

Melatonin Restores Autophagic Flux by Activating the Sirt3/TFEB Signaling Pathway to Attenuate Doxorubicin-Induced Cardiomyopathy
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Experimental Protocols
2.2. Echocardiography
2.3. Histopathology
2.4. Cell Culture and Transduction of GFP-mRFP-LC3 Adenovirus
2.5. siRNA Transfection Experiment
2.6. Western Blot
2.7. Measurement of Mechanical Properties of Cardiomyocytes
2.8. Statistical Analysis
3. Results
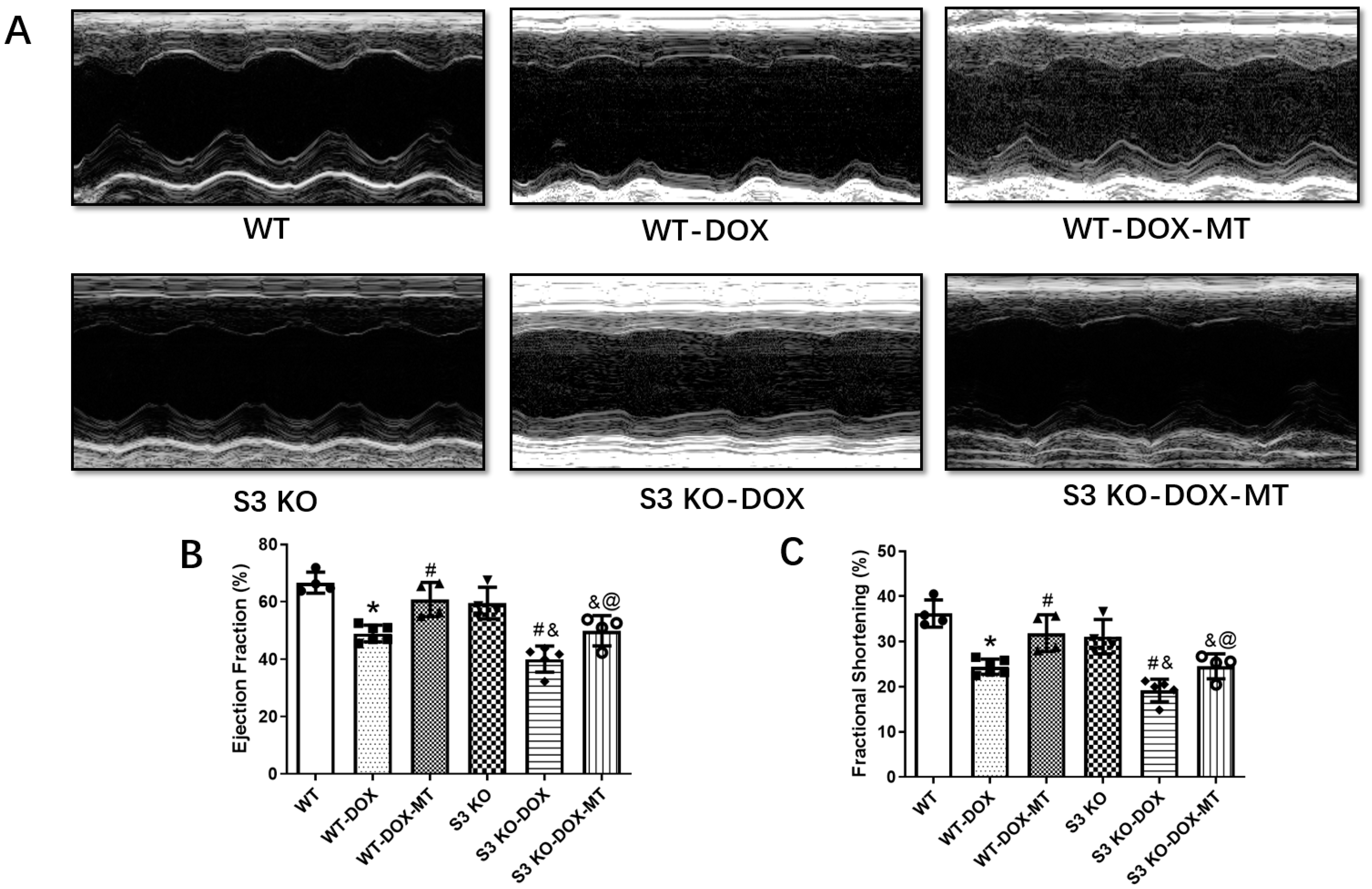
3.1. Melatonin Administration Preserved Cardiac Function in DOX-Treated WT Mice but Not in Sirt3 KO Mice
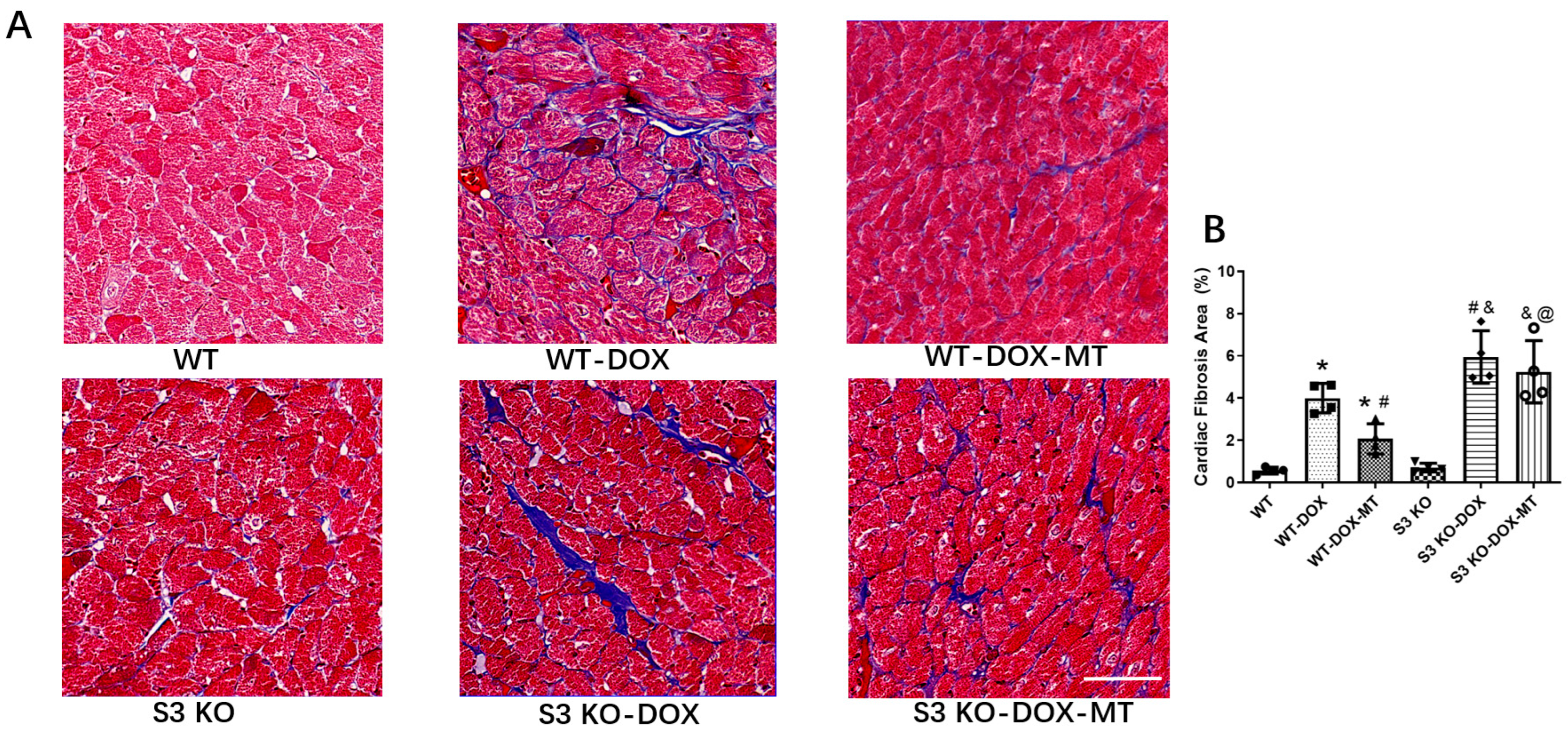
3.2. Melatonin Treatment Failed to Alleviate Myocardial Fibrosis in DOX-Treated Sirt3 Knockout Mice
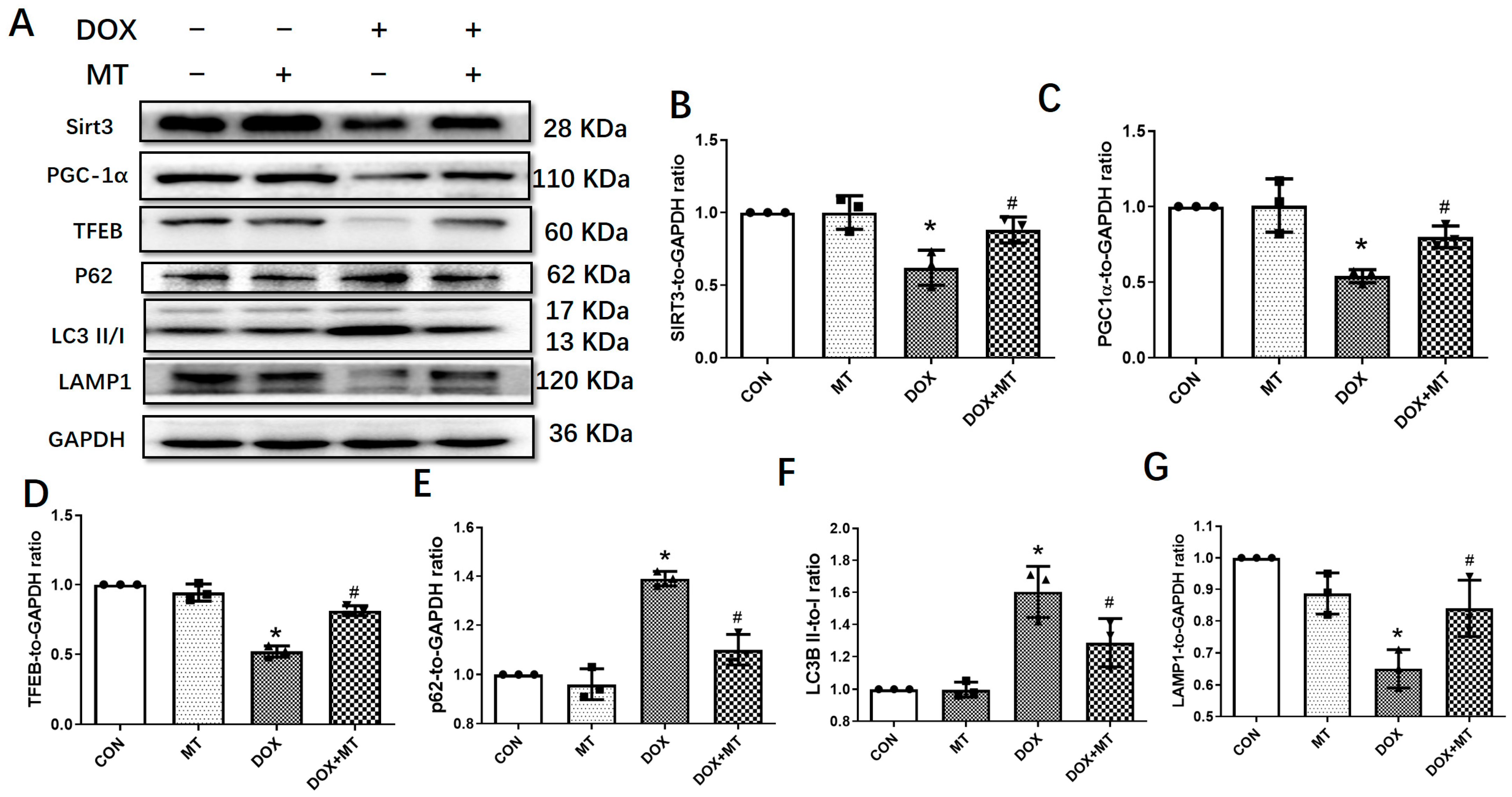
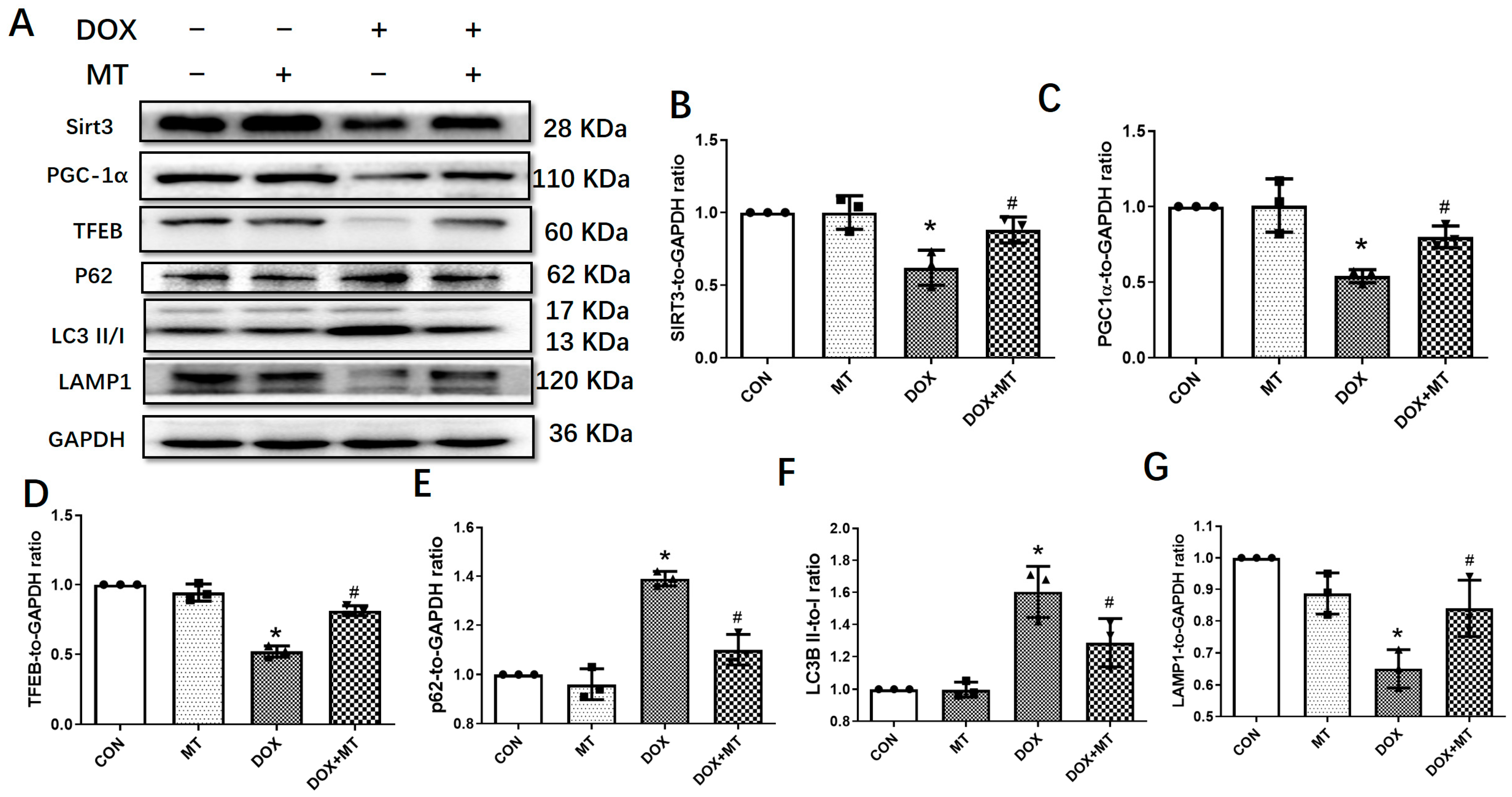
3.3. Melatonin Mitigated Cardiac Dysfunction Induced by DOX by Upregulating Sirt3/TFE-Mediated Autophagic Flux
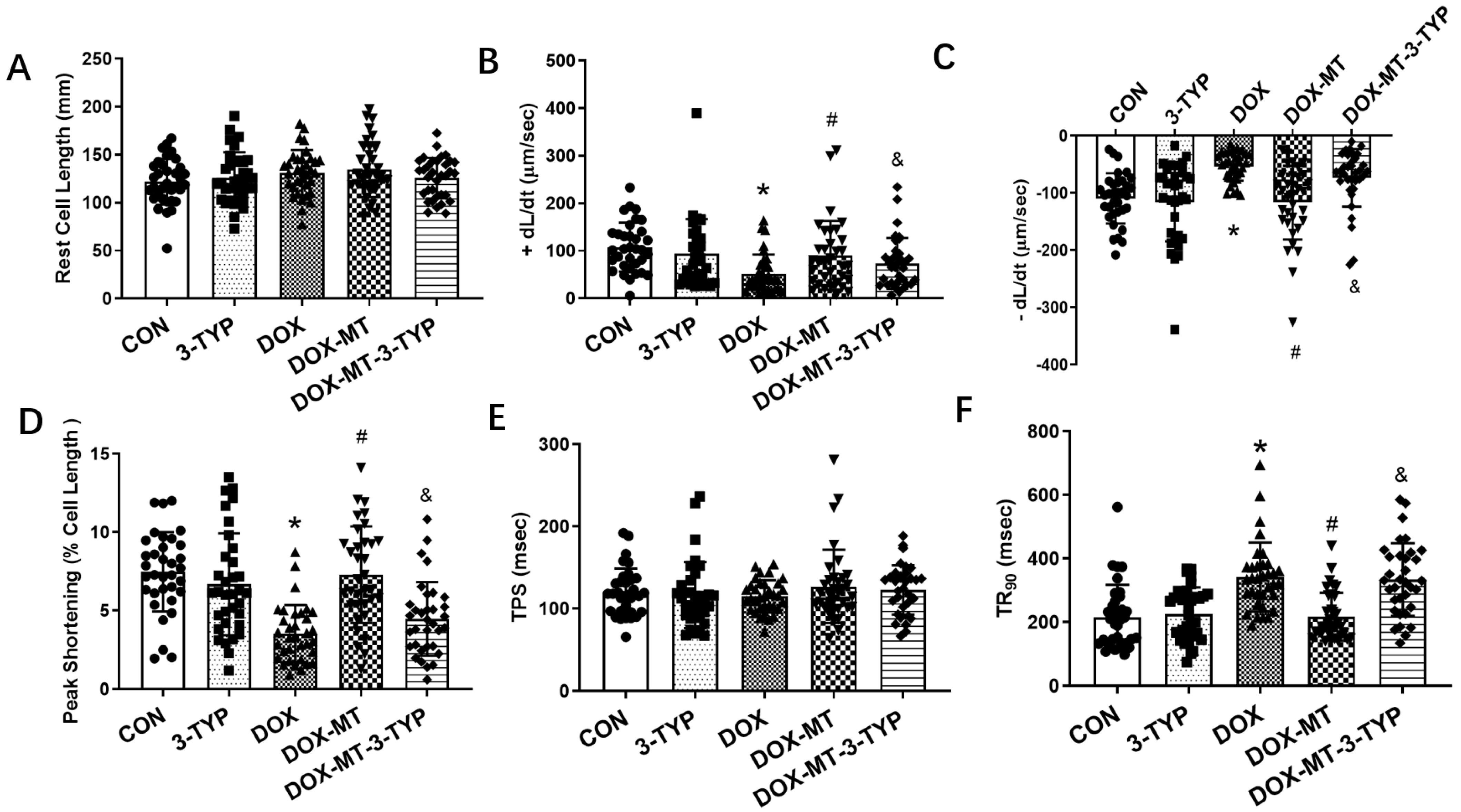
3.4. Melatonin Improved Cardiomyocyte Contractile Properties via the Sirt3 Signaling Pathway in DOX-Induced Cardiotoxicity
3.5. Melatonin Improved the Autophagic Flux via Increasing the Sirt3 Expression in H9c2 Cells
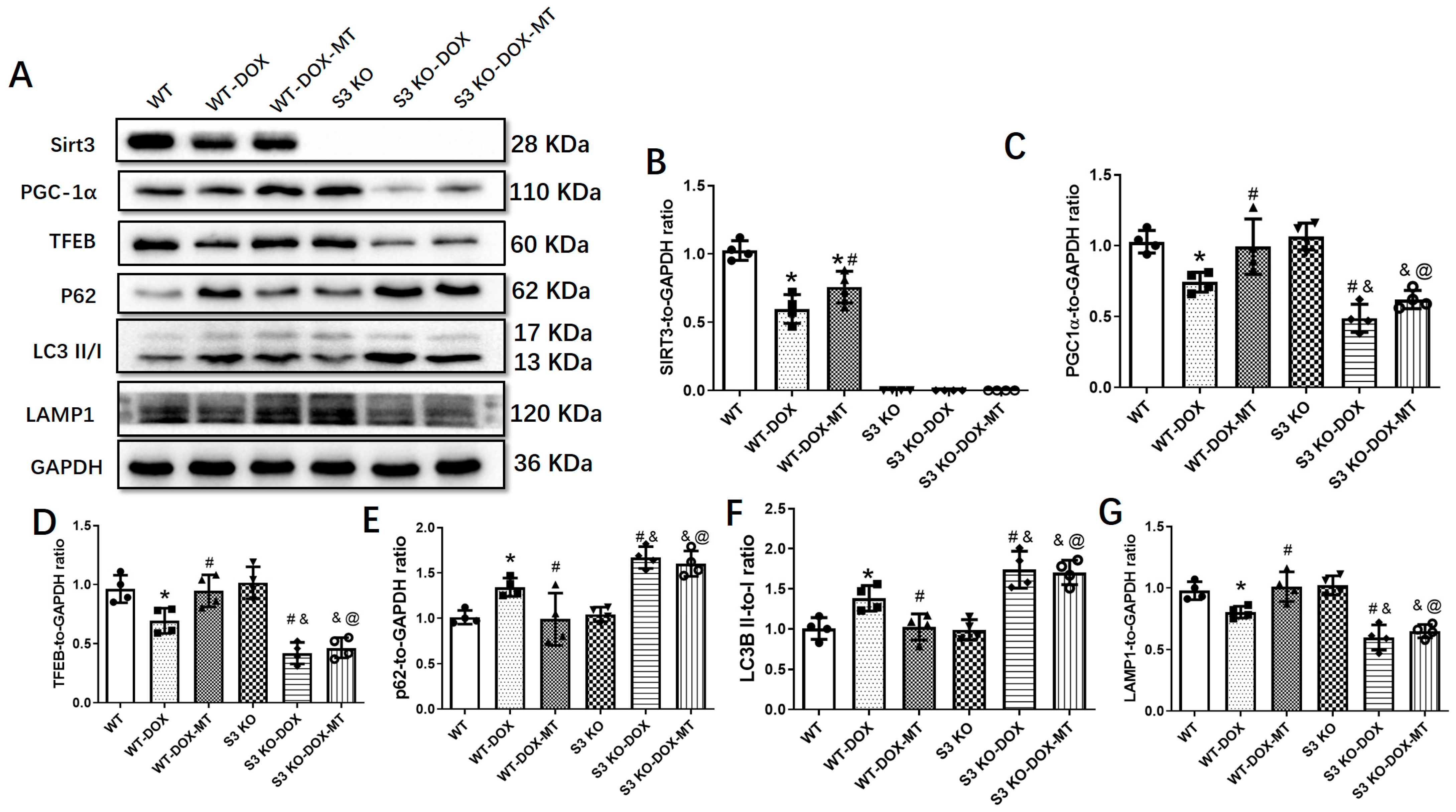
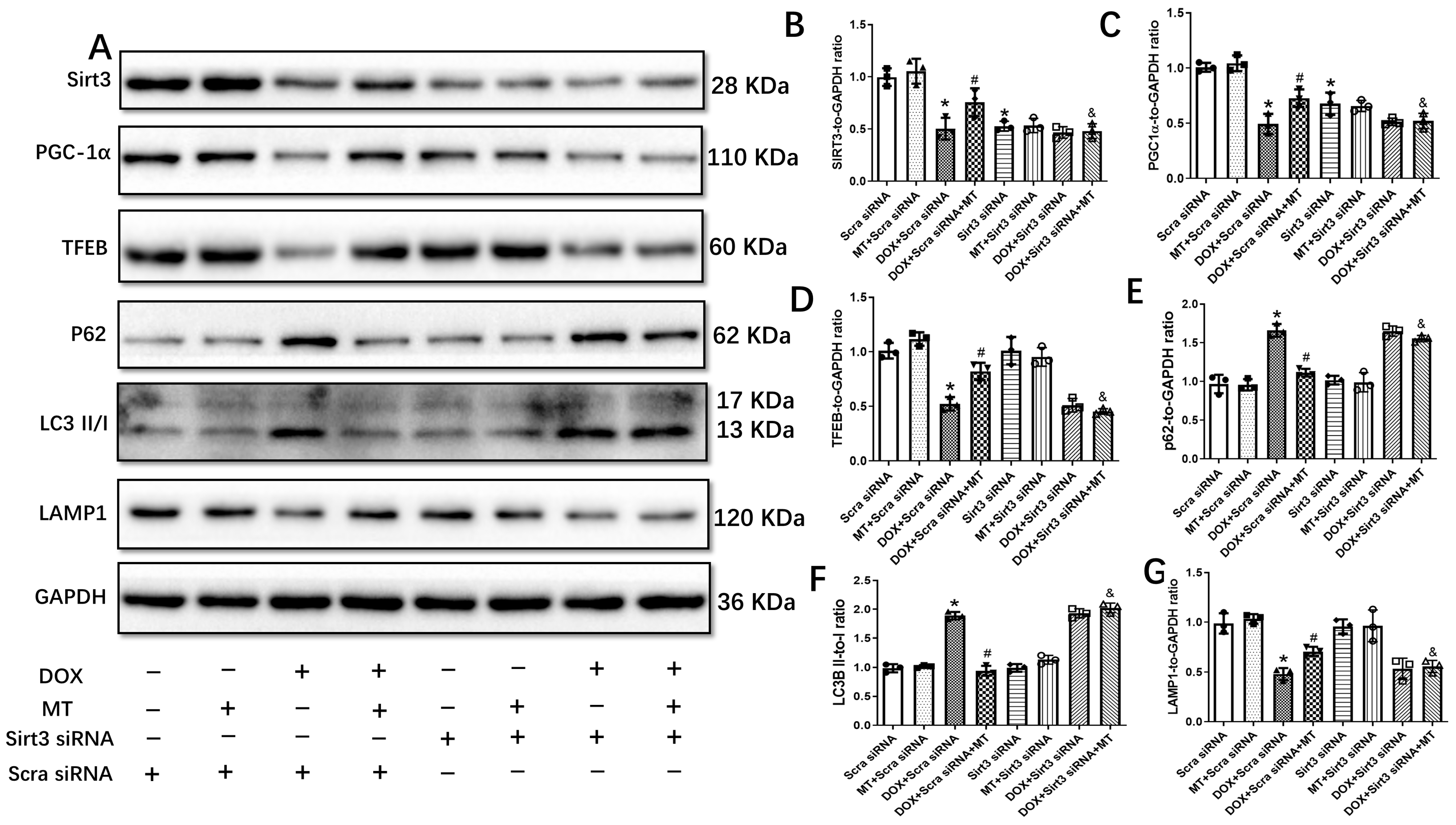
3.6. The Restoration of Melatonin’s Effect on Autophagic Flux Was Abolished by Downregulating the Sirt3/TFEB Signaling Axis in DOX-Treated H9c2 Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gerull, B.; Klaassen, S.; Brodehl, A. The Genetic Landscape of Cardiomyopathies. In Genetic Causes of Cardiac Disease; Erdmann, J., Moretti, A., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 45–91. [Google Scholar]
- Brodehl, A.; Gaertner-Rommel, A.; Klauke, B.; Grewe, S.A.; Schirmer, I.; Peterschröder, A.; Faber, L.; Vorgerd, M.; Gummert, J.; Anselmetti, D.; et al. The novel alphaB-crystallin (CRYAB) mutation p.D109G causes restrictive cardiomyopathy. Hum. Mutat. 2017, 38, 947–952. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Ferrier, R.A.; Hamilton, S.J.; Greenway, S.C.; Brundler, M.-A.; Yu, W.; Gibson, W.T.; McKinnon, M.L.; McGillivray, B.; Alvarez, N.; et al. Mutations in FLNC are Associated with Familial Restrictive Cardiomyopathy. Hum. Mutat. 2016, 37, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Bermúdez-Jiménez, F.J.; Carriel, V.; Brodehl, A.; Alaminos, M.; Campos, A.; Schirmer, I.; Milting, H.; Abril, B.; Álvarez, M.; López-Fernández, S.; et al. Novel Desmin Mutation p.Glu401Asp Impairs Filament Formation, Disrupts Cell Membrane Integrity, and Causes Severe Arrhythmogenic Left Ventricular Cardiomyopathy/Dysplasia. Circulation 2018, 137, 1595–1610. [Google Scholar] [CrossRef] [PubMed]
- Murad, A.M.; Santiago, F.F.; Petroianu, A.; Rocha, P.R.; Rodrigues, M.A.; Rausch, M. Modified therapy with 5-fluorouracil, doxorubicin, and methotrexate in advanced gastric cancer. Cancer 1993, 72, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Paridaens, R.; Biganzoli, L.; Bruning, P.; Klijn, J.G.M.; Gamucci, T.; Houston, S.; Coleman, R.; Schachter, J.; Vreckem, A.V.; Sylvester, R.; et al. Paclitaxel versus Doxorubicin as First-Line Single-Agent Chemotherapy for Metastatic Breast Cancer: A European Organization for Research and Treatment of Cancer Randomized Study with Cross-Over. J. Clin. Oncol. 2000, 18, 724. [Google Scholar] [CrossRef] [PubMed]
- Kuerer, H.M.; Newman, L.A.; Smith, T.L.; Ames, F.C.; Hunt, K.K.; Dhingra, K.; Theriault, R.L.; Singh, G.; Binkley, S.M.; Sneige, N.; et al. Clinical course of breast cancer patients with complete pathologic primary tumor and axillary lymph node response to doxorubicin-based neoadjuvant chemotherapy. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1999, 17, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Layard, M.W.; Basa, P.; Davis, H.L., Jr.; Von Hoff, A.L.; Rozencweig, M.; Muggia, F.M. Risk factors for doxorubicin-induced congestive heart failure. Ann. Intern. Med. 1979, 91, 710–717. [Google Scholar] [CrossRef]
- Swain, S.M.; Whaley, F.S.; Ewer, M.S. Congestive heart failure in patients treated with doxorubicin: A retrospective analysis of three trials. Cancer 2003, 97, 2869–2879. [Google Scholar] [CrossRef]
- L’Ecuyer, T.; Sanjeev, S.; Thomas, R.; Novak, R.; Das, L.; Campbell, W.; Heide, R.V. DNA damage is an early event in doxorubicin-induced cardiac myocyte death. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1273–H1280. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012, 18, 1639–1642. [Google Scholar] [CrossRef]
- Zhou, S.; Starkov, A.; Froberg, M.K.; Leino, R.L.; Wallace, K.B. Cumulative and irreversible cardiac mitochondrial dysfunction induced by doxorubicin. Cancer Res. 2001, 61, 771–777. [Google Scholar] [PubMed]
- Fisher, P.W.; Salloum, F.; Das, A.; Hyder, H.; Kukreja, R.C. Phosphodiesterase-5 inhibition with sildenafil attenuates cardiomyocyte apoptosis and left ventricular dysfunction in a chronic model of doxorubicin cardiotoxicity. Circulation 2005, 111, 1601–1610. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Volden, P.; Timm, D.; Mao, K.; Xu, X.; Liang, Q. Transcription factor GATA4 inhibits doxorubicin-induced autophagy and cardiomyocyte death. J. Biol. Chem. 2010, 285, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.; Cheng, Y.; Zhang, Y.; Zhang, Q.; Wang, S.; Tian, S.; Zou, Y.; Hu, K.; Ren, J.; Ge, J. Aldehyde dehydrogenase 2 ameliorates doxorubicin-induced myocardial dysfunction through detoxification of 4-HNE and suppression of autophagy. J. Mol. Cell. Cardiol. 2014, 71, 92–104. [Google Scholar] [CrossRef]
- Li, D.L.; Wang, Z.V.; Ding, G.; Tan, W.; Luo, X.; Criollo, A.; Xie, M.; Jiang, N.; May, H.; Kyrychenko, V.; et al. Doxorubicin Blocks Cardiomyocyte Autophagic Flux by Inhibiting Lysosome Acidification. Circulation 2016, 133, 1668–1687. [Google Scholar] [CrossRef]
- Cao, Y.; Shen, T.; Huang, X.; Lin, Y.; Chen, B.; Pang, J.; Li, G.; Wang, Q.; Zohrabian, S.; Duan, C.; et al. Astragalus polysaccharide restores autophagic flux and improves cardiomyocyte function in doxorubicin-induced cardiotoxicity. Oncotarget 2017, 8, 4837–4848. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Takemura, G.; Kanamori, H.; Takeyama, T.; Watanabe, T.; Morishita, K.; Ogino, A.; Tsujimoto, A.; Goto, K.; Maruyama, R.; et al. Prior starvation mitigates acute doxorubicin cardiotoxicity through restoration of autophagy in affected cardiomyocytes. Cardiovasc. Res. 2012, 96, 456–465. [Google Scholar] [CrossRef]
- Wang, X.; Wang, X.L.; Chen, H.L.; Wu, D.; Chen, J.X.; Wang, X.X.; Li, R.L.; He, J.H.; Mo, L.; Cen, X.; et al. Ghrelin inhibits doxorubicin cardiotoxicity by inhibiting excessive autophagy through AMPK and p38-MAPK. Biochem. Pharmacol. 2014, 88, 334–350. [Google Scholar] [CrossRef]
- Bartlett, J.J.; Trivedi, P.C.; Yeung, P.; Kienesberger, P.C.; Pulinilkunnil, T. Doxorubicin impairs cardiomyocyte viability by suppressing transcription factor EB expression and disrupting autophagy. Biochem. J. 2016, 473, 3769–3789. [Google Scholar] [CrossRef]
- Lee, K.H.; Cho, H.; Lee, S.; Woo, J.S.; Cho, B.H.; Kang, J.H.; Jeong, Y.M.; Cheng, X.W.; Kim, W. Enhanced-autophagy by exenatide mitigates doxorubicin-induced cardiotoxicity. Int. J. Cardiol. 2017, 232, 40–47. [Google Scholar] [CrossRef]
- Cajochen, C.; Krauchi, K.; Wirz-Justice, A. Role of melatonin in the regulation of human circadian rhythms and sleep. J. Neuroendocrinol. 2003, 15, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Zhai, M.; Liu, Z.; Zhang, B.; Jing, L.; Li, B.; Li, K.; Chen, X.; Zhang, M.; Yu, B.; Ren, K.; et al. Melatonin protects against the pathological cardiac hypertrophy induced by transverse aortic constriction through activating PGC-1beta: In vivo and in vitro studies. J. Pineal Res. 2017, 63, e12433. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Sun, Y.; Cheng, L.; Jin, Z.; Yang, Y.; Zhai, M.; Pei, H.; Wang, X.; Zhang, H.; Meng, Q.; et al. Melatonin receptor-mediated protection against myocardial ischemia/reperfusion injury: Role of SIRT1. J. Pineal Res. 2014, 57, 228–238. [Google Scholar] [CrossRef]
- Yang, Y.; Duan, W.; Jin, Z.; Yi, W.; Yan, J.; Zhang, S.; Wang, N.; Liang, Z.; Li, Y.; Chen, W.; et al. JAK2/STAT3 activation by melatonin attenuates the mitochondrial oxidative damage induced by myocardial ischemia/reperfusion injury. J. Pineal Res. 2013, 55, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Lin, J.; Wang, S.; Cheng, Z.; Hu, J.; Wang, T.; Man, W.; Yin, T.; Guo, W.; Gao, E.; et al. Melatonin protects against diabetic cardiomyopathy through Mst1/Sirt3 signaling. J. Pineal Res. 2017, 63, e12418. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, M.; Ozler, M.; Kurt, Y.G.; Ozturk, B.; Uysal, B.; Ersoz, N.; Yasar, M.; Demirbas, S.; Kurt, B.; Acikel, C.; et al. Efficacy of melatonin, mercaptoethylguanidine and 1400W in doxorubicin- and trastuzumab-induced cardiotoxicity. J. Pineal Res. 2011, 50, 89–96. [Google Scholar] [CrossRef]
- Sahna, E.; Parlakpinar, H.; Ozer, M.K.; Ozturk, F.; Ozugurlu, F.; Acet, A. Melatonin protects against myocardial doxorubicin toxicity in rats: Role of physiological concentrations. J. Pineal Res. 2003, 35, 257–261. [Google Scholar] [CrossRef]
- Govender, J.; Loos, B.; Marais, E.; Engelbrecht, A.M. Mitochondrial catastrophe during doxorubicin-induced cardiotoxicity: A review of the protective role of melatonin. J. Pineal Res. 2014, 57, 367–380. [Google Scholar] [CrossRef]
- Pillai, V.B.; Sundaresan, N.R.; Jeevanandam, V.; Gupta, M.P. Mitochondrial SIRT3 and heart disease. Cardiovasc. Res. 2010, 88, 250–256. [Google Scholar] [CrossRef]
- Lai, Y.C.; Tabima, D.M.; Dube, J.J.; Hughan, K.S.; Vanderpool, R.R.; Goncharov, D.A.; St Croix, C.M.; Garcia-Ocana, A.; Goncharova, E.A.; Tofovic, S.P.; et al. SIRT3-AMP-Activated Protein Kinase Activation by Nitrite and Metformin Improves Hyperglycemia and Normalizes Pulmonary Hypertension Associated with Heart Failure with Preserved Ejection Fraction. Circulation 2016, 133, 717–731. [Google Scholar] [CrossRef]
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S.B.; Isbatan, A.; Gupta, M.P. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Investig. 2009, 119, 2758–2771. [Google Scholar] [CrossRef]
- Zhang, M.; Zhao, Z.; Shen, M.; Zhang, Y.; Duan, J.; Guo, Y.; Zhang, D.; Hu, J.; Lin, J.; Man, W.; et al. Polydatin protects cardiomyocytes against myocardial infarction injury by activating Sirt3. Biochim. Biophys. Acta. Mol. Basis Dis. 2017, 1863, 1962–1972. [Google Scholar] [CrossRef]
- Wang, S.; Zhao, Z.; Fan, Y.; Zhang, M.; Feng, X.; Lin, J.; Hu, J.; Cheng, Z.; Sun, C.; Liu, T.; et al. Mst1 inhibits Sirt3 expression and contributes to diabetic cardiomyopathy through inhibiting Parkin-dependent mitophagy. Biochim. Biophys. Acta. Mol. Basis Dis. 2018, 1865, 1905–1914. [Google Scholar] [CrossRef] [PubMed]
- Pillai, V.B.; Bindu, S.; Sharp, W.; Fang, Y.H.; Kim, G.; Gupta, M.; Samant, S.; Gupta, M.P. Sirt3 protects mitochondrial DNA damage and blocks the development of doxorubicin-induced cardiomyopathy in mice. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H962–H972. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.G.; Cole, L.K.; Xiang, B.; Chen, K.; Ma, X.; Myal, Y.; Hatch, G.M.; Tong, Q.; Dolinsky, V.W. Sirtuin-3 (SIRT3) Protein Attenuates Doxorubicin-induced Oxidative Stress and Improves Mitochondrial Respiration in H9c2 Cardiomyocytes. J. Biol. Chem. 2015, 290, 10981–10993. [Google Scholar] [CrossRef] [PubMed]
- Papadopulos, F.; Spinelli, M.; Valente, S.; Foroni, L.; Orrico, C.; Alviano, F.; Pasquinelli, G. Common tasks in microscopic and ultrastructural image analysis using ImageJ. Ultrastruct. Pathol. 2007, 31, 401–407. [Google Scholar] [CrossRef]
- Yu, W.; Sun, S.; Xu, H.; Li, C.; Ren, J.; Zhang, Y. TBC1D15/RAB7-regulated mitochondria-lysosome interaction confers cardioprotection against acute myocardial infarction-induced cardiac injury. Theranostics 2020, 10, 11244–11263. [Google Scholar] [CrossRef]
- Pei, H.F.; Hou, J.N.; Wei, F.P.; Xue, Q.; Zhang, F.; Peng, C.F.; Yang, Y.; Tian, Y.; Feng, J.; Du, J.; et al. Melatonin attenuates postmyocardial infarction injury via increasing Tom70 expression. J. Pineal Res. 2017, 62, e12371. [Google Scholar] [CrossRef]
- Yang, Y.; Sun, Y.; Yi, W.; Li, Y.; Fan, C.; Xin, Z.; Jiang, S.; Di, S.; Qu, Y.; Reiter, R.J.; et al. A review of melatonin as a suitable antioxidant against myocardial ischemia-reperfusion injury and clinical heart diseases. J. Pineal Res. 2014, 57, 357–366. [Google Scholar] [CrossRef]
- Manchester, L.C.; Coto-Montes, A.; Boga, J.A.; Andersen, L.P.; Zhou, Z.; Galano, A.; Vriend, J.; Tan, D.X.; Reiter, R.J. Melatonin: An ancient molecule that makes oxygen metabolically tolerable. J. Pineal Res. 2015, 59, 403–419. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, S.; Zhao, X.; Wei, T. Melatonin impairs NADPH oxidase assembly and decreases superoxide anion production in microglia exposed to amyloid-beta1-42. J. Pineal Res. 2008, 45, 157–165. [Google Scholar] [CrossRef]
- Chang, H.M.; Ling, E.A.; Lue, J.H.; Wen, C.Y.; Shieh, J.Y. Melatonin attenuates neuronal NADPH-d/NOS expression in the hypoglossal nucleus of adult rats following peripheral nerve injury. Brain Res. 2000, 873, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Li, Y.; Fu, S.; Li, Y.; Wang, C.; Sun, P.; Li, H.; Tian, J.; Du, G.Q. Melatonin alleviates arginine vasopressin-induced cardiomyocyte apoptosis via increasing Mst1-Nrf2 pathway activity to reduce oxidative stress. Biochem. Pharmacol. 2022, 206, 115265. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sun, P.; Li, Y.; Shen, W.; Wang, C.; Zhao, P.; Cui, H.; Xue, J.Y.; Du, G.Q. Melatonin activates the Mst1-Nrf2 signaling to alleviate cardiac hypertrophy in pulmonary arterial hypertension. Eur. J. Pharmacol. 2022, 933, 175262. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chen, Z.; Chua, C.C.; Ma, Y.S.; Youngberg, G.A.; Hamdy, R.; Chua, B.H. Melatonin as an effective protector against doxorubicin-induced cardiotoxicity. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H254–H263. [Google Scholar] [CrossRef]
- Xu, M.; Ashraf, M. Melatonin protection against lethal myocyte injury induced by doxorubicin as reflected by effects on mitochondrial membrane potential. J. Mol. Cell. Cardiol. 2002, 34, 75–79. [Google Scholar] [CrossRef]
- Dziegiel, P.; Murawska-Cialowicz, E.; Jethon, Z.; Januszewska, L.; Podhorska-Okolow, M.; Surowiak, P.; Zawadzki, M.; Rabczynski, J.; Zabel, M. Melatonin stimulates the activity of protective antioxidative enzymes in myocardial cells of rats in the course of doxorubicin intoxication. J. Pineal Res. 2003, 35, 183–187. [Google Scholar] [CrossRef]
- Bai, Y.; Chen, Q.; Sun, Y.P.; Wang, X.; Lv, L.; Zhang, L.P.; Liu, J.S.; Zhao, S.; Wang, X.L. Sulforaphane protection against the development of doxorubicin-induced chronic heart failure is associated with Nrf2 Upregulation. Cardiovasc. Ther. 2017, 35, e12277. [Google Scholar] [CrossRef]
- Gupta, S.K.; Garg, A.; Bar, C.; Chatterjee, S.; Foinquinos, A.; Milting, H.; Streckfuss-Bomeke, K.; Fiedler, J.; Thum, T. Quaking Inhibits Doxorubicin-Mediated Cardiotoxicity through Regulation of Cardiac Circular RNA Expression. Circ. Res. 2018, 122, 246–254. [Google Scholar] [CrossRef]
- Porter, G.A.; Urciuoli, W.R.; Brookes, P.S.; Nadtochiy, S.M. SIRT3 deficiency exacerbates ischemia-reperfusion injury: Implication for aged hearts. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1602–H1609. [Google Scholar] [CrossRef]
- He, X.; Zeng, H.; Chen, J.X. Ablation of SIRT3 causes coronary microvascular dysfunction and impairs cardiac recovery post myocardial ischemia. Int. J. Cardiol. 2016, 215, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Koentges, C.; Pfeil, K.; Schnick, T.; Wiese, S.; Dahlbock, R.; Cimolai, M.C.; Meyer-Steenbuck, M.; Cenkerova, K.; Hoffmann, M.M.; Jaeger, C.; et al. SIRT3 deficiency impairs mitochondrial and contractile function in the heart. Basic Res. Cardiol. 2015, 110, 36. [Google Scholar] [CrossRef] [PubMed]
- Pillai, V.B.; Samant, S.; Sundaresan, N.R.; Raghuraman, H.; Kim, G.; Bonner, M.Y.; Arbiser, J.L.; Walker, D.I.; Jones, D.P.; Gius, D.; et al. Honokiol blocks and reverses cardiac hypertrophy in mice by activating mitochondrial Sirt3. Nat. Commun. 2015, 6, 6656. [Google Scholar] [CrossRef]
- Bartlett, J.J.; Trivedi, P.C.; Pulinilkunnil, T. Autophagic dysregulation in doxorubicin cardiomyopathy. J. Mol. Cell. Cardiol. 2017, 104, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Kyoi, S.; Yamaguchi, O.; Sadoshima, J.; Otsu, K. The role of autophagy in the heart. Cell Death Differ. 2009, 16, 31–38. [Google Scholar] [CrossRef]
- Xu, X.; Bucala, R.; Ren, J. Macrophage migration inhibitory factor deficiency augments doxorubicin-induced cardiomyopathy. J. Am. Heart Assoc. 2013, 2, e000439. [Google Scholar] [CrossRef]
- Shi, T.; Wang, F.; Stieren, E.; Tong, Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J. Biol. Chem. 2005, 280, 13560–13567. [Google Scholar] [CrossRef]
- Zhou, Q.; Wang, Y.; Lu, Z.; Wang, B.; Li, L.; You, M.; Wang, L.; Cao, T.; Zhao, Y.; Li, Q.; et al. Mitochondrial dysfunction caused by SIRT3 inhibition drives proinflammatory macrophage polarization in obesity. Obesity 2023, 31, 1050–1063. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Y.; Ma, J.; Lu, L.; Xiong, X.; Shao, Y.; Ren, J.; Yang, J.; Liu, J. Melatonin Restores Autophagic Flux by Activating the Sirt3/TFEB Signaling Pathway to Attenuate Doxorubicin-Induced Cardiomyopathy. Antioxidants 2023, 12, 1716. https://doi.org/10.3390/antiox12091716
Ma Y, Ma J, Lu L, Xiong X, Shao Y, Ren J, Yang J, Liu J. Melatonin Restores Autophagic Flux by Activating the Sirt3/TFEB Signaling Pathway to Attenuate Doxorubicin-Induced Cardiomyopathy. Antioxidants. 2023; 12(9):1716. https://doi.org/10.3390/antiox12091716
Chicago/Turabian StyleMa, Yanyan, Jipeng Ma, Linhe Lu, Xiang Xiong, Yalan Shao, Jun Ren, Jian Yang, and Jiankang Liu. 2023. "Melatonin Restores Autophagic Flux by Activating the Sirt3/TFEB Signaling Pathway to Attenuate Doxorubicin-Induced Cardiomyopathy" Antioxidants 12, no. 9: 1716. https://doi.org/10.3390/antiox12091716