

Advances in the Use of N-Acetylcysteine in Chronic Respiratory Diseases


Abstract
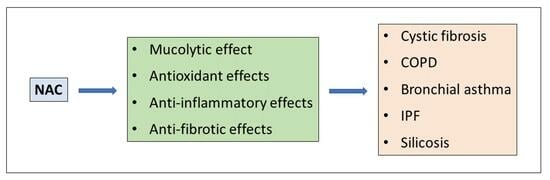
1. Introduction
2. Pharmacological Effects of NAC
2.1. Pharmacokinetics and Pharmacodynamics of NAC
2.2. Adverse Effects and Drug Interactions of NAC
2.3. Mechanisms of Action of NAC
2.3.1. Mucolytic Effect of NAC
2.3.2. NAC as an Antidote
2.3.3. Antioxidant Effects of NAC
2.3.4. Anti-Inflammatory Effects of NAC
2.3.5. Anti-Fibrotic Effects of NAC
2.3.6. Cytoprotective Effects of NAC
3. NAC in Chronic Respiratory Diseases
3.1. NAC in CF
3.1.1. Pathophysiology and Treatment of CF
3.1.2. NAC in In Vitro Studies of CF
3.1.3. NAC in Animal Studies of CF
3.1.4. NAC in Clinical Studies of CF
3.2. NAC in COPD
3.2.1. Pathophysiology and Treatment of COPD
3.2.2. NAC in In Vitro Studies of COPD
3.2.3. NAC in Animal Studies of COPD
3.2.4. NAC in Clinical Studies of COPD
3.3. NAC in Bronchial Asthma
3.3.1. Pathophysiology and Treatment of Asthma
3.3.2. NAC in In Vitro Studies of Asthma
3.3.3. NAC in Animal Studies of Asthma
3.3.4. NAC in Clinical Studies of Asthma
3.4. NAC in IPF
3.4.1. Pathophysiology and Treatment of IPF
3.4.2. NAC in In Vitro Studies of IPF
3.4.3. NAC in Animal Studies of IPF
3.4.4. NAC in Clinical Studies of IPF
3.5. NAC in Lung Silicosis
3.5.1. Pathophysiology and Treatment of Lung Silicosis
3.5.2. NAC in In Vitro Studies of Lung Silicosis
3.5.3. NAC in Animal Studies of Lung Silicosis
3.5.4. NAC in Clinical Studies of Lung Silicosis
4. Limitations and Challenges for the Future
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aldini, G.; Altomare, A.; Baron, G.; Vistoli, G.; Carini, M.; Borsani, L.; Sergio, F. N-Acetylcysteine as an antioxidant and disulphide breaking agent: The reasons why. Free Radic. Res. 2018, 52, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Schwalfenberg, G.K. N-Acetylcysteine: A Review of Clinical Usefulness (an Old Drug with New Tricks). J. Nutr. Metab. 2021, 2021, 9949453. [Google Scholar] [CrossRef]
- Tenório, M.C.D.S.; Graciliano, N.G.; Moura, F.A.; Oliveira, A.C.M.; Goulart, M.O.F. N-Acetylcysteine (NAC): Impacts on Human Health. Antioxidants 2021, 10, 967. [Google Scholar] [CrossRef]
- Diaz Vivancos, P.; Wolff, T.; Markovic, J.; Pallardó, F.V.; Foyer, C.H. A nuclear glutathione cycle within the cell cycle. Biochem. J. 2010, 431, 169–178. [Google Scholar] [CrossRef]
- Demedts, M.; Behr, J.; Buhl, R.; Costabel, U.; Dekhuijzen, R.; Jansen, H.M.; MacNee, W.; Thomeer, M.; Wallaert, B.; Laurent, F.; et al. High-dose acetylcysteine in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2005, 353, 2229–2242. [Google Scholar] [CrossRef] [PubMed]
- Tirouvanziam, R.; Conrad, C.K.; Bottiglieri, T.; Herzenberg, L.A.; Moss, R.B.; Herzenberg, L.A. High-dose oral N-acetylcysteine, a glutathione prodrug, modulates inflammation in cystic fibrosis. Proc. Natl. Acad. Sci. USA 2006, 103, 4628–4633. [Google Scholar] [CrossRef] [PubMed]
- Tse, H.N.; Raiteri, L.; Wong, K.Y.; Yee, K.S.; Ng, L.Y.; Wai, K.Y.; Loo, C.K.; Chan, M.H. High-dose N-acetylcysteine in stable COPD: The 1-year, double-blind, randomized, placebo-controlled HIACE study. Chest 2013, 144, 106–118. [Google Scholar] [CrossRef]
- Zheng, J.P.; Wen, F.Q.; Bai, C.X.; Wan, H.Y.; Kang, J.; Chen, P.; Yao, W.Z.; Ma, L.J.; Li, X.; Raiteri, L.; et al. Twice daily N-acetylcysteine 600 mg for exacerbations of chronic obstructive pulmonary disease (PANTHEON): A randomised, double-blind placebo-controlled trial. Lancet Respir. Med. 2014, 2, 187–194. [Google Scholar] [CrossRef]
- Cazzola, M.; Calzetta, L.; Page, C.; Rogliani, P.; Matera, M.G. Thiol-Based Drugs in Pulmonary Medicine: Much More than Mucolytics. Trends Pharmacol. Sci. 2019, 40, 452–463. [Google Scholar] [CrossRef]
- Holdiness, M.R. Clinical pharmacokinetics of N-acetylcysteine. Clin. Pharmacokinet. 1991, 20, 123–134. [Google Scholar] [CrossRef]
- Borgström, L.; Kågedal, B.; Paulsen, O. Pharmacokinetics of N-acetylcysteine in man. Eur. J. Clin. Pharmacol. 1986, 31, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Johansson, M.; Gabrielsson, J.; Bolme, P. Pharmacokinetics and bioavailability of reduced and oxidized N-acetylcysteine. Eur. J. Clin. Pharmacol. 1988, 34, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Rushworth, G.F.; Megson, I.L. Existing and potential therapeutic uses for N-acetylcysteine: The need for conversion to intracellular glutathione for antioxidant benefits. Pharmacol. Ther. 2014, 141, 150–159. [Google Scholar] [CrossRef]
- Greene, S.C.; Noonan, P.K.; Sanabria, C.; Peacock, W.F. Effervescent N-Acetylcysteine Tablets versus Oral Solution N-Acetylcysteine in Fasting Healthy Adults: An Open-Label, Randomized, Single-Dose, Crossover, Relative Bioavailability Study. Curr. Ther. Res. Clin. Exp. 2016, 83, 1–7. [Google Scholar] [CrossRef]
- Calverley, P.; Rogliani, P.; Papi, A. Safety of N-Acetylcysteine at High Doses in Chronic Respiratory Diseases: A Review. Drug Saf. 2021, 44, 273–290. [Google Scholar] [CrossRef]
- Stey, C.; Steurer, J.; Bachmann, S.; Medici, T.C.; Tramèr, M.R. The effect of oral N-acetylcysteine in chronic bronchitis: A quantitative systematic review. Eur. Respir. J. 2000, 16, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Grandjean, E.M.; Berthet, P.; Ruffmann, R.; Leuenberger, P. Efficacy of oral long-term N-acetylcysteine in chronic bronchopulmonary disease: A meta-analysis of published double-blind, placebo-controlled clinical trials. Clin. Ther. 2000, 22, 209–221. [Google Scholar] [CrossRef]
- Hansen, N.C.; Skriver, A.; Brorsen-Riis, L.; Balsløv, S.; Evald, T.; Maltbaek, N.; Gunnersen, G.; Garsdal, P.; Sander, P.; Pedersen, J.Z.; et al. Orally administered N-acetylcysteine may improve general well-being in patients with mild chronic bronchitis. Respir. Med. 1994, 88, 531–535. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.J.; de Andrade, J.A.; Anstrom, K.J.; King, T.E., Jr.; Raghu, G.; Idiopathic Pulmonary Fibrosis Clinical Research Network. Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2093–2101. [Google Scholar] [CrossRef]
- Johnson, K.; McEvoy, C.E.; Naqvi, S.; Wendt, C.; Reilkoff, R.A.; Kunisaki, K.M.; Wetherbee, E.E.; Nelson, D.; Tirouvanziam, R.; Niewoehner, D.E. High-dose oral N-acetylcysteine fails to improve respiratory health status in patients with chronic obstructive pulmonary disease and chronic bronchitis: A randomized, placebo-controlled trial. Int. J. Chron. Obstruct. Pulmon. Dis. 2016, 11, 799–807. [Google Scholar] [CrossRef]
- Flanagan, R.J.; Meredith, T.J. Use of N-acetylcysteine in clinical toxicology. Am. J. Med. 1991, 91, 131S–139S. [Google Scholar] [CrossRef]
- Kanter, M.Z. Comparison of oral and i.v. acetylcysteine in the treatment of acetaminophen poisoning. Am. J. Health Syst. Pharm. 2006, 63, 1821–1827. [Google Scholar] [CrossRef] [PubMed]
- Yarema, M.; Chopra, P.; Sivilotti, M.L.A.; Johnson, D.; Nettel-Aguirre, A.; Bailey, B.; Victorino, C.; Gosselin, S.; Purssell, R.; Thompson, M.; et al. Anaphylactoid Reactions to Intravenous N-Acetylcysteine during Treatment for Acetaminophen Poisoning. J. Med. Toxicol. 2018, 14, 120–127. [Google Scholar] [CrossRef]
- Feng, F.; Zhang, J.; Wang, Z.; Wu, Q.; Zhou, X. Efficacy and safety of N-acetylcysteine therapy for idiopathic pulmonary fibrosis: An updated systematic review and meta-analysis. Exp. Ther. Med. 2019, 18, 802–816. [Google Scholar] [CrossRef]
- Meyer, A.; Buhl, R.; Kampf, S.; Magnussen, H. Intravenous N-acetylcysteine and lung glutathione of patients with pulmonary fibrosis and normals. Am. J. Respir. Crit. Care Med. 1995, 152, 1055–1060. [Google Scholar] [CrossRef]
- Niemi, T.T.; Munsterhjelm, E.; Pöyhiä, R.; Hynninen, M.S.; Salmenperä, M.T. The effect of N-acetylcysteine on blood coagulation and platelet function in patients undergoing open repair of abdominal aortic aneurysm. Blood Coagul. Fibrinolysis 2006, 17, 29–34. [Google Scholar] [CrossRef]
- URL: Acetylcysteine. DRUGBANK Online. Available online: https://go.drugbank.com/drugs/DB06151 (accessed on 6 June 2023).
- Zhitkovich, A. N-Acetylcysteine: Antioxidant, Aldehyde Scavenger, and More. Chem. Res. Toxicol. 2019, 32, 1318–1319. [Google Scholar] [CrossRef]
- Balsamo, R.; Lanata, L.; Egan, C.G. Mucoactive drugs. Eur. Respir. Rev. 2010, 19, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Fisher, E.S.; Curry, S.C. Evaluation and treatment of acetaminophen toxicity. Adv. Pharmacol. 2019, 85, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H. Acetaminophen: Dose-Dependent Drug Hepatotoxicity and Acute Liver Failure in Patients. Dig. Dis. 2015, 33, 464–471. [Google Scholar] [CrossRef]
- Vrbová, M.; Roušarová, E.; Brůčková, L.; Česla, P.; Roušar, T. Characterization of acetaminophen toxicity in human kidney HK-2 cells. Physiol. Res. 2016, 65, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Akakpo, J.Y.; Umbaugh, D.S.; Ramachandran, A. Novel Therapeutic Approaches against Acetaminophen-induced Liver Injury and Acute Liver Failure. Toxicol. Sci. 2020, 174, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Lauterburg, B.H.; Corcoran, G.B.; Mitchell, J.R. Mechanism of action of N-acetylcysteine in the protection against the hepatotoxicity of acetaminophen in rats in vivo. J. Clin. Investig. 1983, 71, 980–991. [Google Scholar] [CrossRef] [PubMed]
- Aykin-Burns, N.; Franklin, E.A.; Ercal, N. Effects of N-acetylcysteine on lead-exposed PC-12 cells. Arch. Environ. Contam. Toxicol. 2005, 49, 119–123. [Google Scholar] [CrossRef]
- Gawarammana, I.B.; Buckley, N.A. Medical management of paraquat ingestion. Br. J. Clin. Pharmacol. 2011, 72, 745–757. [Google Scholar] [CrossRef]
- Liu, J.; Chen, Y.; Gao, Y.; Walline, J.H.; Lu, X.; Yu, S.; Zhao, L.; Ge, Z.; Li, Y. N-acetylcysteine as a treatment for amatoxin poisoning: A systematic review. Clin. Toxicol. 2020, 58, 1015–1022. [Google Scholar] [CrossRef]
- Samuni, Y.; Goldstein, S.; Dean, O.M.; Berk, M. The chemistry and biological activities of N-acetylcysteine. Biochim. Biophys. Acta 2013, 1830, 4117–4129. [Google Scholar] [CrossRef]
- Sen, C.K.; Packer, L. Thiol homeostasis and supplements in physical exercise. Am. J. Clin. Nutr. 2000, 72, 653S–669S. [Google Scholar] [CrossRef]
- Deponte, M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta 2013, 1830, 3217–3266. [Google Scholar] [CrossRef]
- Lopert, P.; Day, B.J.; Patel, M. Thioredoxin reductase deficiency potentiates oxidative stress, mitochondrial dysfunction and cell death in dopaminergic cells. PLoS ONE 2012, 7, e50683. [Google Scholar] [CrossRef]
- Morris, G.; Walker, A.J.; Walder, K.; Berk, M.; Marx, W.; Carvalho, A.F.; Maes, M.; Puri, B.K. Increasing Nrf2 Activity as a Treatment Approach in Neuropsychiatry. Mol. Neurobiol. 2021, 58, 2158–2182. [Google Scholar] [CrossRef]
- Ji, L.; Liu, R.; Zhang, X.D.; Chen, H.L.; Bai, H.; Wang, X.; Zhao, H.L.; Liang, X.; Hai, C.X. N-acetylcysteine attenuates phosgene-induced acute lung injury via up-regulation of Nrf2 expression. Inhal. Toxicol. 2010, 22, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Oter, S.; Jin, S.; Cucullo, L.; Dorman, H.J. Oxidants and antioxidants: Friends or foes? Oxid. Antioxid. Med. Sci. 2012, 1, 1–4. [Google Scholar] [CrossRef] [PubMed]
- McCarty, M.F.; DiNicolantonio, J.J. An increased need for dietary cysteine in support of glutathione synthesis may underlie the increased risk for mortality associated with low protein intake in the elderly. Age 2015, 37, 96. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Liu, C.; Suliburk, J.W.; Minard, C.G.; Muthupillai, R.; Chacko, S.; Hsu, J.W.; Jahoor, F.; Sekhar, R.V. Supplementing Glycine and N-acetylcysteine (GlyNAC) in Aging HIV Patients Improves Oxidative Stress, Mitochondrial Dysfunction, Inflammation, Endothelial Dysfunction, Insulin Resistance, Genotoxicity, Strength, and Cognition: Results of an Open-Label Clinical Trial. Biomedicines 2020, 8, 390. [Google Scholar] [CrossRef]
- Kumar, P.; Liu, C.; Hsu, J.W.; Chacko, S.; Minard, C.; Jahoor, F.; Sekhar, R.V. Glycine and N-acetylcysteine (GlyNAC) supplementation in older adults improves glutathione deficiency, oxidative stress, mitochondrial dysfunction, inflammation, insulin resistance, endothelial dysfunction, genotoxicity, muscle strength, and cognition: Results of a pilot clinical trial. Clin. Transl. Med. 2021, 11, e372. [Google Scholar] [CrossRef]
- Raghu, G.; Berk, M.; Campochiaro, P.A.; Jaeschke, H.; Marenzi, G.; Richeldi, L.; Wen, F.Q.; Nicoletti, F.; Calverley, P.M.A. The Multifaceted Therapeutic Role of N-Acetylcysteine (NAC) in Disorders Characterized by Oxidative Stress. Curr. Neuropharmacol. 2021, 19, 1202–1224. [Google Scholar] [CrossRef]
- Matera, M.G.; Calzetta, L.; Cazzola, M. Oxidation pathway and exacerbations in COPD: The role of NAC. Expert. Rev. Respir. Med. 2016, 10, 89–97. [Google Scholar] [CrossRef]
- Sun, J.; Song, P.; Wang, Y.; Chen, Y. Clinical efficacy of acetylcysteine combined with tetrandrine tablets in the treatment of silicosis and the effect on serum IL-6 and TNF-α. Exp. Ther. Med. 2019, 18, 3383–3388. [Google Scholar] [CrossRef]
- Kumar, P.; Osahon, O.; Vides, D.B.; Hanania, N.; Minard, C.G.; Sekhar, R.V. Severe Glutathione Deficiency, Oxidative Stress and Oxidant Damage in Adults Hospitalized with COVID-19: Implications for GlyNAC (Glycine and N-Acetylcysteine) Supplementation. Antioxidants 2021, 11, 50. [Google Scholar] [CrossRef]
- Alam, M.S.; Hasan, M.N.; Maowa, Z.; Khatun, F.; Nazir, K.H.M.N.H.; Alam, M.Z. N-acetylcysteine reduces severity and mortality in COVID-19 patients: A systematic review and meta-analysis. J. Adv. Vet. Anim. Res. 2023, 10, 157–168. [Google Scholar] [CrossRef]
- Meliante, P.G.; Zoccali, F.; Cascone, F.; Di Stefano, V.; Greco, A.; de Vincentiis, M.; Petrella, C.; Fiore, M.; Minni, A.; Barbato, C. Molecular Pathology, Oxidative Stress, and Biomarkers in Obstructive Sleep Apnea. Int. J. Mol. Sci. 2023, 24, 5478. [Google Scholar] [CrossRef] [PubMed]
- Oliva, A.; Pallecchi, L.; Rossolini, G.M.; Travaglino, F.; Zanatta, P. Rationale and evidence for the adjunctive use of N-acetylcysteine in multidrug-resistant infections. Eur. Rev. Med. Pharmacol. Sci. 2023, 27, 4316–4325. [Google Scholar] [CrossRef] [PubMed]
- Wilder, T.; Ryba, D.M.; Wieczorek, D.F.; Wolska, B.M.; Solaro, R.J. N-acetylcysteine reverses diastolic dysfunction and hypertrophy in familial hypertrophic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1720–H1730. [Google Scholar] [CrossRef] [PubMed]
- Sekhar, R.V. GlyNAC (Glycine and N-Acetylcysteine) Supplementation Improves Impaired Mitochondrial Fuel Oxidation and Lowers Insulin Resistance in Patients with Type 2 Diabetes: Results of a Pilot Study. Antioxidants 2022, 11, 154. [Google Scholar] [CrossRef]
- Ye, M.; Lin, W.; Zheng, J.; Lin, S. N-acetylcysteine for chronic kidney disease: A systematic review and meta-analysis. Am. J. Transl. Res. 2021, 13, 2472–2485. [Google Scholar] [PubMed]
- Sandhu, J.K.; Waqar, A.; Jain, A.; Joseph, C.; Srivastava, K.; Ochuba, O.; Alkayyali, T.; Ruo, S.W.; Poudel, S. Oxidative Stress in Polycystic Ovarian Syndrome and the Effect of Antioxidant N-Acetylcysteine on Ovulation and Pregnancy Rate. Cureus 2021, 13, e17887. [Google Scholar] [CrossRef]
- Zafarullah, M.; Li, W.Q.; Sylvester, J.; Ahmad, M. Molecular mechanisms of N-acetylcysteine actions. Cell. Mol. Life Sci. 2003, 60, 6–20. [Google Scholar] [CrossRef]
- Vlahopoulos, S.; Boldogh, I.; Casola, A.; Brasier, A.R. Nuclear factor-κB-dependent induction of interleukin-8 gene expression by tumor necrosis factor α: Evidence for an antioxidant sensitive activating pathway distinct from nuclear translocation. Blood 1999, 94, 1878–1889. [Google Scholar] [CrossRef]
- Hashimoto, S.; Gon, Y.; Matsumoto, K.; Takeshita, I.; Horie, T. N-acetylcysteine attenuates TNF-α-induced p38 MAP kinase activation and p38 MAP kinase-mediated IL-8 production by human pulmonary vascular endothelial cells. Br. J. Pharmacol. 2001, 132, 270–276. [Google Scholar] [CrossRef]
- Hashimoto, S.; Gon, Y.; Matsumoto, K.; Takeshita, I.; MacHino, T.; Horie, T. Intracellular glutathione regulates tumour necrosis factor-α-induced p38 MAP kinase activation and RANTES production by human bronchial epithelial cells. Clin. Exp. Allergy 2001, 31, 144–151. [Google Scholar] [CrossRef] [PubMed]
- De Flora, S.; Balansky, R.; La Maestra, S. Antioxidants and COVID-19. J. Prev. Med. Hyg. 2021, 62, E34–E45. [Google Scholar] [CrossRef]
- Kinnula, V.L.; Fattman, C.L.; Tan, R.J.; Oury, T.D. Oxidative stress in pulmonary fibrosis: A possible role for redox modulatory therapy. Am. J. Respir. Crit. Care Med. 2005, 172, 417–422. [Google Scholar] [CrossRef]
- Kliment, C.R.; Oury, T.D. Oxidative stress, extracellular matrix targets, and idiopathic pulmonary fibrosis. Free Radic. Biol. Med. 2010, 49, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Nardi, J.; Nascimento, S.; Göethel, G.; Gauer, B.; Sauer, E.; Fão, N.; Cestonaro, L.; Peruzzi, C.; Souza, J.; Garcia, S.C. Inflammatory and oxidative stress parameters as potential early biomarkers for silicosis. Clin. Chim. Acta 2018, 484, 305–313. [Google Scholar] [CrossRef]
- Adamcakova, J.; Mokra, D. New Insights into Pathomechanisms and Treatment Possibilities for Lung Silicosis. Int. J. Mol. Sci. 2021, 22, 4162. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.M.; Liu, Y.; Forman, H.J.; Olman, M.; Tarpey, M.M. Glutathione regulates transforming growth factor-β-stimulated collagen production in fibroblasts. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, L121–L128. [Google Scholar] [CrossRef] [PubMed]
- Felton, V.M.; Borok, Z.; Willis, B.C. N-acetylcysteine inhibits alveolar epithelial-mesenchymal transition. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L805–L812. [Google Scholar] [CrossRef]
- Sugiura, H.; Ichikawa, T.; Liu, X.; Kobayashi, T.; Wang, X.Q.; Kawasaki, S.; Togo, S.; Kamio, K.; Mao, L.; Ann, Y.; et al. N-acetyl-L-cysteine inhibits TGF-β1-induced profibrotic responses in fibroblasts. Pulm. Pharmacol. Ther. 2009, 22, 487–491. [Google Scholar] [CrossRef]
- Hagiwara, S.I.; Ishii, Y.; Kitamura, S. Aerosolized administration of N-acetylcysteine attenuates lung fibrosis induced by bleomycin in mice. Am. J. Respir. Crit. Care Med. 2000, 162, 225–231. [Google Scholar] [CrossRef]
- Cortijo, J.; Cerdá-Nicolás, M.; Serrano, A.; Bioque, G.; Estrela, J.M.; Santangelo, F.; Esteras, A.; Llombart-Bosch, A.; Morcillo, E.J. Attenuation by oral N-acetylcysteine of bleomycin-induced lung injury in rats. Eur. Respir. J. 2001, 17, 1228–1235. [Google Scholar] [CrossRef] [PubMed]
- Mata, M.; Ruíz, A.; Cerdá, M.; Martinez-Losa, M.; Cortijo, J.; Santangelo, F.; Serrano-Mollar, A.; Llombart-Bosch, A.; Morcillo, E.J. Oral N-acetylcysteine reduces bleomycin-induced lung damage and mucin Muc5ac expression in rats. Eur. Respir. J. 2003, 22, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Mollar, A.; Closa, D.; Prats, N.; Blesa, S.; Martinez-Losa, M.; Cortijo, J.; Estrela, J.M.; Morcillo, E.J.; Bulbena, O. In vivo antioxidant treatment protects against bleomycin-induced lung damage in rats. Br. J. Pharmacol. 2003, 138, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yang, X.; Li, W.; Li, J.; Su, X.; Chen, L.; Yan, G. N-acetylcysteine downregulation of lysyl oxidase activity alleviating bleomycin-induced pulmonary fibrosis in rats. Respiration 2012, 84, 509–517. [Google Scholar] [CrossRef]
- Zhang, H.; Yin, G.; Jiang, H.; Zhang, C. High-dose N-acetylcysteine decreases silica-induced lung fibrosis in the rat. J. Int. Med. Res. 2013, 41, 1179–1186. [Google Scholar] [CrossRef]
- Zhang, L.; He, Y.L.; Li, Q.Z.; Hao, X.H.; Zhang, Z.F.; Yuan, J.X.; Bai, Y.P.; Jin, Y.L.; Liu, N.; Chen, G.; et al. N-acetylcysteine alleviated silica-induced lung fibrosis in rats by down-regulation of ROS and mitochondrial apoptosis signaling. Toxicol. Mech. Methods 2014, 24, 212–219. [Google Scholar] [CrossRef]
- Oldham, J.M.; Ma, S.F.; Martinez, F.J.; Anstrom, K.J.; Raghu, G.; Schwartz, D.A.; Valenzi, E.; Witt, L.; Lee, C.; Vij, R.; et al. TOLLIP, MUC5B, and the Response to N-Acetylcysteine among Individuals with Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2015, 192, 1475–1482. [Google Scholar] [CrossRef]
- Behr, J.; Bendstrup, E.; Crestani, B.; Günther, A.; Olschewski, H.; Sköld, C.M.; Wells, A.; Wuyts, W.; Koschel, D.; Kreuter, M.; et al. Safety and tolerability of acetylcysteine and pirfenidone combination therapy in idiopathic pulmonary fibrosis: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Respir. Med. 2016, 4, 445–453. [Google Scholar] [CrossRef]
- Sakamoto, S.; Muramatsu, Y.; Satoh, K.; Ishida, F.; Kikuchi, N.; Sano, G.; Sugino, K.; Isobe, K.; Takai, Y.; Homma, S. Effectiveness of combined therapy with pirfenidone and inhaled N-acetylcysteine for advanced idiopathic pulmonary fibrosis: A case-control study. Respirology 2015, 20, 445–452. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Y.; Zhang, S.; Li, J.; Fang, H. Effects of tetrandrine combined with acetylcysteine on exercise tolerance, pulmonary function and serum TNF-β1 and MMP-7 in silicosis patients. Exp. Ther. Med. 2020, 19, 2195–2201. [Google Scholar] [CrossRef]
- Fass, D.; Thorpe, C. Chemistry and Enzymology of Disulfide Cross-Linking in Proteins. Chem. Rev. 2018, 118, 1169–1198. [Google Scholar] [CrossRef]
- van Zandwijk, N. N-acetylcysteine (NAC) and glutathione (GSH): Antioxidant and chemopreventive properties, with special reference to lung cancer. J. Cell. Biochem. Suppl. 1995, 22, 24–32. [Google Scholar] [CrossRef]
- De Flora, S.; Izzotti, A.; D’Agostini, F.; Balansky, R.M. Mechanisms of N-acetylcysteine in the prevention of DNA damage and cancer, with special reference to smoking-related end-points. Carcinogenesis 2001, 22, 999–1013. [Google Scholar] [CrossRef]
- Yang, Y.M.; Conaway, C.C.; Chiao, J.W.; Wang, C.X.; Amin, S.; Whysner, J.; Dai, W.; Reinhardt, J.; Chung, F.L. Inhibition of benzo(a)pyrene-induced lung tumorigenesis in A/J mice by dietary N-acetylcysteine conjugates of benzyl and phenethyl isothiocyanates during the postinitiation phase is associated with activation of mitogen-activated protein kinases and p53 activity and induction of apoptosis. Cancer Res. 2002, 62, 2–7. [Google Scholar] [PubMed]
- Boucher, R.C. An overview of the pathogenesis of cystic fibrosis lung disease. Adv. Drug Deliv. Rev. 2002, 54, 1359–1371. [Google Scholar] [CrossRef] [PubMed]
- Kunzelmann, K.; Schreiber, R.; Hadorn, H.B. Bicarbonate in cystic fibrosis. J. Cyst. Fibros. 2017, 16, 653–662. [Google Scholar] [CrossRef]
- De Boeck, K. Cystic fibrosis in the year 2020: A disease with a new face. Acta Paediatr. 2020, 109, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Hollinger, M.; Lachowicz-Scroggins, M.E.; Kerr, S.C.; Dunican, E.M.; Daniel, B.M.; Ghosh, S.; Erzurum, S.C.; Willard, B.; Hazen, S.L.; et al. Oxidation increases mucin polymer cross-links to stiffen airway mucus gels. Sci. Transl. Med. 2015, 7, 276ra27. [Google Scholar] [CrossRef]
- Bartling, T.R.; Drumm, M.L. Oxidative stress causes IL8 promoter hyperacetylation in cystic fibrosis airway cell models. Am. J. Respir. Cell Mol. Biol. 2009, 40, 58–65. [Google Scholar] [CrossRef]
- Guerini, M.; Condrò, G.; Friuli, V.; Maggi, L.; Perugini, P. N-acetylcysteine (NAC) and Its Role in Clinical Practice Management of Cystic Fibrosis (CF): A Review. Pharmaceuticals 2022, 15, 217. [Google Scholar] [CrossRef]
- Dauletbaev, N.; Viel, K.; Buhl, R.; Wagner, T.O.; Bargon, J. Glutathione and glutathione peroxidase in sputum samples of adult patients with cystic fibrosis. J. Cyst. Fibros. 2004, 3, 119–124. [Google Scholar] [CrossRef]
- Luciani, A.; Villella, V.R.; Esposito, S.; Brunetti-Pierri, N.; Medina, D.; Settembre, C.; Gavina, M.; Pulze, L.; Giardino, I.; Pettoello-Mantovani, M.; et al. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat. Cell Biol. 2010, 12, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Rowe, S.M.; Heltshe, S.L.; Gonska, T.; Donaldson, S.H.; Borowitz, D.; Gelfond, D.; Sagel, S.D.; Khan, U.; Mayer-Hamblett, N.; Van Dalfsen, J.M.; et al. Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 175–184. [Google Scholar] [CrossRef]
- Kiefer, A.; Plattner, E.; Ruppel, R.; Weiss, C.; Zhou-Suckow, Z.; Mall, M.; Renner, M.; Müller, H. DMBT1 is upregulated in cystic fibrosis, affects ciliary motility, and is reduced by acetylcysteine. Mol. Cell. Pediatr. 2022, 9, 4. [Google Scholar] [CrossRef] [PubMed]
- Guerini, M.; Grisoli, P.; Pane, C.; Perugini, P. Microstructured Lipid Carriers (MLC) Based on N-Acetylcysteine and Chitosan Preventing Pseudomonas aeruginosa Biofilm. Int. J. Mol. Sci. 2021, 22, 891. [Google Scholar] [CrossRef] [PubMed]
- Aiyer, A.; Das, T.; Whiteley, G.S.; Glasbey, T.; Kriel, F.H.; Farrell, J.; Manos, J. The Efficacy of an N-Acetylcysteine-Antibiotic Combination Therapy on Achromobacter xylosoxidans in a Cystic Fibrosis Sputum/Lung Cell Model. Biomedicines 2022, 10, 2886. [Google Scholar] [CrossRef]
- Valzano, F.; Boncompagni, S.R.; Micieli, M.; Di Maggio, T.; Di Pilato, V.; Colombini, L.; Santoro, F.; Pozzi, G.; Rossolini, G.M.; Pallecchi, L. Activity of N-Acetylcysteine Alone and in Combination with Colistin against Pseudomonas aeruginosa Biofilms and Transcriptomic Response to N-Acetylcysteine Exposure. Microbiol. Spectr. 2022, 10, e0100622. [Google Scholar] [CrossRef]
- Ehre, C.; Rushton, Z.L.; Wang, B.; Hothem, L.N.; Morrison, C.B.; Fontana, N.C.; Markovetz, M.R.; Delion, M.F.; Kato, T.; Villalon, D.; et al. An Improved Inhaled Mucolytic to Treat Airway Muco-obstructive Diseases. Am. J. Respir. Crit. Care Med. 2019, 199, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Dauletbaev, N.; Fischer, P.; Aulbach, B.; Gross, J.; Kusche, W.; Thyroff-Friesinger, U.; Wagner, T.O.; Bargon, J. A phase II study on safety and efficacy of high-dose N-acetylcysteine in patients with cystic fibrosis. Eur. J. Med. Res. 2009, 14, 352–358. [Google Scholar] [CrossRef]
- App, E.M.; Baran, D.; Dab, I.; Malfroot, A.; Coffiner, M.; Vanderbist, F.; King, M. Dose-finding and 24-h monitoring for efficacy and safety of aerosolized Nacystelyn in cystic fibrosis. Eur. Respir. J. 2002, 19, 294–302. [Google Scholar] [CrossRef]
- Skov, M.; Pressler, T.; Lykkesfeldt, J.; Poulsen, H.E.; Jensen, P.Ø.; Johansen, H.K.; Qvist, T.; Kræmer, D.; Høiby, N.; Ciofu, O. The effect of short-term, high-dose oral N-acetylcysteine treatment on oxidative stress markers in cystic fibrosis patients with chronic P. aeruginosa infection—A pilot study. J. Cyst. Fibros. 2015, 14, 211–218. [Google Scholar] [CrossRef]
- Conrad, C.; Lymp, J.; Thompson, V.; Dunn, C.; Davies, Z.; Chatfield, B.; Nichols, D.; Clancy, J.; Vender, R.; Egan, M.E.; et al. Long-term treatment with oral N-acetylcysteine: Affects lung function but not sputum inflammation in cystic fibrosis subjects. A phase II randomized placebo-controlled trial. J. Cyst. Fibros. 2015, 14, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Oxidative Stress in Chronic Obstructive Pulmonary Disease. Antioxidants 2022, 11, 965. [Google Scholar] [CrossRef] [PubMed]
- Vogelmeier, C.F.; Román-Rodríguez, M.; Singh, D.; Han, M.K.; Rodríguez-Roisin, R.; Ferguson, G.T. Goals of COPD treatment: Focus on symptoms and exacerbations. Respir. Med. 2020, 166, 105938. [Google Scholar] [CrossRef]
- Boutten, A.; Goven, D.; Artaud-Macari, E.; Boczkowski, J.; Bonay, M. NRF2 targeting: A promising therapeutic strategy in chronic obstructive pulmonary disease. Trends Mol. Med. 2011, 17, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. The cytokine network in COPD. Am. J. Respir. Cell Mol. Biol. 2009, 41, 631–638. [Google Scholar] [CrossRef]
- Barnes, P.J. Cellular and molecular mechanisms of asthma and COPD. Clin. Sci. 2017, 131, 1541–1558. [Google Scholar] [CrossRef]
- Barnes, P.J.; Baker, J.; Donnelly, L.E. Cellular senescence as a mechanism and target in chronic lung diseases. Am. J. Respir. Crit. Care Med. 2019, 200, 556–564. [Google Scholar] [CrossRef]
- Cloonan, S.M.; Kim, K.; Esteves, P.; Trian, T.; Barnes, P.J. Mitochondrial dysfunction in lung ageing and disease. Eur. Respir. Rev. 2020, 29, 200165. [Google Scholar] [CrossRef]
- Dransfield, M.T.; Wilhelm, A.M.; Flanagan, B.; Courville, C.; Tidwell, S.L.; Raju, S.V.; Gaggar, A.; Steele, C.; Tang, L.P.; Liu, B.; et al. Acquired cystic fibrosis transmembrane conductance regulator dysfunction in the lower airways in COPD. Chest 2013, 144, 498–506. [Google Scholar] [CrossRef]
- MacNee, W.; Bridgeman, M.M.; Marsden, M.; Drost, E.; Lannan, S.; Selby, C.; Donaldson, K. The effects of N-acetylcysteine and glutathione on smoke-induced changes in lung phagocytes and epithelial cells. Am. J. Med. 1991, 91, 60S–66S. [Google Scholar] [CrossRef]
- van der Deen, M.; Homan, S.; Timmer-Bosscha, H.; Scheper, R.J.; Timens, W.; Postma, D.S.; de Vries, E.G. Effect of COPD treatments on MRP1-mediated transport in bronchial epithelial cells. Int. J. Chron. Obstruct. Pulmon. Dis. 2008, 3, 469–475. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Milara, J.; Juan, G.; Peiró, T.; Serrano, A.; Cortijo, J. Neutrophil activation in severe, early-onset COPD patients versus healthy non-smoker subjects in vitro: Effects of antioxidant therapy. Respiration 2012, 83, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Messier, E.M.; Day, B.J.; Bahmed, K.; Kleeberger, S.R.; Tuder, R.M.; Bowler, R.P.; Chu, H.W.; Mason, R.J.; Kosmider, B. N-acetylcysteine protects murine alveolar type II cells from cigarette smoke injury in a nuclear erythroid 2-related factor-2-independent manner. Am. J. Respir. Cell Mol. Biol. 2013, 48, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Aridgides, D.S.; Mellinger, D.L.; Armstrong, D.A.; Hazlett, H.F.; Dessaint, J.A.; Hampton, T.H.; Atkins, G.T.; Carroll, J.L.; Ashare, A. Functional and metabolic impairment in cigarette smoke-exposed macrophages is tied to oxidative stress. Sci. Rep. 2019, 9, 9624. [Google Scholar] [CrossRef]
- Xu, L.; Cai, B.Q.; Zhu, Y.J. Pathogenesis of cigarette smoke-induced chronic obstructive pulmonary disease and therapeutic effects of glucocorticoids and N-acetylcysteine in rats. Chin. Med. J. 2004, 117, 1611–1619. [Google Scholar] [PubMed]
- Cai, S.; Chen, P.; Zhang, C.; Chen, J.B.; Wu, J. Oral N-acetylcysteine attenuates pulmonary emphysema and alveolar septal cell apoptosis in smoking-induced COPD in rats. Respirology 2009, 14, 354–359. [Google Scholar] [CrossRef]
- Li, F.; Wiegman, C.; Seiffert, J.M.; Zhu, J.; Clarke, C.; Chang, Y.; Bhavsar, P.; Adcock, I.; Zhang, J.; Zhou, X.; et al. Effects of N-acetylcysteine in ozone-induced chronic obstructive pulmonary disease model. PLoS ONE 2013, 8, e80782. [Google Scholar] [CrossRef]
- Zhu, L.; Xu, F.; Kang, X.; Zhou, J.; Yao, Q.; Lin, Y.; Zhang, W. The antioxidant N-acetylcysteine promotes immune response and inhibits epithelial-mesenchymal transition to alleviate pulmonary fibrosis in chronic obstructive pulmonary disease by suppressing the VWF/p38 MAPK axis. Mol. Med. 2021, 27, 97. [Google Scholar] [CrossRef]
- Black, P.N.; Morgan-Day, A.; McMillan, T.E.; Poole, P.J.; Young, R.P. Randomised, controlled trial of N-acetylcysteine for treatment of acute exacerbations of chronic obstructive pulmonary disease [ISRCTN21676344]. BMC. Pulm. Med. 2004, 4, 13. [Google Scholar] [CrossRef]
- Decramer, M.; Rutten-van Mölken, M.; Dekhuijzen, P.N.; Troosters, T.; van Herwaarden, C.; Pellegrino, R.; van Schayck, C.P.; Olivieri, D.; Del Donno, M.; De Backer, W.; et al. Effects of N-acetylcysteine on outcomes in chronic obstructive pulmonary disease (Bronchitis Randomized on NAC Cost-Utility Study, BRONCUS): A randomised placebo-controlled trial. Lancet 2005, 365, 1552–1560. [Google Scholar] [CrossRef]
- Stav, D.; Raz, M. Effect of N-acetylcysteine on air trapping in COPD: A randomized placebo-controlled study. Chest 2009, 136, 381–386. [Google Scholar] [CrossRef]
- Zhang, L.; Xiong, Y.; Du, L. Efficacy and Safety of N-Acetylcysteine for Chronic Obstructive Pulmonary Disease and Chronic Bronchitis. BioMed Res. Int. 2022, 2022, 9133777. [Google Scholar] [CrossRef]
- Liu, X.; Hu, Z.; Zhou, H. N-Acetylcysteine Improves Inflammatory Response in COPD Patients by Regulating Th17/Treg Balance through Hypoxia Inducible Factor-1α Pathway. BioMed Res. Int. 2021, 2021, 6372128. [Google Scholar] [CrossRef]
- Cazzola, M.; Calzetta, L.; Facciolo, F.; Rogliani, P.; Matera, M.G. Pharmacological investigation on the anti-oxidant and anti-inflammatory activity of N-acetylcysteine in an ex vivo model of COPD exacerbation. Respir. Res. 2017, 18, 26. [Google Scholar] [CrossRef]
- Calzetta, L.; Rogliani, P.; Facciolo, F.; Rinaldi, B.; Cazzola, M.; Matera, M.G. N-Acetylcysteine protects human bronchi by modulating the release of neurokinin A in an ex vivo model of COPD exacerbation. Biomed. Pharmacother. 2018, 103, 1–8. [Google Scholar] [CrossRef]
- Papi, A.; Brightling, C.; Pedersen, S.E.; Reddel, H.K. Asthma. Lancet 2018, 391, 783–800. [Google Scholar] [CrossRef]
- Kuruvilla, M.E.; Lee, F.E.; Lee, G.B. Understanding Asthma Phenotypes, Endotypes, and Mechanisms of Disease. Clin. Rev. Allergy Immunol. 2019, 56, 219–233. [Google Scholar] [CrossRef]
- Sahiner, U.M.; Birben, E.; Erzurum, S.; Sackesen, C.; Kalayci, Ö. Oxidative stress in asthma: Part of the puzzle. Pediatr. Allergy Immunol. 2018, 29, 789–800. [Google Scholar] [CrossRef]
- Rahman, I.; Biswas, S.K.; Kode, A. Oxidant and antioxidant balance in the airways and airway diseases. Eur. J. Pharmacol. 2006, 533, 222–239. [Google Scholar] [CrossRef]
- Romieu, I.; Sienra-Monge, J.J.; Ramírez-Aguilar, M.; Téllez-Rojo, M.M.; Moreno-Macías, H.; Reyes-Ruiz, N.I.; del Río-Navarro, B.E.; Ruiz-Navarro, M.X.; Hatch, G.; Slade, R.; et al. Antioxidant supplementation and lung functions among children with asthma exposed to high levels of air pollutants. Am. J. Respir. Crit. Care Med. 2002, 166, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.G.; Garg, M.L.; Smart, J.M.; Scott, H.A.; Barker, D.; Gibson, P.G. Manipulating antioxidant intake in asthma: A randomized controlled trial. Am. J. Clin. Nutr. 2012, 96, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, B.; Berthon, B.S.; Jensen, M.E.; McLoughlin, R.F.; Wark, P.A.B.; Nichol, K.; Williams, E.J.; Baines, K.J.; Collison, A.; Starkey, M.R.; et al. The Effects of Increasing Fruit and Vegetable Intake in Children with Asthma on the Modulation of Innate Immune Responses. Nutrients 2022, 14, 3087. [Google Scholar] [CrossRef]
- Wuyts, W.A.; Vanaudenaerde, B.M.; Dupont, L.J.; Demedts, M.G.; Verleden, G.M. N-acetylcysteine reduces chemokine release via inhibition of p38 MAPK in human airway smooth muscle cells. Eur. Respir. J. 2003, 22, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Losa, M.; Cortijo, J.; Juan, G.; O’Connor, J.E.; Sanz, M.J.; Santangelo, F.; Morcillo, E.J. Inhibitory effects of N-acetylcysteine on the functional responses of human eosinophils in vitro. Clin. Exp. Allergy 2007, 37, 714–722. [Google Scholar] [CrossRef]
- Lin, Y.C.; Lin, Y.C.; Tsai, M.L.; Liao, W.T.; Hung, C.H. TSLP regulates mitochondrial ROS-induced mitophagy via histone modification in human monocytes. Cell Biosci. 2022, 12, 32. [Google Scholar] [CrossRef]
- Blesa, S.; Cortijo, J.; Mata, M.; Serrano, A.; Closa, D.; Santangelo, F.; Estrela, J.M.; Suchankova, J.; Morcillo, E.J. Oral N-acetylcysteine attenuates the rat pulmonary inflammatory response to antigen. Eur. Respir. J. 2003, 21, 394–400. [Google Scholar] [CrossRef]
- Eftekhari, P.; Hajizadeh, S.; Raoufy, M.R.; Masjedi, M.R.; Yang, M.; Hansbro, N.; Li, J.J.; Foster, P.S. Preventive effect of N-acetylcysteine in a mouse model of steroid resistant acute exacerbation of asthma. EXCLI J. 2013, 12, 184–192. [Google Scholar]
- Silveira, J.S.; Antunes, G.L.; Kaiber, D.B.; da Costa, M.S.; Marques, E.P.; Ferreira, F.S.; Gassen, R.B.; Breda, R.V.; Wyse, A.T.S.; Pitrez, P.; et al. Reactive oxygen species are involved in eosinophil extracellular traps release and in airway inflammation in asthma. J. Cell. Physiol. 2019, 234, 23633–23646. [Google Scholar] [CrossRef]
- Lee, P.H.; Hong, J.; Jang, A.S. N-acetylcysteine decreases airway inflammation and responsiveness in asthma by modulating claudin 18 expression. Korean J. Intern. Med. 2020, 35, 1229–1237. [Google Scholar] [CrossRef]
- Liu, X.; Yi, M.; Jin, R.; Feng, X.; Ma, L.; Wang, Y.; Shan, Y.; Yang, Z.; Zhao, B. Correlation between oxidative stress and NF-κB signaling pathway in the obesity-asthma mice. Mol. Biol. Rep. 2020, 47, 3735–3744. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Yao, L.; Shi, J.; Li, J.; Xu, C. Protective effects of N-acetylcysteine on a chemical-induced murine model of asthma. J. Asthma 2021, 58, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Bylin, G.; Hedenstierna, G.; Lagerstrand, L.; Wagner, P.D. No influence of acetylcysteine on gas exchange and spirometry in chronic asthma. Eur. J. Respir. Dis. 1987, 71, 102–107. [Google Scholar] [PubMed]
- Aliyali, M.; Poorhasan Amiri, A.; Sharifpoor, A.; Zalli, F. Effects of N-acetylcysteine on asthma exacerbation. Iran. J. Allergy Asthma Immunol. 2010, 9, 103–109. [Google Scholar]
- Kurtoglu, Y.E.; Navath, R.S.; Wang, B.; Kannan, S.; Romero, R.; Kannan, R.M. Poly(amidoamine) dendrimer-drug conjugates with disulfide linkages for intracellular drug delivery. Biomaterials 2009, 30, 2112–2121. [Google Scholar] [CrossRef]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef]
- Luppi, F.; Kalluri, M.; Faverio, P.; Kreuter, M.; Ferrara, G. Idiopathic pulmonary fibrosis beyond the lung: Understanding disease mechanisms to improve diagnosis and management. Respir. Res. 2021, 22, 109. [Google Scholar] [CrossRef]
- Pardo, A.; Selman, M. Lung Fibroblasts, Aging, and Idiopathic Pulmonary Fibrosis. Ann. Am. Thorac. Soc. 2016, 13 (Suppl. S5), S417–S421. [Google Scholar] [CrossRef]
- Noble, P.W.; Barkauskas, C.E.; Jiang, D. Pulmonary fibrosis: Patterns and perpetrators. J. Clin. Investig. 2012, 122, 2756–2762. [Google Scholar] [CrossRef]
- Cantin, A.M.; Hubbard, R.C.; Crystal, R.G. Glutathione deficiency in the epithelial lining fluid of the lower respiratory tract in idiopathic pulmonary fibrosis. Am. Rev. Respir. Dis. 1989, 139, 370–372. [Google Scholar] [CrossRef]
- Otoupalova, E.; Smith, S.; Cheng, G.; Thannickal, V.J. Oxidative Stress in Pulmonary Fibrosis. Compr. Physiol. 2020, 10, 509–547. [Google Scholar] [CrossRef] [PubMed]
- Walters, D.M.; Cho, H.Y.; Kleeberger, S.R. Oxidative stress and antioxidants in the pathogenesis of pulmonary fibrosis: A potential role for Nrf2. Antioxid. Redox Signal. 2008, 10, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wei, J.; Deng, H.; Zheng, L.; Yang, H.; Lv, X. The Role of Nrf2 in Pulmonary Fibrosis: Molecular Mechanisms and Treatment Approaches. Antioxidants 2022, 11, 1685. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Kang, H.; Chen, S. From Basic Research to Clinical Practice: Considerations for Treatment Drugs for Silicosis. Int. J. Mol. Sci. 2023, 24, 8333. [Google Scholar] [CrossRef]
- Glass, D.S.; Grossfeld, D.; Renna, H.A.; Agarwala, P.; Spiegler, P.; DeLeon, J.; Reiss, A.B. Idiopathic pulmonary fibrosis: Current and future treatment. Clin. Respir. J. 2022, 16, 84–96. [Google Scholar] [CrossRef]
- Estornut, C.; Milara, J.; Bayarri, M.A.; Belhadj, N.; Cortijo, J. Targeting Oxidative Stress as a Therapeutic Approach for Idiopathic Pulmonary Fibrosis. Front. Pharmacol. 2022, 12, 794997. [Google Scholar] [CrossRef]
- Cu, A.; Ye, Q.; Sarria, R.; Nakamura, S.; Guzman, J.; Costabel, U. N-acetylcysteine inhibits TNF-α, sTNFR, and TGF-β1 release by alveolar macrophages in idiopathic pulmonary fibrosis in vitro. Sarcoidosis Vasc. Diffus. Lung Dis. 2009, 26, 147–154. [Google Scholar]
- Radomska-Leśniewska, D.M.; Skopińska-Rózewska, E.; Jankowska-Steifer, E.; Sobiecka, M.; Sadowska, A.M.; Hevelke, A.; Malejczyk, J. N-acetylcysteine inhibits IL-8 and MMP-9 release and ICAM-1 expression by bronchoalveolar cells from interstitial lung disease patients. Pharmacol. Rep. 2010, 62, 131–138. [Google Scholar] [CrossRef]
- Shahzeidi, S.; Sarnstrand, B.; Jeffery, P.K.; McAnulty, R.J.; Laurent, G.J. Oral N-acetylcysteine reduces bleomycin-induced collagen deposition in the lungs of mice. Eur. Respir. J. 1991, 4, 845–852. [Google Scholar] [CrossRef]
- Yildirim, Z.; Kotuk, M.; Iraz, M.; Kuku, I.; Ulu, R.; Armutcu, F.; Ozen, S. Attenuation of bleomycin-induced lung fibrosis by oral sulfhydryl containing antioxidants in rats: Erdosteine and N-acetylcysteine. Pulm. Pharmacol. Ther. 2005, 18, 367–373. [Google Scholar] [CrossRef]
- Teixeira, K.C.; Soares, F.S.; Rocha, L.G.; Silveira, P.C.; Silva, L.A.; Valença, S.S.; Dal Pizzol, F.; Streck, E.L.; Pinho, R.A. Attenuation of bleomycin-induced lung injury and oxidative stress by N-acetylcysteine plus deferoxamine. Pulm. Pharmacol. Ther. 2008, 21, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.C.; Tian, L.Y.; Cheng, W. Efficacy study of edaravone and acetylcysteine towards bleomycin-induced rat pulmonary fibrosis. Int. J. Clin. Exp. Med. 2015, 8, 8730–8739. [Google Scholar] [PubMed]
- Yu, W.; Guo, F.; Song, X. Effects and mechanisms of pirfenidone, prednisone and acetylcysteine on pulmonary fibrosis in rat idiopathic pulmonary fibrosis models. Pharm. Biol. 2017, 55, 450–455. [Google Scholar] [CrossRef]
- Chen, H.; Chen, H.; Liang, J.; Gu, X.; Zhou, J.; Xie, C.; Lv, X.; Wang, R.; Li, Q.; Mao, Z.; et al. TGF-β1/IL-11/MEK/ERK signaling mediates senescence-associated pulmonary fibrosis in a stress-induced premature senescence model of Bmi-1 deficiency. Exp. Mol. Med. 2020, 52, 130–151. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.; Buhl, R.; Magnussen, H. The effect of oral N-acetylcysteine on lung glutathione levels in idiopathic pulmonary fibrosis. Eur. Respir. J. 1994, 7, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Behr, J.; Maier, K.; Degenkolb, B.; Krombach, F.; Vogelmeier, C. Antioxidative and clinical effects of high-dose N-acetylcysteine in fibrosing alveolitis. Adjunctive therapy to maintenance immunosuppression. Am. J. Respir. Crit. Care Med. 1997, 156, 1897–1901. [Google Scholar] [CrossRef]
- Homma, S.; Azuma, A.; Taniguchi, H.; Ogura, T.; Mochiduki, Y.; Sugiyama, Y.; Nakata, K.; Yoshimura, K.; Takeuchi, M.; Kudoh, S.; et al. Efficacy of inhaled N-acetylcysteine monotherapy in patients with early stage idiopathic pulmonary fibrosis. Respirology 2012, 17, 467–477. [Google Scholar] [CrossRef]
- Okuda, R.; Matsushima, H.; Oba, T.; Kawabe, R.; Matsubayashi, M.; Amano, M.; Nishizawa, T.; Honda, K. Efficacy and safety of inhaled N-acetylcysteine in idiopathic pulmonary fibrosis: A prospective, single-arm study. Respir. Investig. 2016, 54, 156–161. [Google Scholar] [CrossRef]
- Muramatsu, Y.; Sugino, K.; Ishida, F.; Tatebe, J.; Morita, T.; Homma, S. Effect of inhaled N-acetylcysteine monotherapy on lung function and redox balance in idiopathic pulmonary fibrosis. Respir. Investig. 2016, 54, 170–178. [Google Scholar] [CrossRef]
- Izumi, S.; Iikura, M.; Hirano, S. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N. Engl. J. Med. 2012, 367, 870. [Google Scholar] [CrossRef]
- Sakamoto, S.; Kataoka, K.; Kondoh, Y.; Kato, M.; Okamoto, M.; Mukae, H.; Bando, M.; Suda, T.; Yatera, K.; Tanino, Y.; et al. Pirfenidone plus inhaled N-acetylcysteine for idiopathic pulmonary fibrosis: A randomised trial. Eur. Respir. J. 2021, 57, 2000348. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Song, Z.; Guan, Y.; Zhang, J.; Zou, J.; Sun, Y.; Shao, H. Clinical efficacy and quality of life effect of acetylcysteine plus pirfenidone in patients with pulmonary fibrosis. Am. J. Transl. Res. 2022, 14, 5660–5668. [Google Scholar] [PubMed]
- Barnes, H.; Goh, N.S.L.; Leong, T.L.; Hoy, R. Silica-associated lung disease: An old-world exposure in modern industries. Respirology 2019, 24, 1165–1175. [Google Scholar] [CrossRef] [PubMed]
- Vallyathan, V.; Castranova, V.; Pack, D.; Leonard, S.; Shumaker, J.; Hubbs, A.F.; Shoemaker, D.A.; Ramsey, D.M.; Pretty, J.R.; McLaurin, J.L.; et al. Freshly fractured quartz inhalation leads to enhanced lung injury and inflammation. Potential role of free radicals. Am. J. Respir. Crit. Care Med. 1995, 152, 1003–1009. [Google Scholar] [CrossRef]
- Hamilton, R.F., Jr.; Thakur, S.A.; Holian, A. Silica binding and toxicity in alveolar macrophages. Free Radic. Biol. Med. 2008, 44, 1246–1258. [Google Scholar] [CrossRef]
- Porter, D.W.; Ye, J.; Ma, J.; Barger, M.; Robinson, V.A.; Ramsey, D.; McLaurin, J.; Khan, A.; Landsittel, D.; Teass, A.; et al. Time course of pulmonary response of rats to inhalation of crystalline silica: NF-κB activation, inflammation, cytokine production, and damage. Inhal. Toxicol. 2002, 14, 349–367. [Google Scholar] [CrossRef]
- Harijith, A.; Ebenezer, D.L.; Natarajan, V. Reactive oxygen species at the crossroads of inflammasome and inflammation. Front. Physiol. 2014, 5, 352. [Google Scholar] [CrossRef]
- Seo, M.S.; Kim, J.K.; Lim, Y.; Kang, S.W.; Cho, Y.J.; Lee, W.K.; Kim, H.J.; Cho, K.K.; Lee, K.H.; Rhee, S.G. Rapid degradation of PrxI and PrxII induced by silica in Rat2 cells. Biochem. Biophys. Res. Commun. 1999, 265, 541–544. [Google Scholar] [CrossRef]
- Biswas, R.; Bunderson-Schelvan, M.; Holian, A. Potential role of the inflammasome-derived inflammatory cytokines in pulmonary fibrosis. Pulm. Med. 2011, 2011, 105707. [Google Scholar] [CrossRef]
- Liu, X.; Wang, J.; Dou, P.; Zhang, X.; Ran, X.; Liu, L.; Dou, D. The Ameliorative Effects of Arctiin and Arctigenin on the Oxidative Injury of Lung Induced by Silica via TLR-4/NLRP3/TGF-β Signaling Pathway. Oxid. Med. Cell. Longev. 2021, 2021, 5598980. [Google Scholar] [CrossRef]
- Ma, Y.; Liang, Q.; Wang, F.; Yan, K.; Sun, M.; Lin, L.; Li, T.; Duan, J.; Sun, Z. Silica nanoparticles induce pulmonary autophagy dysfunction and epithelial-to-mesenchymal transition via p62/NF-κB signaling pathway. Ecotoxicol. Environ. Saf. 2022, 232, 113303. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.C.; Du, Y.; Zhang, X.F.; Guan, L.; Liu, X.M.; Zeng, M. SiO2 dust induces inflammation and pulmonary fibrosis in rat lungs through activation of ASMase/ceramide pathway. J. Appl. Toxicol. 2023. ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Shi, F.; Wang, X.; Yang, P.; Sun, K.; Zhang, L.; Hao, X.; Li, X.; Li, J.; Jin, Y. Silica dust exposure induces pulmonary fibrosis through autophagy signaling. Environ. Toxicol. 2021, 36, 1269–1277. [Google Scholar] [CrossRef]
- Li, T.; Yang, X.; Xu, H.; Liu, H. Early Identification, Accurate Diagnosis, and Treatment of Silicosis. Can. Respir. J. 2022, 2022, 3769134. [Google Scholar] [CrossRef] [PubMed]
- Barrett, E.G.; Johnston, C.; Oberdörster, G.; Finkelstein, J.N. Antioxidant treatment attenuates cytokine and chemokine levels in murine macrophages following silica exposure. Toxicol. Appl. Pharmacol. 1999, 158, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.L.; Go, Y.H.; Hur, K.C.; Castranova, V. Silica-induced nuclear factor-κB activation: Involvement of reactive oxygen species and protein tyrosine kinase activation. J. Toxicol. Environ. Health A 2000, 60, 27–46. [Google Scholar] [CrossRef]
- Hu, S.; Zhao, H.; Al-Humadi, N.H.; Yin, X.J.; Ma, J.K. Silica-induced apoptosis in alveolar macrophages: Evidence of in vivo thiol depletion and the activation of mitochondrial pathway. J. Toxicol. Environ. Health A 2006, 69, 1261–1284. [Google Scholar] [CrossRef]
- Hu, S.; Zhao, H.; Yin, X.J.; Ma, J.K. Role of mitochondria in silica-induced apoptosis of alveolar macrophages: Inhibition of apoptosis by rhodamine 6G and N-acetyl-L-cysteine. J. Toxicol. Environ. Health A 2007, 70, 1403–1415. [Google Scholar] [CrossRef]
- Tang, M.; Yang, Z.; Liu, J.; Zhang, X.; Guan, L.; Liu, X.; Zeng, M. Combined intervention with N-acetylcysteine and desipramine alleviated silicosis development by regulating the Nrf2/HO-1 and ASMase/ceramide signaling pathways. Ecotoxicol. Environ. Saf. 2022, 242, 113914. [Google Scholar] [CrossRef]
- Huang, H.; Chen, M.; Liu, F.; Wu, H.; Wang, J.; Chen, J.; Liu, M.; Li, X. N-acetylcysteine tiherapeutically protects against pulmonary fibrosis in a mouse model of silicosis. Biosci. Rep. 2019, 39, BSR20190681. [Google Scholar] [CrossRef]
- Guo, X.; Qi, J.; Li, H.; Xing, Z. Clinical efficacy of acetylcysteine combined with tetrandrine tablets on patients with silicosis and its effect on exercise tolerance and pulmonary function. Exp. Ther. Med. 2020, 20, 1285–1290. [Google Scholar] [CrossRef] [PubMed]
- Zosky, G.R.; Sly, P.D. Animal models of asthma. Clin. Exp. Allergy 2007, 37, 973–988. [Google Scholar] [CrossRef] [PubMed]
- Mouratis, M.A.; Aidinis, V. Modeling pulmonary fibrosis with bleomycin. Curr. Opin. Pulm. Med. 2011, 17, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Fricker, M.; Deane, A.; Hansbro, P.M. Animal models of chronic obstructive pulmonary disease. Expert Opin. Drug Discov. 2014, 9, 629–645. [Google Scholar] [CrossRef] [PubMed]
- Kianmeher, M.; Ghorani, V.; Boskabady, M.H. Animal Model of Asthma, Various Methods and Measured Parameters: A Methodological Review. Iran. J. Allergy Asthma Immunol. 2016, 15, 445–465. [Google Scholar]
- Lavelle, G.M.; White, M.M.; Browne, N.; McElvaney, N.G.; Reeves, E.P. Animal Models of Cystic Fibrosis Pathology: Phenotypic Parallels and Divergences. BioMed Res. Int. 2016, 2016, 5258727. [Google Scholar] [CrossRef]
- Jones, B.; Donovan, C.; Liu, G.; Gomez, H.M.; Chimankar, V.; Harrison, C.L.; Wiegman, C.H.; Adcock, I.M.; Knight, D.A.; Hirota, J.A.; et al. Animal models of COPD: What do they tell us? Respirology 2017, 22, 21–32. [Google Scholar] [CrossRef]
- Sadowska, A.M.; Manuel-Y-Keenoy, B.; De Backer, W.A. Antioxidant and anti-inflammatory efficacy of NAC in the treatment of COPD: Discordant in vitro and in vivo dose-effects: A review. Pulm. Pharmacol. Ther. 2007, 20, 9–22. [Google Scholar] [CrossRef]
- Kim, V.; Evans, C.M.; Dickey, B.F. Dawn of a New Era in the Diagnosis and Treatment of Airway Mucus Dysfunction. Am. J. Respir. Crit. Care Med. 2019, 199, 133–134. [Google Scholar] [CrossRef]
- Bonser, L.R.; Erle, D.J. Airway Mucus and Asthma: The Role of MUC5AC and MUC5B. J. Clin. Med. 2017, 6, 112. [Google Scholar] [CrossRef]
- Caramori, G.; Di Gregorio, C.; Carlstedt, I.; Casolari, P.; Guzzinati, I.; Adcock, I.M.; Barnes, P.J.; Ciaccia, A.; Cavallesco, G.; Chung, K.F.; et al. Mucin expression in peripheral airways of patients with chronic obstructive pulmonary disease. Histopathology 2004, 45, 477–484. [Google Scholar] [CrossRef]
- Henke, M.O.; Renner, A.; Huber, R.M.; Seeds, M.C.; Rubin, B.K. MUC5AC and MUC5B Mucins Are Decreased in Cystic Fibrosis Airway Secretions. Am. J. Respir. Cell Mol. Biol. 2004, 31, 86–91. [Google Scholar] [CrossRef]
- Henke, M.O.; John, G.; Germann, M.; Lindemann, H.; Rubin, B.K. MUC5AC and MUC5B mucins increase in cystic fibrosis airway secretions during pulmonary exacerbation. Am. J. Respir. Crit. Care Med. 2007, 175, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Addante, A.; Raymond, W.; Gitlin, I.; Charbit, A.; Orain, X.; Scheffler, A.W.; Kuppe, A.; Duerr, J.; Daniltchenko, M.; Drescher, M.; et al. A novel thiol-saccharide mucolytic for the treatment of muco-obstructive lung diseases. Eur. Respir. J. 2023, 61, 2202022. [Google Scholar] [CrossRef] [PubMed]
- Pangeni, R.; Meng, T.; Poudel, S.; Sharma, D.; Hutsell, H.; Ma, J.; Rubin, B.K.; Longest, W.; Hindle, M.; Xu, Q. Airway mucus in pulmonary diseases: Muco-adhesive and muco-penetrating particles to overcome the airway mucus barriers. Int. J. Pharm. 2023, 634, 122661. [Google Scholar] [CrossRef] [PubMed]
- De Nardo, D.; De Nardo, C.M.; Latz, E. New insights into mechanisms controlling the NLRP3 inflammasome and its role in lung disease. Am. J. Pathol. 2014, 184, 42–54. [Google Scholar] [CrossRef]
- Lee, S.; Suh, G.Y.; Ryter, S.W.; Choi, A.M. Regulation and Function of the Nucleotide Binding Domain Leucine-Rich Repeat-Containing Receptor, Pyrin Domain-Containing-3 Inflammasome in Lung Disease. Am. J. Respir. Cell Mol. Biol. 2016, 54, 151–160. [Google Scholar] [CrossRef]
- Thomas, P.G.; Dash, P.; Aldridge, J.R., Jr.; Ellebedy, A.H.; Reynolds, C.; Funk, A.J.; Martin, W.J.; Lamkanfi, M.; Webby, R.J.; Boyd, K.L.; et al. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity 2009, 30, 566–575. [Google Scholar] [CrossRef]
- Liu, B.; He, R.; Zhang, L.; Hao, B.; Jiang, W.; Wang, W.; Geng, Q. Inflammatory Caspases Drive Pyroptosis in Acute Lung Injury. Front. Pharmacol. 2021, 12, 631256. [Google Scholar] [CrossRef]
- Vora, S.M.; Lieberman, J.; Wu, H. Inflammasome activation at the crux of severe COVID-19. Nat. Rev. Immunol. 2021, 21, 694–703. [Google Scholar] [CrossRef]
- Zhang, W.J.; Chen, S.J.; Zhou, S.C.; Wu, S.Z.; Wang, H. Inflammasomes and Fibrosis. Front. Immunol. 2021, 12, 643149. [Google Scholar] [CrossRef] [PubMed]
- Lam, M.; Mansell, A.; Tate, M.D. Another One Fights the Dust: Targeting the NLRP3 Inflammasome for the Treatment of Silicosis. Am. J. Respir. Cell Mol. Biol. 2022, 66, 601–611. [Google Scholar] [CrossRef]
- Colunga Biancatelli, R.M.L.; Solopov, P.A.; Catravas, J.D. The Inflammasome NLR Family Pyrin Domain-Containing Protein 3 (NLRP3) as a Novel Therapeutic Target for Idiopathic Pulmonary Fibrosis. Am. J. Pathol. 2022, 192, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Cantin, A.M. Cystic Fibrosis Lung Disease and Immunometabolism. Targeting the NLRP3 Inflammasome. Am. J. Respir. Crit. Care Med. 2019, 200, 1335–1337. [Google Scholar] [CrossRef] [PubMed]
- Graustein, A.D.; Berrington, W.R.; Buckingham, K.J.; Nguyen, F.K.; Joudeh, L.L.; Rosenfeld, M.; Bamshad, M.J.; Gibson, R.L.; Hawn, T.R.; Emond, M.J. Inflammasome Genetic Variants, Macrophage Function, and Clinical Outcomes in Cystic Fibrosis. Am. J. Respir. Cell Mol. Biol. 2021, 65, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Theofani, E.; Semitekolou, M.; Morianos, I.; Samitas, K.; Xanthou, G. Targeting NLRP3 Inflammasome Activation in Severe Asthma. J. Clin. Med. 2019, 8, 1615. [Google Scholar] [CrossRef]
- Wu, Y.; Di, X.; Zhao, M.; Li, H.; Bai, L.; Wang, K. The role of the NLRP3 inflammasome in chronic inflammation in asthma and chronic obstructive pulmonary disease. Immun. Inflamm. Dis. 2022, 10, e750. [Google Scholar] [CrossRef]
- Qiu, Z.; He, Y.; Ming, H.; Lei, S.; Leng, Y.; Xia, Z.Y. Lipopolysaccharide (LPS) Aggravates High Glucose- and Hypoxia/Reoxygenation-Induced Injury through Activating ROS-Dependent NLRP3 Inflammasome-Mediated Pyroptosis in H9C2 Cardiomyocytes. J. Diabetes Res. 2019, 2019, 8151836. [Google Scholar] [CrossRef]
- Hindman, B.; Ma, Q. Carbon nanotubes and crystalline silica stimulate robust ROS production, inflammasome activation, and IL-1β secretion in macrophages to induce myofibroblast transformation. Arch. Toxicol. 2019, 93, 887–907. [Google Scholar] [CrossRef]
- Xie, C.; Ge, M.; Jin, J.; Xu, H.; Mao, L.; Geng, S.; Wu, J.; Zhu, J.; Li, X.; Zhong, C. Mechanism investigation on Bisphenol S-induced oxidative stress and inflammation in murine RAW264.7 cells: The role of NLRP3 inflammasome, TLR4, Nrf2 and MAPK. J. Hazard. Mater. 2020, 394, 122549. [Google Scholar] [CrossRef]
- Milara, J.; Martínez-Expósito, F.; Montero, P.; Roger, I.; Bayarri, M.A.; Ribera, P.; Oishi-Konari, M.N.; Alba-García, J.R.; Zapater, E.; Cortijo, J. N-acetylcysteine Reduces Inflammasome Activation Induced by SARS-CoV-2 Proteins In Vitro. Int. J. Mol. Sci. 2022, 23, 14518. [Google Scholar] [CrossRef]
- Fernández-Lázaro, D.; Domínguez-Ortega, C.; Busto, N.; Santamaría-Peláez, M.; Roche, E.; Gutiérez-Abejón, E.; Mielgo-Ayuso, J. Influence of N-Acetylcysteine Supplementation on Physical Performance and Laboratory Biomarkers in Adult Males: A Systematic Review of Controlled Trials. Nutrients 2023, 15, 2463. [Google Scholar] [CrossRef] [PubMed]
- Paschalis, V.; Theodorou, A.A.; Margaritelis, N.V.; Kyparos, A.; Nikolaidis, M.G. N-acetylcysteine supplementation increases exercise performance and reduces oxidative stress only in individuals with low levels of glutathione. Free Radic. Biol. Med. 2018, 115, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Tyuryaeva, I.; Lyublinskaya, O. Expected and Unexpected Effects of Pharmacological Antioxidants. Int. J. Mol. Sci. 2023, 24, 9303. [Google Scholar] [CrossRef] [PubMed]
- URL: FDA Policy Regarding N-acetyl-L-cysteine: Guidance for Industry. Available online: https://www.fda.gov/media/157784/download (accessed on 23 August 2023).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animal Model | Species | NAC Dose/Way of Delivery | Major Findings | Study |
|---|---|---|---|---|
| Model of CF | WT and βENaC-overexpressing mice | NAC (15 μL, 50 mg/kg), i.t. instillation in WT mice; NAC (200 mM, nasal nebulizations) in βENaC-overexpressing mice | WT mice: no reduction in Muc5b, rapid NAC clearance, ↑ total cells and neutrophils in BALF, epithelial injury; βENaC-overexpressing mice: no reduction in AW mucus obstruction | [99] |
| Cigarette smoke (CS)-induced model of COPD | Sprague Dawley rats | NAC (800 mg/kg, once a day, i.g.), from 30 days of CS exposure | ↓ lung damage, emphysema, and alveolar septal cell apoptosis by partial reversing of ↓ VEGF | [118] |
| Ozone-induced model of COPD | C57/BL6 mice | NAC (100 mg/kg, i.p.) before each exposure (preventive) or after completion of exposure (therapeutic), for 6 weeks | Preventive NAC: ↓ BALF macrophages, ↓ AW smooth muscle mass. Therapeutic NAC: reversed AW hyperresponsiveness, ↓ AW smooth muscle mass and apoptotic cells | [119] |
| Cigarette smoke (CS)-induced model of COPD | Rats | NAC (800 mg/kg, once a day, i.g.), from 2 days before the model establishment | ↓ inflammation, enhanced lung functions, ↓ fibrotic changes | [120] |
| OVA-induced model of asthma | Norway rats | NAC (3 mM/kg, once a day, p.o.), pretreatment daily from 7 days before challenge | ↓ AW hyperresponsiveness, ↓ lipid peroxidation, ↓ oxidized GSH, ↓ TNFα, iNOS, ICAM-1, and MUC5AC | [138] |
| OVA-induced model of asthma | Balb/c mice | NAC (320 mg/kg, i.p.), 30 min before and 12 h after each challenge | ↓ AW hyperresponsiveness, ↓ neutrophils and eosinophils in BALF, ↓ IL-13 and IL-5 | [139] |
| OVA-induced model of asthma | Balb/cJ mice | NAC (15 mg/100 g body weight, i.n.), 45 min before three i.n. challenges | ↓ AW inflammation, ↓ mitochondrial damage, ↓ goblet cell hyperplasia, ↓ eosinophil extracellular traps, ↓ oxidative stress in lungs | [140] |
| OVA-induced model of asthma | Balb/c mice | NAC (100 mg/kg), inhalation 1 h after each challenge for 6 days | ↓ AW hyperresponsiveness, ↓ AW inflammation, ↓ claudin 18 | [141] |
| Obesity-associated model of asthma | C57BL/6 J mice | NAC (200 mg/kg, i.g.) | ↓ inflammation and oxidative stress, ↓ IKK-β and NF-κB-P65, ↑ IκB-α, ↓ MDA | [142] |
| Chemical-induced model of asthma | Balb/c mice | NAC (100 mg/kg, i.p.), after each challenge | ↓ AW hyperresponsiveness, ↓ AW inflammation, ↓ goblet cell metaplasia, ↓ inflammatory cell counts in BALF, ↓ IL-4 and IL-5, ↓ oxidative stress | [143] |
| Bleomycin-induced model of IPF | Sprague Dawley rats | NAC (3 mM/kg, p.o.), daily from 1 week prior to i.t. bleomycin instillation and for 14 d postinstillation | ↓ collagen and ↓ inflammatory cells in the lungs, ↑ total GSH and taurine in BALF, no effect on lung edema formation | [72] |
| Bleomycin-induced model of IPF | Sprague Dawley rats | NAC (3 mM/kg, p.o.), daily from 1 week prior to i.t. bleomycin instillation and for 14 d postinstillation | ↓ collagen and fibrotic area in the lungs, ↓ MUC5ac expression, ↓ TNFα and MPO, ↓ mucus hypersecretion | [73] |
| Bleomycin-induced model of IPF | Sprague Dawley rats | NAC (3 mM/kg, p.o.), daily from 1 week prior to i.t. bleomycin instillation and for 14 d postinstillation | ↓ hydroxyproline, ↓ depletion of GSH peroxidase, prevention of ↑ in MPO, NO, and MDA | [161] |
| Bleomycin-induced model of IPF | Sprague Dawley rats | NAC (490 mg/kg/day, p.o.), daily from bleomycin instillation | ↓ lysyl oxidase activity, ↑ GSH, ↓ lung fibrosis, ↓ TGF-β1 and α-SMA expression | [75] |
| Bleomycin-induced model of IPF | Sprague Dawley rats | NAC (300 mg/kg/day, i.p.), daily from 1 week prior to i.t. bleomycin instillation and for 15 d postinstillation | ↑ GSH/GSSG ratio, ↓ NO, ↓ lipid hydroperoxides, ↓ lung weight, ↓ hydroxyproline, ↓ inflammatory cytokines, MPO, and LDH, ↓ deposition of collagen | [74] |
| Bleomycin-induced model of IPF | CF1 mice | NAC (20 mg/kg/day, i.p.), from day 0 of bleomycin instillation, for 60 days | ↓ lung injury, ↓ LDH, neutrophils, total cell counts, and protein in BALF | [162] |
| Bleomycin-induced model of IPF | Wistar rats | NAC (250 mg/kg/day, i.g.), from day 0 of bleomycin instillation, for 31 days | ↓ lung inflammation and fibrosis, ↓ MDA, ↑ SOD | [163] |
| Bleomycin-induced model of IPF | Wistar rats | NAC (4 mg/kg, 3 times/day, i.g.), from 1 day after bleomycin instillation, for 45 days | ↓ lung inflammation and fibrosis, ↓ TGF-β1, TNFα and PDGF expressions, ↑ caveolin-1 | [164] |
| Model of silicosis | Wistar rats | NAC (500 mg/kg/day, p.o.), for 7 days before and up to 28 days after silica instillation | ↓ fibrosis score, ↓ hydroxyproline, MDA, TNFα, IL-8, and hsCRP | [76] |
| Model of silicosis | Sprague Dawley rats | NAC (600 mg/kg/day, by gavage), from day 0 of silica instillation, for 28 days | ↓ ROS, ↓ changes in mitochondrial transmembrane potential, ↓ fibrotic changes, ↓ markers of apoptosis | [77] |
| Model of silicosis | C57BL/6J mice | NAC (1.73 mg/20 g, 3.46 mg/20 g, or 5.19 mg/20 g, by gavage), from day 0 of silica instillation, observation for 5 months | ↓ lung injury, fibrosis, and inflammation, ↓ MDA, ↑ GSH peroxidase, SOD, total antioxidant activity, and E-cadherin, ↓ oxidizing enzymes, vimentin, and cytochrome C | [191] |
| Diagnosis | No. of Patients | Treatment | Outcomes in NAC-Treated Patients | Study |
|---|---|---|---|---|
| CF | 21 in total (11 low-dose NAC, 10 high-dose NAC) | Oral NAC (700 mg daily or 2800 mg daily p.o.), for 12 weeks | High-dose NAC well-tolerated, trend to ↑ extracellular GSH | [100] |
| CF | 22 in total (10 in study 1, 12 in study 2) | Inhalational NAC: Study 1: from 2 (4 mg) to 8 puffs (16 mg) of NAC, or 12 puffs (24 mg) of NAC for 5 days; Study 2: single dose 12 puffs, 24 h monitoring | Study 1: dose-dependent ↓ in sputum viscoelasticity, ↓ in sputum solids; Study 2: ↓ mucus rigidity, max. at 4 h | [101] |
| CF | 21 in total (11 NAC, 10 control) | Oral NAC (2400 mg/day in 2 doses), for 30 days | ↓ oxidized vit. C, ↑ levels of vit. C, slight improvement in lung function | [102] |
| CF | 70 in total (36 NAC, 34 placebo) | Oral NAC (900 mg, 3 times a day) for 24 weeks | Stable lung function, but no effect on neutrophilic inflammation | [103] |
| CF | 5 in total | Inhalational NAC (20%, 1.27 M), 90 min observation | No effect on mucus properties | [99] |
| COPD | 50 in total (25 NAC, 25 placebo) (ISRCTN21676344) | Oral NAC (600 mg, twice daily), for 7 days (addition to corticosteroids and bronchodilators) | No effect on lung functions or median length of stay in hospital | [121] |
| COPD | 523 in total (256 NAC, 267 placebo) (BRONCUS trial) | Oral NAC (600 mg/day), patients followed for 3 years | No prevention of deterioration in lung functions, no prevention of exacerbations | [122] |
| COPD | 24 in total (12 NAC, 12 placebo) (NCT00476736) | Oral NAC (1200 mg/day), for 6 weeks | Beneficial effect on physical performance | [123] |
| COPD | 120 in total (58 NAC, 62 placebo) (HIACE trial, NCT01136239) | Oral NAC (600 mg bid or placebo after 4-week run-in), for 1 year | Enhanced small AW function, ↓ exacerbation frequency | [7] |
| COPD | 1006 in total (504 NAC, 502 placebo) (PANTHEON trial, ChiCTR-TRC-09000460) | Oral NAC (600 mg, twice daily), for 1 year | Prevention of acute exacerbations | [8] |
| COPD/chronic bronchitis | 45 in total (23 NAC, 22 placebo) (NCT01599884) | Oral NAC (1800 mg twice daily), for 8 weeks | No clinical benefit, study prematurely terminated | [20] |
| COPD | 100 in total (50 NAC, 50 placebo) | Oral NAC (900 mg twice daily), for 3 months | Absence of clinical improvement | [124] |
| COPD | 121 in total (60 NAC, 61 control) | Oral NAC (0.2 g × 10 bags, 0.4 g each time, 3 times/day), for 6 months | ↑ proportion of Treg in CD4+ T cells and ↓ Th17/Treg ratio | [125] |
| Asthma | 25 in total | Oral NAC (200 mg, 3 times daily), for 9 weeks | No effect on spirometric, lung mechanics or gas exchange variables, or frequency of clinical symptoms | [144] |
| Asthma | 50 in total (25 NAC, 25 placebo) | Oral NAC (600 mg/day), for 5 days | No effect on clinical symptoms | [145] |
| IPF | 14 in total (8 NAC, 6 controls) | Intravenous NAC (600 mg, 1800 mg and 4800 mg), in weekly intervals | ↑ GSH in low and moderate dose in IPF patients, but no effect on GSH in controls, no adverse effects | [25] |
| IPF | 31 in total (17 NAC, 14 controls) | Oral NAC (600 mg, 3 times/day), for 5 days | ↑ GSH in BALF/ELF, treatment well-tolerated | [166] |
| IPF | 18 in total | Oral NAC (600 mg, 3 times/daily), for 12 weeks | ↑ GSH in BALF/ELF, ↓ methionine sulfoxide content of BALF proteins, improved lung functions | [167] |
| IPF | 76 in total (38 NAC, 38 controls) | Inhalational NAC (352.4 mg, twice daily), observation for 48 weeks | No differences in lung functions | [168] |
| IPF | 28 in total | Inhalational NAC (352.4 mg, twice daily), observation for 26 weeks | Prevented a ↓ in FVC in mild-to moderate IPF, better effect in more severe IPF | [169] |
| IPF | 22 in total | Inhalational NAC (352.4 mg, twice daily), observation for 12 months | Prevented ↓ in FVC, ↓ oxidative imbalance, better effect in more severe oxidative stress | [170] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mokra, D.; Mokry, J.; Barosova, R.; Hanusrichterova, J. Advances in the Use of N-Acetylcysteine in Chronic Respiratory Diseases. Antioxidants 2023, 12, 1713. https://doi.org/10.3390/antiox12091713
Mokra D, Mokry J, Barosova R, Hanusrichterova J. Advances in the Use of N-Acetylcysteine in Chronic Respiratory Diseases. Antioxidants. 2023; 12(9):1713. https://doi.org/10.3390/antiox12091713
Chicago/Turabian StyleMokra, Daniela, Juraj Mokry, Romana Barosova, and Juliana Hanusrichterova. 2023. "Advances in the Use of N-Acetylcysteine in Chronic Respiratory Diseases" Antioxidants 12, no. 9: 1713. https://doi.org/10.3390/antiox12091713
APA StyleMokra, D., Mokry, J., Barosova, R., & Hanusrichterova, J. (2023). Advances in the Use of N-Acetylcysteine in Chronic Respiratory Diseases. Antioxidants, 12(9), 1713. https://doi.org/10.3390/antiox12091713