
Fenofibrate Decreases Ethanol-Induced Neuroinflammation and Oxidative Stress and Reduces Alcohol Relapse in Rats by a PPAR-α-Dependent Mechanism

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Treatments
2.2. Quantification of the Expression of Proinflammatory Cytokines and Oxidative Stress Markers
2.3. Determination of Oxidized Glutathione (GSSG) and Reduced Glutathione (GSH) Levels
2.4. Determination of Microglia Immunoreactivity
2.5. Statistical Analyses
3. Results
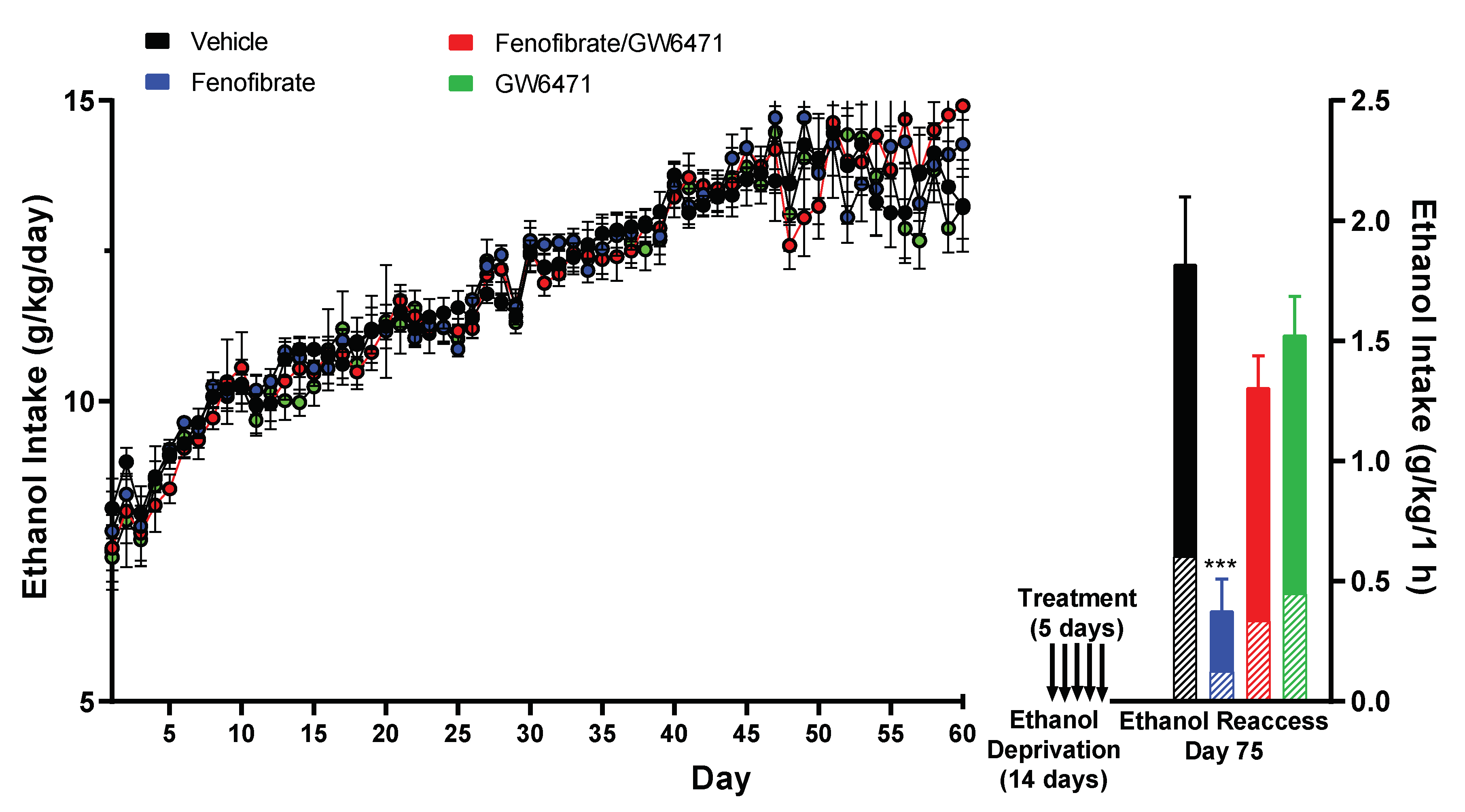
3.1. Effect of Fenofibrate on Relapse-like Alcohol Consumption
3.2. Effect of Fenofibrate on Ethanol-Induced Expression of Proinflammatory Cytokines and an Oxidative Stress Marker
3.3. Effect of Fenofibrate on the Levels of the Antioxidant Glutathione
3.4. Effect of Fenofibrate on Ethanol-Induced Microglial Reactivity
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Maisel, N.C.; Blodgett, J.C.; Wilbourne, P.L.; Humphreys, K.; Finney, J.W. Meta-Analysis of Naltrexone and Acamprosate for Treating Alcohol Use Disorders: When Are These Medications Most Helpful? Addiction 2013, 108, 275–293. [Google Scholar] [CrossRef]
- Plosker, G.L. Acamprosate: A Review of Its Use in Alcohol Dependence. Drugs 2015, 75, 1255–1268. [Google Scholar] [CrossRef]
- Holleck, J.L.; Merchant, N.; Gunderson, C.G. Symptom-Triggered Therapy for Alcohol Withdrawal Syndrome: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. J. Gen. Intern. Med. 2019, 34, 1018–1024. [Google Scholar] [CrossRef]
- Coller, J.K.; Hutchinson, M.R. Implications of Central Immune Signaling Caused by Drugs of Abuse: Mechanisms, Mediators and New Therapeutic Approaches for Prediction and Treatment of Drug Dependence. Pharmacol. Ther. 2012, 134, 219–245. [Google Scholar] [CrossRef]
- Flores-Bastías, O.; Karahanian, E. Neuroinflammation Produced by Heavy Alcohol Intake Is Due to Loops of Interactions between Toll-like 4 and TNF Receptors, Peroxisome Proliferator-Activated Receptors and the Central Melanocortin System: A Novel Hypothesis and New Therapeutic Avenues. Neuropharmacology 2018, 128, 401–407. [Google Scholar] [CrossRef]
- Crews, F.T.; Zou, J.; Qin, L. Induction of Innate Immune Genes in Brain Create the Neurobiology of Addiction. Brain. Behav. Immun. 2011, 25 (Suppl. S1), S4–S12. [Google Scholar] [CrossRef]
- Cao, Q.; Mak, K.M.; Lieber, C.S. Cytochrome P4502E1 Primes Macrophages to Increase TNF-Alpha Production in Response to Lipopolysaccharide. Am. J. Physiol.-Gastrointest. Liver Physiol. 2005, 289, G95–G107. [Google Scholar] [CrossRef]
- Chandel, N.S.; Trzyna, W.C.; McClintock, D.S.; Schumacker, P.T. Role of Oxidants in NF-Kappa B Activation and TNF-Alpha Gene Transcription Induced by Hypoxia and Endotoxin. J. Immunol. 2000, 165, 1013–1021. [Google Scholar] [CrossRef]
- Qin, L.; He, J.; Hanes, R.N.; Pluzarev, O.; Hong, J.-S.; Crews, F.T. Increased Systemic and Brain Cytokine Production and Neuroinflammation by Endotoxin Following Ethanol Treatment. J. Neuroinflamm. 2008, 5, 10. [Google Scholar] [CrossRef]
- Ferrier, L.; Bérard, F.; Debrauwer, L.; Chabo, C.; Langella, P.; Buéno, L.; Fioramonti, J. Impairment of the Intestinal Barrier by Ethanol Involves Enteric Microflora and Mast Cell Activation in Rodents. Am. J. Pathol. 2006, 168, 1148–1154. [Google Scholar] [CrossRef] [PubMed]
- Crews, F.T.; Vetreno, R.P. Mechanisms of Neuroimmune Gene Induction in Alcoholism. Psychopharmacology 2016, 233, 1543–1557. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.-S.; Knapp, D.J.; Crews, F.T. Systemic LPS Causes Chronic Neuroinflammation and Progressive Neurodegeneration. Glia 2007, 55, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Crews, F.T.; Sarkar, D.K.; Qin, L.; Zou, J.; Boyadjieva, N.; Vetreno, R.P. Neuroimmune Function and the Consequences of Alcohol Exposure. Alcohol Res. 2015, 37, 331–341,344–351. [Google Scholar]
- Clapp, P.; Bhave, S.V.; Hoffman, P.L. How Adaptation of the Brain to Alcohol Leads to Dependence: A Pharmacological Perspective. Alcohol Res. Health J. Natl. Inst. Alcohol Abus. Alcohol. 2008, 31, 310–339. [Google Scholar]
- Koob, G.F. A Role for GABA Mechanisms in the Motivational Effects of Alcohol. Biochem. Pharmacol. 2004, 68, 1515–1525. [Google Scholar] [CrossRef]
- Hou, R.; Baldwin, D.S. A Neuroimmunological Perspective on Anxiety Disorders. Hum. Psychopharmacol. 2012, 27, 6–14. [Google Scholar] [CrossRef]
- Simen, B.B.; Duman, C.H.; Simen, A.A.; Duman, R.S. TNFalpha Signaling in Depression and Anxiety: Behavioral Consequences of Individual Receptor Targeting. Biol. Psychiatry 2006, 59, 775–785. [Google Scholar] [CrossRef]
- Koo, J.W.; Duman, R.S. Interleukin-1 Receptor Null Mutant Mice Show Decreased Anxiety-like Behavior and Enhanced Fear Memory. Neurosci. Lett. 2009, 456, 39–43. [Google Scholar] [CrossRef]
- Reissner, K.J.; Kalivas, P.W. Using Glutamate Homeostasis as a Target for Treating Addictive Disorders. Behav. Pharmacol. 2010, 21, 514–522. [Google Scholar] [CrossRef]
- Rao, P.S.S.; Bell, R.L.; Engleman, E.A.; Sari, Y. Targeting Glutamate Uptake to Treat Alcohol Use Disorders. Front. Neurosci. 2015, 9, 144. [Google Scholar] [CrossRef]
- Dahchour, A.; De Witte, P. Taurine Blocks the Glutamate Increase in the Nucleus Accumbens Microdialysate of Ethanol-Dependent Rats. Pharmacol. Biochem. Behav. 2000, 65, 345–350. [Google Scholar] [CrossRef]
- Scofield, M.D.; Heinsbroek, J.A.; Gipson, C.D.; Kupchik, Y.M.; Spencer, S.; Smith, A.C.W.; Roberts-Wolfe, D.; Kalivas, P.W. The Nucleus Accumbens: Mechanisms of Addiction across Drug Classes Reflect the Importance of Glutamate Homeostasis. Pharmacol. Rev. 2016, 68, 816–871. [Google Scholar] [CrossRef]
- Berger, J.; Moller, D.E. The Mechanisms of Action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef] [PubMed]
- Schoonjans, K.; Staels, B.; Auwerx, J. Role of the Peroxisome Proliferator-Activated Receptor (PPAR) in Mediating the Effects of Fibrates and Fatty Acids on Gene Expression. J. Lipid Res. 1996, 37, 907–925. [Google Scholar] [CrossRef] [PubMed]
- Cignarella, A.; Bellosta, S.; Corsini, A.; Bolego, C. Hypolipidemic Therapy for the Metabolic Syndrome. Pharmacol. Res. 2006, 53, 492–500. [Google Scholar] [CrossRef]
- Karahanian, E.; Quintanilla, M.E.; Fernandez, K.; Israel, Y. Fenofibrate—A Lipid-Lowering Drug—Reduces Voluntary Alcohol Drinking in Rats. Alcohol 2014, 48, 665–670. [Google Scholar] [CrossRef]
- Rivera-Meza, M.; Muñoz, D.; Jerez, E.; Quintanilla, M.E.; Salinas-Luypaert, C.; Fernandez, K.; Karahanian, E. Fenofibrate Administration Reduces Alcohol and Saccharin Intake in Rats: Possible Effects at Peripheral and Central Levels. Front. Behav. Neurosci. 2017, 11, 133. [Google Scholar] [CrossRef]
- Blednov, Y.A.; Benavidez, J.M.; Black, M.; Ferguson, L.B.; Schoenhard, G.L.; Goate, A.M.; Edenberg, H.J.; Wetherill, L.; Hesselbrock, V.; Foroud, T.; et al. Peroxisome Proliferator-Activated Receptors α and γ Are Linked with Alcohol Consumption in Mice and Withdrawal and Dependence in Humans. Alcohol. Clin. Exp. Res. 2015, 39, 136–145. [Google Scholar] [CrossRef]
- Blednov, Y.A.; Black, M.; Benavidez, J.M.; Stamatakis, E.E.; Harris, R.A. PPAR Agonists: II. Fenofibrate and Tesaglitazar Alter Behaviors Related to Voluntary Alcohol Consumption. Alcohol. Clin. Exp. Res. 2016, 40, 563–571. [Google Scholar] [CrossRef]
- Haile, C.N.; Kosten, T.A. The Peroxisome Proliferator-Activated Receptor Alpha Agonist Fenofibrate Attenuates Alcohol Self-Administration in Rats. Neuropharmacology 2017, 116, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Barson, J.R.; Karatayev, O.; Chang, G.-Q.; Johnson, D.F.; Bocarsly, M.E.; Hoebel, B.G.; Leibowitz, S.F. Positive Relationship between Dietary Fat, Ethanol Intake, Triglycerides, and Hypothalamic Peptides: Counteraction by Lipid-Lowering Drugs. Alcohol 2009, 43, 433–441. [Google Scholar] [CrossRef]
- Bordet, R.; Ouk, T.; Petrault, O.; Gelé, P.; Gautier, S.; Laprais, M.; Deplanque, D.; Duriez, P.; Staels, B.; Fruchart, J.C.; et al. PPAR: A New Pharmacological Target for Neuroprotection in Stroke and Neurodegenerative Diseases. Biochem. Soc. Trans. 2006, 34 Pt 6, 1341–1346. [Google Scholar] [CrossRef]
- Chen, X.R.; Besson, V.C.; Palmier, B.; Garcia, Y.; Plotkine, M.; Marchand-Leroux, C. Neurological Recovery-Promoting, Anti-Inflammatory, and Anti-Oxidative Effects Afforded by Fenofibrate, a PPAR Alpha Agonist, in Traumatic Brain Injury. J. Neurotrauma 2007, 24, 1119–1131. [Google Scholar] [CrossRef]
- Collino, M.; Aragno, M.; Mastrocola, R.; Benetti, E.; Gallicchio, M.; Dianzani, C.; Danni, O.; Thiemermann, C.; Fantozzi, R. Oxidative Stress and Inflammatory Response Evoked by Transient Cerebral Ischemia/Reperfusion: Effects of the PPAR-Alpha Agonist WY14643. Free Radic. Biol. Med. 2006, 41, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Poynter, M.E.; Daynes, R.A. Peroxisome Proliferator-Activated Receptor Alpha Activation Modulates Cellular Redox Status, Represses Nuclear Factor-KappaB Signaling, and Reduces Inflammatory Cytokine Production in Aging. J. Biol. Chem. 1998, 273, 32833–32841. [Google Scholar] [CrossRef] [PubMed]
- Shehata, A.H.F.; Ahmed, A.-S.F.; Abdelrehim, A.B.; Heeba, G.H. The Impact of Single and Combined PPAR-α and PPAR-γ Activation on the Neurological Outcomes Following Cerebral Ischemia Reperfusion. Life Sci. 2020, 252, 117679. [Google Scholar] [CrossRef]
- Villavicencio-Tejo, F.; Flores-Bastías, O.; Marambio-Ruiz, L.; Pérez-Reytor, D.; Karahanian, E. Fenofibrate (a PPAR-α Agonist) Administered during Ethanol Withdrawal Reverts Ethanol-Induced Astrogliosis and Restores the Levels of Glutamate Transporter in Ethanol-Administered Adolescent Rats. Front. Pharmacol. 2021, 12, 653175. [Google Scholar] [CrossRef] [PubMed]
- Israel, Y.; Karahanian, E.; Ezquer, F.; Morales, P.; Ezquer, M.; Rivera-Meza, M.; Herrera-Marschitz, M.; Quintanilla, M.E. Acquisition, Maintenance and Relapse-Like Alcohol Drinking: Lessons from the UChB Rat Line. Front. Behav. Neurosci. 2017, 11, 57. [Google Scholar] [CrossRef]
- Karahanian, E.; Rivera-Meza, M.; Tampier, L.; Quintanilla, M.E.; Herrera-Marschitz, M.; Israel, Y. Long-Term Inhibition of Ethanol Intake by the Administration of an Aldehyde Dehydrogenase-2 (ALDH2)-Coding Lentiviral Vector into the Ventral Tegmental Area of Rats. Addict. Biol. 2015, 20, 336–344. [Google Scholar] [CrossRef]
- Dhaher, R.; McConnell, K.K.; Rodd, Z.A.; McBride, W.J.; Bell, R.L. Daily Patterns of Ethanol Drinking in Adolescent and Adult, Male and Female, High Alcohol Drinking (HAD) Replicate Lines of Rats. Pharmacol. Biochem. Behav. 2012, 102, 540–548. [Google Scholar] [CrossRef]
- Loi, B.; Colombo, G.; Maccioni, P.; Carai, M.A.M.; Franconi, F.; Gessa, G.L. High Alcohol Intake in Female Sardinian Alcohol-Preferring Rats. Alcohol 2014, 48, 345–351. [Google Scholar] [CrossRef]
- Gavzan, H.; Hashemi, F.; Babaei, J.; Sayyah, M. A Role for Peroxisome Proliferator-Activated Receptor α in Anticonvulsant Activity of Docosahexaenoic Acid against Seizures Induced by Pentylenetetrazole. Neurosci. Lett. 2018, 681, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Ezquer, F.; Quintanilla, M.E.; Morales, P.; Santapau, D.; Ezquer, M.; Kogan, M.J.; Salas-Huenuleo, E.; Herrera-Marschitz, M.; Israel, Y. Intranasal Delivery of Mesenchymal Stem Cell-Derived Exosomes Reduces Oxidative Stress and Markedly Inhibits Ethanol Consumption and Post-Deprivation Relapse Drinking. Addict. Biol. 2019, 24, 994–1007. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Choi, A.M.K. Heme Oxygenase-1: Redox Regulation of a Stress Protein in Lung and Cell Culture Models. Antioxid. Redox Signal. 2005, 7, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Moreno, S.; Cerù, M.P. In Search for Novel Strategies towards Neuroprotection and Neuroregeneration: Is PPARα a Promising Therapeutic Target? Neural Regen. Res. 2015, 10, 1409–1412. [Google Scholar] [CrossRef]
- Wójtowicz, S.; Strosznajder, A.K.; Jeżyna, M.; Strosznajder, J.B. The Novel Role of PPAR Alpha in the Brain: Promising Target in Therapy of Alzheimer’s Disease and Other Neurodegenerative Disorders. Neurochem. Res. 2020, 45, 972–988. [Google Scholar] [CrossRef]
- Galan-Rodriguez, B.; Suarez, J.; Gonzalez-Aparicio, R.; Bermudez-Silva, F.J.; Maldonado, R.; Robledo, P.; Rodriguez de Fonseca, F.; Fernandez-Espejo, E. Oleoylethanolamide Exerts Partial and Dose-Dependent Neuroprotection of Substantia Nigra Dopamine Neurons. Neuropharmacology 2009, 56, 653–664. [Google Scholar] [CrossRef]
- Quintanilla, M.E.; Ezquer, F.; Morales, P.; Ezquer, M.; Olivares, B.; Santapau, D.; Herrera-Marschitz, M.; Israel, Y. N-Acetylcysteine and Acetylsalicylic Acid Inhibit Alcohol Consumption by Different Mechanisms: Combined Protection. Front. Behav. Neurosci. 2020, 14, 122. [Google Scholar] [CrossRef]
- Lo Verme, J.; Fu, J.; Astarita, G.; La Rana, G.; Russo, R.; Calignano, A.; Piomelli, D. The Nuclear Receptor Peroxisome Proliferator-Activated Receptor-Alpha Mediates the Anti-Inflammatory Actions of Palmitoylethanolamide. Mol. Pharmacol. 2005, 67, 15–19. [Google Scholar] [CrossRef]
- Fu, J.; Gaetani, S.; Oveisi, F.; Lo Verme, J.; Serrano, A.; Rodríguez De Fonseca, F.; Rosengarth, A.; Luecke, H.; Di Giacomo, B.; Tarzia, G.; et al. Oleylethanolamide Regulates Feeding and Body Weight through Activation of the Nuclear Receptor PPAR-Alpha. Nature 2003, 425, 90–93. [Google Scholar] [CrossRef]
- Sayd, A.; Antón, M.; Alén, F.; Caso, J.R.; Pavón, J.; Leza, J.C.; Rodríguez de Fonseca, F.; García-Bueno, B.; Orio, L. Systemic Administration of Oleoylethanolamide Protects from Neuroinflammation and Anhedonia Induced by LPS in Rats. Int. J. Neuropsychopharmacol. 2014, 18, pyu111. [Google Scholar] [CrossRef] [PubMed]
- Stahel, P.F.; Smith, W.R.; Bruchis, J.; Rabb, C.H. Peroxisome Proliferator-Activated Receptors: “Key” Regulators of Neuroinflammation after Traumatic Brain Injury. PPAR Res. 2008, 2008, 538141. [Google Scholar] [CrossRef] [PubMed]
- Bilbao, A.; Serrano, A.; Cippitelli, A.; Pavón, F.J.; Giuffrida, A.; Suárez, J.; García-Marchena, N.; Baixeras, E.; Gómez de Heras, R.; Orio, L.; et al. Role of the Satiety Factor Oleoylethanolamide in Alcoholism. Addict. Biol. 2016, 21, 859–872. [Google Scholar] [CrossRef] [PubMed]
- Orio, L.; Alen, F.; Pavón, F.J.; Serrano, A.; García-Bueno, B. Oleoylethanolamide, Neuroinflammation, and Alcohol Abuse. Front. Mol. Neurosci. 2019, 11, 490. [Google Scholar] [CrossRef]
- Kane, C.J.M.; Phelan, K.D.; Douglas, J.C.; Wagoner, G.; Johnson, J.W.; Xu, J.; Phelan, P.S.; Drew, P.D. Effects of Ethanol on Immune Response in the Brain: Region-Specific Changes in Adolescent versus Adult Mice. Alcohol. Clin. Exp. Res. 2014, 38, 384–391. [Google Scholar] [CrossRef]
- Bell, R.L.; Lopez, M.F.; Cui, C.; Egli, M.; Johnson, K.W.; Franklin, K.M.; Becker, H.C. Ibudilast Reduces Alcohol Drinking in Multiple Animal Models of Alcohol Dependence. Addict. Biol. 2015, 20, 38–42. [Google Scholar] [CrossRef]
- Mirza, R.; Sharma, B. Benefits of Fenofibrate in Prenatal Valproic Acid-Induced Autism Spectrum Disorder Related Phenotype in Rats. Brain Res. Bull. 2019, 147, 36–46. [Google Scholar] [CrossRef]
- Barbiero, J.K.; Santiago, R.; Tonin, F.S.; Boschen, S.; da Silva, L.M.; Werner, M.F.; da Cunha, C.; Lima, M.M.; Vital, M.A. PPAR-α agonist fenofibrate protects against the damaging effects of MPTP in a rat model of Parkinson’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 53, 35–44. [Google Scholar] [CrossRef]
- Caillaud, M.; Patel, N.H.; White, A.; Wood, M.; Contreras, K.M.; Toma, W.; Alkhlaif, Y.; Roberts, J.L.; Tran, T.H.; Jackson, A.B.; et al. Targeting Peroxisome Proliferator-Activated Receptor-α (PPAR-α) to reduce paclitaxel-induced peripheral neuropathy. Brain Behav. Immun. 2021, 93, 172–185. [Google Scholar] [CrossRef]
- Chistyakov, D.V.; Astakhova, A.A.; Goriainov, S.V.; Sergeeva, M.G. Comparison of PPAR Ligands as Modulators of Resolution of Inflammation, via Their Influence on Cytokines and Oxylipins Release in Astrocytes. Int. J. Mol. Sci. 2020, 21, 9577. [Google Scholar] [CrossRef]
- Blednov, Y.A.; Black, M.; Benavidez, J.M.; Stamatakis, E.E.; Harris, R.A. PPAR Agonists: I. Role of Receptor Subunits in Alcohol Consumption in Male and Female Mice. Alcohol. Clin. Exp. Res. 2016, 40, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Dludla, P.V.; Nkambule, B.B.; Mazibuko-Mbeje, S.E.; Nyambuya, T.M.; Silvestri, S.; Orlando, P.; Mxinwa, V.; Louw, J.; Tiano, L. The impact of dimethyl sulfoxide on oxidative stress and cytotoxicity in various experimental models. In Toxicology; Patel, V.B., Preedy, V.R., Eds.; Academic Press: Cambridge, MA, USA, 2021; Chapter 25; pp. 243–261. ISBN 9780128190920. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibáñez, C.; Acuña, T.; Quintanilla, M.E.; Pérez-Reytor, D.; Morales, P.; Karahanian, E. Fenofibrate Decreases Ethanol-Induced Neuroinflammation and Oxidative Stress and Reduces Alcohol Relapse in Rats by a PPAR-α-Dependent Mechanism. Antioxidants 2023, 12, 1758. https://doi.org/10.3390/antiox12091758
Ibáñez C, Acuña T, Quintanilla ME, Pérez-Reytor D, Morales P, Karahanian E. Fenofibrate Decreases Ethanol-Induced Neuroinflammation and Oxidative Stress and Reduces Alcohol Relapse in Rats by a PPAR-α-Dependent Mechanism. Antioxidants. 2023; 12(9):1758. https://doi.org/10.3390/antiox12091758
Chicago/Turabian StyleIbáñez, Cristina, Tirso Acuña, María Elena Quintanilla, Diliana Pérez-Reytor, Paola Morales, and Eduardo Karahanian. 2023. "Fenofibrate Decreases Ethanol-Induced Neuroinflammation and Oxidative Stress and Reduces Alcohol Relapse in Rats by a PPAR-α-Dependent Mechanism" Antioxidants 12, no. 9: 1758. https://doi.org/10.3390/antiox12091758