. Cancer Res. 2001, 61, 4365–4370. [Google Scholar]
- Lerat, H.; Honda, M.; Beard, M.R.; Loesch, K.; Sun, J.; Yang, Y.; Okuda, M.; Gosert, R.; Xiao, S.Y.; Weinman, S.A.; et al. Steatosis and liver cancer in transgenic mice expressing the structural and nonstructural proteins of hepatitis C virus. Gastroenterology 2002, 122, 352–365. [Google Scholar] [CrossRef]
- Furutani, T.; Hino, K.; Okuda, M.; Gondo, T.; Nishina, S.; Kitase, A.; Korenaga, M.; Xiao, S.Y.; Weinman, S.A.; Lemon, S.M.; et al. Hepatic iron overload induces hepatocellular carcinoma in transgenic mice expressing the hepatitis C virus polyprotein. Gastroenterology 2006, 130, 2087–2098. [Google Scholar] [CrossRef]
- Idalsoaga, F.; Kulkarni, A.V.; Mousa, O.Y.; Arrese, M.; Arab, J.P. Non-alcoholic Fatty Liver Disease and Alcohol-Related Liver Disease: Two Intertwined Entities. Front. Med. 2020, 7, 448. [Google Scholar] [CrossRef] [PubMed]
- Koo, S.H. Nonalcoholic fatty liver disease: Molecular mechanisms for the hepatic steatosis. Clin. Mol. Hepatol. 2013, 19, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, J.; Viggiano, T.R.; McGill, D.B.; Oh, B.J. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin. Proc. 1980, 55, 434–438. [Google Scholar] [PubMed]
- Zhang, P.; Wang, W.; Mao, M.; Gao, R.; Shi, W.; Li, D.; Calderone, R.; Sui, B.; Tian, X.; Meng, X. Similarities and Differences: A Comparative Review of the Molecular Mechanisms and Effectors of NAFLD and AFLD. Front. Physiol. 2021, 12, 710285. [Google Scholar] [CrossRef] [PubMed]
- Dulai, P.S.; Singh, S.; Patel, J.; Soni, M.; Prokop, L.J.; Younossi, Z.; Sebastiani, G.; Ekstedt, M.; Hagstrom, H.; Nasr, P.; et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta-analysis. Hepatology 2017, 65, 1557–1565. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J.; International Consensus, P. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P.; et al. A multi-society Delphi consensus statement on new fatty liver disease nomenclature. Hepatology 2023. [Google Scholar] [CrossRef]
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P.; et al. A multi-society Delphi consensus statement on new fatty liver disease nomenclature. J. Hepatol. 2023. [Google Scholar] [CrossRef]
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P.; et al. A multi-society Delphi consensus statement on new fatty liver disease nomenclature. Ann. Hepatol. 2023, 101133. [Google Scholar] [CrossRef]
- Campagna, R.; Vignini, A. NAD(+) Homeostasis and NAD(+)-Consuming Enzymes: Implications for Vascular Health. Antioxidants 2023, 12, 376. [Google Scholar] [CrossRef]
- Song, Q.; Chen, Y.; Wang, J.; Hao, L.; Huang, C.; Griffiths, A.; Sun, Z.; Zhou, Z.; Song, Z. ER stress-induced upregulation of NNMT contributes to alcohol-related fatty liver development. J. Hepatol. 2020, 73, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Ma, Y.; Lai, S.; Dou, X.; Li, S. NNMT aggravates hepatic steatosis, but alleviates liver injury in alcoholic liver disease. J. Hepatol. 2021, 74, 1248–1250. [Google Scholar] [CrossRef]
- Shin, J.H.; Park, C.W.; Yoon, G.; Hong, S.M.; Choi, K.Y. NNMT depletion contributes to liver cancer cell survival by enhancing autophagy under nutrient starvation. Oncogenesis 2018, 7, 58. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Zhou, X.; Wang, Y.; Huang, X.; Yang, J.; Zeng, J.; Li, G.; Xie, X.; Zhang, J. Nicotinamide N-methyltransferase inhibits autophagy induced by oxidative stress through suppressing the AMPK pathway in breast cancer cells. Cancer Cell Int. 2020, 20, 191. [Google Scholar] [CrossRef] [PubMed]
- Campagna, R.; Mateuszuk, L.; Wojnar-Lason, K.; Kaczara, P.; Tworzydlo, A.; Kij, A.; Bujok, R.; Mlynarski, J.; Wang, Y.; Sartini, D.; et al. Nicotinamide N-methyltransferase in endothelium protects against oxidant stress-induced endothelial injury. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 119082. [Google Scholar] [CrossRef]
- Rolo, A.P.; Teodoro, J.S.; Palmeira, C.M. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2012, 52, 59–69. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Seki, S.; Kitada, T.; Yamada, T.; Sakaguchi, H.; Nakatani, K.; Wakasa, K. In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases. J. Hepatol. 2002, 37, 56–62. [Google Scholar] [CrossRef]
- Seki, S.; Kitada, T.; Sakaguchi, H. Clinicopathological significance of oxidative cellular damage in non-alcoholic fatty liver diseases. Hepatol. Res. 2005, 33, 132–134. [Google Scholar] [CrossRef]
- Perez-Carreras, M.; Del Hoyo, P.; Martin, M.A.; Rubio, J.C.; Martin, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003, 38, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Campbell-Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Sebastiani, G.; Saeed, S.; Lebouche, B.; de Pokomandy, A.; Szabo, J.; Haraoui, L.P.; Routy, J.P.; Wong, P.; Deschenes, M.; Ghali, P.; et al. Vitamin E is an effective treatment for nonalcoholic steatohepatitis in HIV mono-infected patients. AIDS 2020, 34, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef]
- Lavine, J.E.; Schwimmer, J.B.; Van Natta, M.L.; Molleston, J.P.; Murray, K.F.; Rosenthal, P.; Abrams, S.H.; Scheimann, A.O.; Sanyal, A.J.; Chalasani, N.; et al. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: The TONIC randomized controlled trial. JAMA 2011, 305, 1659–1668. [Google Scholar] [CrossRef]
- Karedath, J.; Javed, H.; Ahsan Talpur, F.; Lal, B.; Kumari, A.; Kivan, H.; Anirudh Chunchu, V.; Hirani, S. Effect of Vitamin E on Clinical Outcomes in Patients with Non-alcoholic Fatty Liver Disease: A Meta-Analysis. Cureus 2022, 14, e32764. [Google Scholar] [CrossRef]
- Theodoridis, X.; Kalopitas, G.; Vadarlis, A.; Bakaloudi, D.R.; Gkiourtzis, N.; Dionysopoulos, G.; Karanika, E.; Tsekitsidi, E.; Chourdakis, M. Comparative efficacy of different treatment modalities in the management of pediatric non-alcoholic fatty liver disease: A systematic review and network meta-analysis. Pharmacol. Ther. 2022, 240, 108294. [Google Scholar] [CrossRef]
- Leclercq, I.A.; Farrell, G.C.; Field, J.; Bell, D.R.; Gonzalez, F.J.; Robertson, G.R. CYP2E1 and CYP4A as microsomal catalysts of lipid peroxides in murine nonalcoholic steatohepatitis. J. Clin. Investig. 2000, 105, 1067–1075. [Google Scholar] [CrossRef]
- Chowdhry, S.; Nazmy, M.H.; Meakin, P.J.; Dinkova-Kostova, A.T.; Walsh, S.V.; Tsujita, T.; Dillon, J.F.; Ashford, M.L.; Hayes, J.D. Loss of Nrf2 markedly exacerbates nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2010, 48, 357–371. [Google Scholar] [CrossRef]
- Handa, P.; Maliken, B.D.; Nelson, J.E.; Morgan-Stevenson, V.; Messner, D.J.; Dhillon, B.K.; Klintworth, H.M.; Beauchamp, M.; Yeh, M.M.; Elfers, C.T.; et al. Reduced adiponectin signaling due to weight gain results in nonalcoholic steatohepatitis through impaired mitochondrial biogenesis. Hepatology 2014, 60, 133–145. [Google Scholar] [CrossRef]
- Juarez-Fernandez, M.; Goikoetxea-Usandizaga, N.; Porras, D.; Garcia-Mediavilla, M.V.; Bravo, M.; Serrano-Macia, M.; Simon, J.; Delgado, T.C.; Lachiondo-Ortega, S.; Martinez-Florez, S.; et al. Enhanced mitochondrial activity reshapes a gut microbiota profile that delays NASH progression. Hepatology 2023, 77, 1654–1669. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Kuang, H.; Ansari, S.; Liu, T.; Gong, J.; Wang, S.; Zhao, X.Y.; Ji, Y.; Li, C.; Guo, L.; et al. Landscape of Intercellular Crosstalk in Healthy and NASH Liver Revealed by Single-Cell Secretome Gene Analysis. Mol. Cell 2019, 75, 644–660. [Google Scholar] [CrossRef] [PubMed]
- Daemen, S.; Gainullina, A.; Kalugotla, G.; He, L.; Chan, M.M.; Beals, J.W.; Liss, K.H.; Klein, S.; Feldstein, A.E.; Finck, B.N.; et al. Dynamic Shifts in the Composition of Resident and Recruited Macrophages Influence Tissue Remodeling in NASH. Cell Rep. 2021, 34, 108626. [Google Scholar] [CrossRef] [PubMed]
- Corradini, E.; Pietrangelo, A. Iron and steatohepatitis. J. Gastroenterol. Hepatol. 2012, 27 (Suppl. 2), 42–46. [Google Scholar] [CrossRef]
- Padda, R.S.; Gkouvatsos, K.; Guido, M.; Mui, J.; Vali, H.; Pantopoulos, K. A high-fat diet modulates iron metabolism but does not promote liver fibrosis in hemochromatotic Hjv−/− mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G251–G261. [Google Scholar] [CrossRef]
- Wagner, J.; Fillebeen, C.; Haliotis, T.; Charlebois, E.; Katsarou, A.; Mui, J.; Vali, H.; Pantopoulos, K. Mouse models of hereditary hemochromatosis do not develop early liver fibrosis in response to a high fat diet. PLoS ONE 2019, 14, e0221455. [Google Scholar] [CrossRef]
- Sebastiani, G.; Gkouvatsos, K.; Maffettone, C.; Busatto, G.; Guido, M.; Pantopoulos, K. Accelerated CCl4-Induced Liver Fibrosis in Hjv-/- Mice, Associated with an Oxidative Burst and Precocious Profibrogenic Gene Expression. PLoS ONE 2011, 6, e25138. [Google Scholar] [CrossRef]
- Handa, P.; Morgan-Stevenson, V.; Maliken, B.D.; Nelson, J.E.; Washington, S.; Westerman, M.; Yeh, M.M.; Kowdley, K.V. Iron overload results in hepatic oxidative stress, immune cell activation, and hepatocellular ballooning injury, leading to nonalcoholic steatohepatitis in genetically obese mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G117–G127. [Google Scholar] [CrossRef]
- Handa, P.; Thomas, S.; Morgan-Stevenson, V.; Maliken, B.D.; Gochanour, E.; Boukhar, S.; Yeh, M.M.; Kowdley, K.V. Iron alters macrophage polarization status and leads to steatohepatitis and fibrogenesis. J. Leukoc. Biol. 2019, 105, 1015–1026. [Google Scholar] [CrossRef]
- Gao, H.; Jin, Z.; Bandyopadhyay, G.; Wang, G.; Zhang, D.; Rocha, K.C.E.; Liu, X.; Zhao, H.; Kisseleva, T.; Brenner, D.A.; et al. Aberrant iron distribution via hepatocyte-stellate cell axis drives liver lipogenesis and fibrosis. Cell Metab. 2022, 34, 1201–1213. [Google Scholar] [CrossRef]
- Nelson, J.E.; Wilson, L.; Brunt, E.M.; Yeh, M.M.; Kleiner, D.E.; Unalp-Arida, A.; Kowdley, K.V.; Nonalcoholic Steatohepatitis Clinical Research, N. Relationship between the pattern of hepatic iron deposition and histological severity in nonalcoholic fatty liver disease. Hepatology 2011, 53, 448–457. [Google Scholar] [CrossRef]
- Adams, P.C.; Jeffrey, G.; Ryan, J. Haemochromatosis. Lancet 2023, 401, 1811–1821. [Google Scholar] [CrossRef] [PubMed]
- Houglum, K.; Ramm, G.A.; Crawford, D.H.; Witztum, J.L.; Powell, L.W.; Chojkier, M. Excess iron induces hepatic oxidative stress and transforming growth factor beta1 in genetic hemochromatosis. Hepatology 1997, 26, 605–610. [Google Scholar] [CrossRef]
- Hussain, S.P.; Raja, K.; Amstad, P.A.; Sawyer, M.; Trudel, L.J.; Wogan, G.N.; Hofseth, L.J.; Shields, P.G.; Billiar, T.R.; Trautwein, C.; et al. Increased p53 mutation load in nontumorous human liver of wilson disease and hemochromatosis: Oxyradical overload diseases. Proc. Natl. Acad. Sci. USA 2000, 97, 12770–12775. [Google Scholar] [CrossRef] [PubMed]
- Lebeau, A.; Frank, J.; Biesalski, H.K.; Weiss, G.; Srai, S.K.; Simpson, R.J.; McKie, A.T.; Bahram, S.; Gilfillan, S.; Schumann, K. Long-term sequelae of HFE deletion in C57BL/6 × 129/O1a mice, an animal model for hereditary haemochromatosis. Eur. J. Clin. Investig. 2002, 32, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Bloomer, S.A.; Brown, K.E. Iron-Induced Liver Injury: A Critical Reappraisal. Int. J. Mol. Sci. 2019, 20, 2132. [Google Scholar] [CrossRef]
- Czlonkowska, A.; Litwin, T.; Dusek, P.; Ferenci, P.; Lutsenko, S.; Medici, V.; Rybakowski, J.K.; Weiss, K.H.; Schilsky, M.L. Wilson disease. Nat. Rev. Dis. Primers 2018, 4, 21. [Google Scholar] [CrossRef]
- Nagasaka, H.; Inoue, I.; Inui, A.; Komatsu, H.; Sogo, T.; Murayama, K.; Murakami, T.; Yorifuji, T.; Asayama, K.; Katayama, S.; et al. Relationship between oxidative stress and antioxidant systems in the liver of patients with Wilson disease: Hepatic manifestation in Wilson disease as a consequence of augmented oxidative stress. Pediatr. Res. 2006, 60, 472–477. [Google Scholar] [CrossRef]
- Sanchez-Monteagudo, A.; Ripolles, E.; Berenguer, M.; Espinos, C. Wilson’s Disease: Facing the Challenge of Diagnosing a Rare Disease. Biomedicines 2021, 9, 1100. [Google Scholar] [CrossRef]
- Torres-Duran, M.; Lopez-Campos, J.L.; Barrecheguren, M.; Miravitlles, M.; Martinez-Delgado, B.; Castillo, S.; Escribano, A.; Baloira, A.; Navarro-Garcia, M.M.; Pellicer, D.; et al. Alpha-1 antitrypsin deficiency: Outstanding questions and future directions. Orphanet J. Rare Dis. 2018, 13, 114. [Google Scholar] [CrossRef]
- Karatas, E.; Bouchecareilh, M. Alpha 1-Antitrypsin Deficiency: A Disorder of Proteostasis-Mediated Protein Folding and Trafficking Pathways. Int. J. Mol. Sci. 2020, 21, 1493. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Fischer, H.P. Hereditary hemochromatosis, alpha-1-antitrypsin deficiency and Wilson’s disease. Pathogenesis, clinical findings and pathways to diagnosis. Pathologe 2008, 29, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Bouchecareilh, M. Alpha-1 Antitrypsin Deficiency-Mediated Liver Toxicity: Why Do Some Patients Do Poorly? What Do We Know So Far? Chronic Obstr. Pulm. Dis. 2020, 7, 172–181. [Google Scholar] [CrossRef]
- Marcus, N.Y.; Blomenkamp, K.; Ahmad, M.; Teckman, J.H. Oxidative stress contributes to liver damage in a murine model of alpha-1-antitrypsin deficiency. Exp. Biol. Med. 2012, 237, 1163–1172. [Google Scholar] [CrossRef] [PubMed]
- Acharya, P.; Chouhan, K.; Weiskirchen, S.; Weiskirchen, R. Cellular Mechanisms of Liver Fibrosis. Front. Pharmacol. 2021, 12, 671640. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Brenner, D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Diehl, A.M. Evidence for and against epithelial-to-mesenchymal transition in the liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G881–G890. [Google Scholar] [CrossRef]
- Arriazu, E.; Ruiz de Galarreta, M.; Cubero, F.J.; Varela-Rey, M.; Perez de Obanos, M.P.; Leung, T.M.; Lopategi, A.; Benedicto, A.; Abraham-Enachescu, I.; Nieto, N. Extracellular matrix and liver disease. Antioxid. Redox Signal. 2014, 21, 1078–1097. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, Y.; Sun, B. The Molecular Mechanisms of Liver Fibrosis and Its Potential Therapy in Application. Int. J. Mol. Sci. 2022, 23, 12572. [Google Scholar] [CrossRef]
- Shi, Y.; Massague, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Abreu, J.G.; Ketpura, N.I.; Reversade, B.; De Robertis, E.M. Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGF-beta. Nat. Cell Biol. 2002, 4, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.J.; Yu, J.P.; Li, D.; Huang, Y.H.; Chen, Z.X.; Wang, X.Z. Effects of cytokines on carbon tetrachloride-induced hepatic fibrogenesis in rats. World J. Gastroenterol. 2004, 10, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Lipson, K.E.; Wong, C.; Teng, Y.; Spong, S. CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenes. Tissue Repair 2012, 5, S24. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Wang, K.; Aoyama, T.; Grivennikov, S.I.; Paik, Y.; Scholten, D.; Cong, M.; Iwaisako, K.; Liu, X.; Zhang, M.; et al. Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology 2012, 143, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Meyer, C.; Muller, A.; Herweck, F.; Li, Q.; Mullenbach, R.; Mertens, P.R.; Dooley, S.; Weng, H.L. IL-13 induces connective tissue growth factor in rat hepatic stellate cells via TGF-beta-independent Smad signaling. J. Immunol. 2011, 187, 2814–2823. [Google Scholar] [CrossRef]
- Tan, Z.; Liu, Q.; Jiang, R.; Lv, L.; Shoto, S.S.; Maillet, I.; Quesniaux, V.; Tang, J.; Zhang, W.; Sun, B.; et al. Interleukin-33 drives hepatic fibrosis through activation of hepatic stellate cells. Cell Mol. Immunol. 2018, 15, 388–398. [Google Scholar] [CrossRef]
- Winwood, P.J.; Schuppan, D.; Iredale, J.P.; Kawser, C.A.; Docherty, A.J.; Arthur, M.J. Kupffer cell-derived 95-kd type IV collagenase/gelatinase B: Characterization and expression in cultured cells. Hepatology 1995, 22, 304–315. [Google Scholar]
- Ying, H.Z.; Chen, Q.; Zhang, W.Y.; Zhang, H.H.; Ma, Y.; Zhang, S.Z.; Fang, J.; Yu, C.H. PDGF signaling pathway in hepatic fibrosis pathogenesis and therapeutics (Review). Mol. Med. Rep. 2017, 16, 7879–7889. [Google Scholar] [CrossRef]
- Sanchez-Valle, V.; Chavez-Tapia, N.C.; Uribe, M.; Mendez-Sanchez, N. Role of oxidative stress and molecular changes in liver fibrosis: A review. Curr. Med. Chem. 2012, 19, 4850–4860. [Google Scholar] [CrossRef]
- Luangmonkong, T.; Suriguga, S.; Mutsaers, H.A.M.; Groothuis, G.M.M.; Olinga, P.; Boersema, M. Targeting Oxidative Stress for the Treatment of Liver Fibrosis. Rev. Physiol. Biochem. Pharmacol. 2018, 175, 71–102. [Google Scholar] [CrossRef]
- Bao, Y.L.; Wang, L.; Pan, H.T.; Zhang, T.R.; Chen, Y.H.; Xu, S.J.; Mao, X.L.; Li, S.W. Animal and Organoid Models of Liver Fibrosis. Front. Physiol. 2021, 12, 666138. [Google Scholar] [CrossRef] [PubMed]
- Arroyave-Ospina, J.C.; Wu, Z.; Geng, Y.; Moshage, H. Role of Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Implications for Prevention and Therapy. Antioxidants 2021, 10, 174. [Google Scholar] [CrossRef]
- Han, K.H.; Hashimoto, N.; Fukushima, M. Relationships among alcoholic liver disease, antioxidants, and antioxidant enzymes. World J. Gastroenterol. 2016, 22, 37–49. [Google Scholar] [CrossRef]
- Sun, Y.; Pu, L.Y.; Lu, L.; Wang, X.H.; Zhang, F.; Rao, J.H. N-acetylcysteine attenuates reactive-oxygen-species-mediated endoplasmic reticulum stress during liver ischemia-reperfusion injury. World J. Gastroenterol. 2014, 20, 15289–15298. [Google Scholar] [CrossRef] [PubMed]
- Singal, A.K.; Jampana, S.C.; Weinman, S.A. Antioxidants as therapeutic agents for liver disease. Liver Int. 2011, 31, 1432–1448. [Google Scholar] [CrossRef]
- D’Amico, G.; Morabito, A.; D’Amico, M.; Pasta, L.; Malizia, G.; Rebora, P.; Valsecchi, M.G. Clinical states of cirrhosis and competing risks. J. Hepatol. 2018, 68, 563–576. [Google Scholar] [CrossRef] [PubMed]
- Gines, P.; Krag, A.; Abraldes, J.G.; Sola, E.; Fabrellas, N.; Kamath, P.S. Liver cirrhosis. Lancet 2021, 398, 1359–1376. [Google Scholar] [CrossRef]
- Samonakis, D.N.; Koulentaki, M.; Coucoutsi, C.; Augoustaki, A.; Baritaki, C.; Digenakis, E.; Papiamonis, N.; Fragaki, M.; Matrella, E.; Tzardi, M.; et al. Clinical outcomes of compensated and decompensated cirrhosis: A long term study. World J. Hepatol. 2014, 6, 504–512. [Google Scholar] [CrossRef]
- Tarao, K.; Nozaki, A.; Ikeda, T.; Sato, A.; Komatsu, H.; Komatsu, T.; Taguri, M.; Tanaka, K. Real impact of liver cirrhosis on the development of hepatocellular carcinoma in various liver diseases-meta-analytic assessment. Cancer Med. 2019, 8, 1054–1065. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Huang, J.; Lok, V.; Ngai, C.H.; Chu, C.; Patel, H.K.; Thoguluva Chandraseka, V.; Zhang, L.; Chen, P.; Wang, S.; Lao, X.Q.; et al. Disease Burden, Risk Factors, and Recent Trends of Liver Cancer: A Global Country-Level Analysis. Liver Cancer 2021, 10, 330–345. [Google Scholar] [CrossRef] [PubMed]
- Rumgay, H.; Ferlay, J.; de Martel, C.; Georges, D.; Ibrahim, A.S.; Zheng, R.; Wei, W.; Lemmens, V.; Soerjomataram, I. Global, regional and national burden of primary liver cancer by subtype. Eur. J. Cancer 2022, 161, 108–118. [Google Scholar] [CrossRef]
- Li, Y.; Xu, A.; Jia, S.; Huang, J. Recent advances in the molecular mechanism of sex disparity in hepatocellular carcinoma. Oncol. Lett. 2019, 17, 4222–4228. [Google Scholar] [CrossRef]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed]
- Tarocchi, M.; Galli, A. Oxidative stress as a mechanism for hepatocellular carcinoma. In Liver Pathophysiology: Therapies and Antioxidants; Muriel, P., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 279–287. [Google Scholar]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef] [PubMed]
- Edamoto, Y.; Hara, A.; Biernat, W.; Terracciano, L.; Cathomas, G.; Riehle, H.M.; Matsuda, M.; Fujii, H.; Scoazec, J.Y.; Ohgaki, H. Alterations of RB1, p53 and Wnt pathways in hepatocellular carcinomas associated with hepatitis C, hepatitis B and alcoholic liver cirrhosis. Int. J. Cancer 2003, 106, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Feng, Z.; Eveleigh, J.; Iyer, G.; Pan, J.; Amin, S.; Chung, F.L.; Tang, M.S. The major lipid peroxidation product, trans-4-hydroxy-2-nonenal, preferentially forms DNA adducts at codon 249 of human p53 gene, a unique mutational hotspot in hepatocellular carcinoma. Carcinogenesis 2002, 23, 1781–1789. [Google Scholar] [CrossRef]
- Lim, S.O.; Gu, J.M.; Kim, M.S.; Kim, H.S.; Park, Y.N.; Park, C.K.; Cho, J.W.; Park, Y.M.; Jung, G. Epigenetic changes induced by reactive oxygen species in hepatocellular carcinoma: Methylation of the E-cadherin promoter. Gastroenterology 2008, 135, 2128–2140.e8. [Google Scholar] [CrossRef]
- Takaki, A.; Yamamoto, K. Control of oxidative stress in hepatocellular carcinoma: Helpful or harmful? World J. Hepatol. 2015, 7, 968–979. [Google Scholar] [CrossRef]
- Stickel, F.; Schuppan, D.; Hahn, E.G.; Seitz, H.K. Cocarcinogenic effects of alcohol in hepatocarcinogenesis. Gut 2002, 51, 132–139. [Google Scholar] [CrossRef]
- Refolo, M.G.; Messa, C.; Guerra, V.; Carr, B.I.; D’Alessandro, R. Inflammatory Mechanisms of HCC Development. Cancers 2020, 12, 641. [Google Scholar] [CrossRef] [PubMed]
- Thadathil, N.; Selvarani, R.; Mohammed, S.; Nicklas, E.H.; Tran, A.L.; Kamal, M.; Luo, W.; Brown, J.L.; Lawrence, M.M.; Borowik, A.K.; et al. Senolytic treatment reduces cell senescence and necroptosis in Sod1 knockout mice that is associated with reduced inflammation and hepatocellular carcinoma. Aging Cell 2022, 21, e13676. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Chen, L.; Leung, L.; Yen, T.S.B.; Lee, C.; Chan, J.Y. Liver-specific inactivation of the Nrf1 gene in adult mouse leads to nonalcoholic steatohepatitis and hepatic neoplasia. Proc. Natl. Acad. Sci. USA 2005, 102, 4120–4125. [Google Scholar] [CrossRef]
- Wang, R.; Yin, C.; Li, X.X.; Yang, X.Z.; Yang, Y.; Zhang, M.Y.; Wang, H.Y.; Zheng, X.F. Reduced SOD2 expression is associated with mortality of hepatocellular carcinoma patients in a mutant p53-dependent manner. Aging 2016, 8, 1184–1200. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Zhao, X.P.; Wang, L.Y.; Gao, S.; Zhao, J.; Fan, Y.C.; Wang, K. Glutathione S-transferase P1 correlated with oxidative stress in hepatocellular carcinoma. Int. J. Med. Sci. 2013, 10, 683–690. [Google Scholar] [CrossRef]
- Elmberg, M.; Hultcrantz, R.; Ekbom, A.; Brandt, L.; Olsson, S.; Olsson, R.; Lindgren, S.; Loof, L.; Stal, P.; Wallerstedt, S.; et al. Cancer risk in patients with hereditary hemochromatosis and in their first-degree relatives. Gastroenterology 2003, 125, 1733–1741. [Google Scholar] [CrossRef]
- Nahon, P.; Sutton, A.; Rufat, P.; Ziol, M.; Thabut, G.; Schischmanoff, P.O.; Vidaud, D.; Charnaux, N.; Couvert, P.; Ganne-Carrie, N.; et al. Liver iron, HFE gene mutations, and hepatocellular carcinoma occurrence in patients with cirrhosis. Gastroenterology 2008, 134, 102–110. [Google Scholar] [CrossRef]
- Atkins, J.L.; Pilling, L.C.; Masoli, J.A.H.; Kuo, C.L.; Shearman, J.D.; Adams, P.C.; Melzer, D. Association of Hemochromatosis HFE p.C282Y Homozygosity with Hepatic Malignancy. JAMA 2020, 324, 2048–2057. [Google Scholar] [CrossRef]
- Marrogi, A.J.; Khan, M.A.; van Gijssel, H.E.; Welsh, J.A.; Rahim, H.; Demetris, A.J.; Kowdley, K.V.; Hussain, S.P.; Nair, J.; Bartsch, H.; et al. Oxidative stress and p53 mutations in the carcinogenesis of iron overload-associated hepatocellular carcinoma. J. Natl. Cancer Inst. 2001, 93, 1652–1655. [Google Scholar] [CrossRef]
- Muto, Y.; Moroishi, T.; Ichihara, K.; Nishiyama, M.; Shimizu, H.; Eguchi, H.; Moriya, K.; Koike, K.; Mimori, K.; Mori, M.; et al. Disruption of FBXL5-mediated cellular iron homeostasis promotes liver carcinogenesis. J. Exp. Med. 2019, 216, 950–965. [Google Scholar] [CrossRef]
- Asare, G.A.; Paterson, A.C.; Kew, M.C.; Khan, S.; Mossanda, K.S. Iron-free neoplastic nodules and hepatocellular carcinoma without cirrhosis in Wistar rats fed a diet high in iron. J. Pathol. 2006, 208, 82–90. [Google Scholar] [CrossRef]
- Choi, J.; Corder, N.L.; Koduru, B.; Wang, Y. Oxidative stress and hepatic Nox proteins in chronic hepatitis C and hepatocellular carcinoma. Free Radic. Biol. Med. 2014, 72, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Vandierendonck, A.; Degroote, H.; Vanderborght, B.; Verhelst, X.; Geerts, A.; Devisscher, L.; Van Vlierberghe, H. NOX1 inhibition attenuates the development of a pro-tumorigenic environment in experimental hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2021, 40, 40. [Google Scholar] [CrossRef] [PubMed]
- Eun, H.S.; Cho, S.Y.; Joo, J.S.; Kang, S.H.; Moon, H.S.; Lee, E.S.; Kim, S.H.; Lee, B.S. Gene expression of NOX family members and their clinical significance in hepatocellular carcinoma. Sci. Rep. 2017, 7, 11060. [Google Scholar] [CrossRef]
- Ha, S.Y.; Paik, Y.H.; Yang, J.W.; Lee, M.J.; Bae, H.; Park, C.K. NADPH Oxidase 1 and NADPH Oxidase 4 Have Opposite Prognostic Effects for Patients with Hepatocellular Carcinoma after Hepatectomy. Gut Liver 2016, 10, 826–835. [Google Scholar] [CrossRef]
- Senturk, S.; Mumcuoglu, M.; Gursoy-Yuzugullu, O.; Cingoz, B.; Akcali, K.C.; Ozturk, M. Transforming growth factor-beta induces senescence in hepatocellular carcinoma cells and inhibits tumor growth. Hepatology 2010, 52, 966–974. [Google Scholar] [CrossRef]
- Sancho, P.; Fabregat, I. NADPH oxidase NOX1 controls autocrine growth of liver tumor cells through up-regulation of the epidermal growth factor receptor pathway. J. Biol. Chem. 2010, 285, 24815–24824. [Google Scholar] [CrossRef]
- Liang, S.; Ma, H.Y.; Zhong, Z.; Dhar, D.; Liu, X.; Xu, J.; Koyama, Y.; Nishio, T.; Karin, D.; Karin, G.; et al. NADPH Oxidase 1 in Liver Macrophages Promotes Inflammation and Tumor Development in Mice. Gastroenterology 2019, 156, 1156–1172. [Google Scholar] [CrossRef] [PubMed]
- Pavlovic, N.; Heindryckx, F. Exploring the Role of Endoplasmic Reticulum Stress in Hepatocellular Carcinoma through mining of the Human Protein Atlas. Biology 2021, 10, 640. [Google Scholar] [CrossRef]
- Wei, J.; Fang, D. Endoplasmic Reticulum Stress Signaling and the Pathogenesis of Hepatocarcinoma. Int. J. Mol. Sci. 2021, 22, 1799. [Google Scholar] [CrossRef]
- Nakagawa, H.; Umemura, A.; Taniguchi, K.; Font-Burgada, J.; Dhar, D.; Ogata, H.; Zhong, Z.; Valasek, M.A.; Seki, E.; Hidalgo, J.; et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 2014, 26, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Zeeshan, H.M.; Lee, G.H.; Kim, H.R.; Chae, H.J. Endoplasmic Reticulum Stress and Associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef]
- Santos, C.X.; Nabeebaccus, A.A.; Shah, A.M.; Camargo, L.L.; Filho, S.V.; Lopes, L.R. Endoplasmic reticulum stress and Nox-mediated reactive oxygen species signaling in the peripheral vasculature: Potential role in hypertension. Antioxid. Redox Signal. 2014, 20, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Zhou, X.; Cao, T.; Chen, E.; Li, Y.; Lei, W.; Hu, Y.; He, B.; Liu, S. Endoplasmic Reticulum Stress and Oxidative Stress in Inflammatory Diseases. DNA Cell Biol. 2022, 41, 924–934. [Google Scholar] [CrossRef]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, X.; Tang, H.; Yu, J.; Zu, X.; Xie, Z.; Yang, X.; Hu, J.; Tan, F.; Li, Q.; et al. Nuclear factor erythroid 2 (NF-E2)-related factor 2 (Nrf2) in autophagy-induced hepatocellular carcinoma. Clin. Chim. Acta 2020, 506, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, C.; Zhang, L.; Yang, Q.; Zhou, S.; Wen, Q.; Wang, J. Nrf2 is a potential prognostic marker and promotes proliferation and invasion in human hepatocellular carcinoma. BMC Cancer 2015, 15, 531. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allameh, A.; Niayesh-Mehr, R.; Aliarab, A.; Sebastiani, G.; Pantopoulos, K. Oxidative Stress in Liver Pathophysiology and Disease. Antioxidants 2023, 12, 1653. https://doi.org/10.3390/antiox12091653
Allameh A, Niayesh-Mehr R, Aliarab A, Sebastiani G, Pantopoulos K. Oxidative Stress in Liver Pathophysiology and Disease. Antioxidants. 2023; 12(9):1653. https://doi.org/10.3390/antiox12091653
Chicago/Turabian StyleAllameh, Abdolamir, Reyhaneh Niayesh-Mehr, Azadeh Aliarab, Giada Sebastiani, and Kostas Pantopoulos. 2023. "Oxidative Stress in Liver Pathophysiology and Disease" Antioxidants 12, no. 9: 1653. https://doi.org/10.3390/antiox12091653
APA StyleAllameh, A., Niayesh-Mehr, R., Aliarab, A., Sebastiani, G., & Pantopoulos, K. (2023). Oxidative Stress in Liver Pathophysiology and Disease. Antioxidants, 12(9), 1653. https://doi.org/10.3390/antiox12091653