
Mitochondrial H2O2 Is a Central Mediator of Diclofenac-Induced Hepatocellular Injury

 , , and
, , and
Abstract
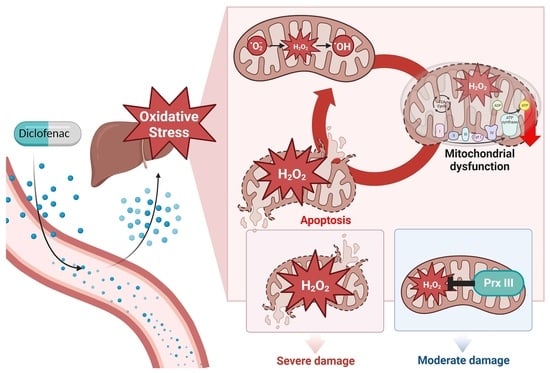
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Reagents and Antibodies
2.3. Cell Culture and Infection
2.4. Establishment of HepG2 Cells Expressing Small Hairpin RNA Targeting PrxIII
2.5. Western Blotting
2.6. Determination of Mitochondrial H2O2
2.7. Flow Cytometry Analyses
2.8. Oxygen Consumption Rate (OCR) Measurement
2.9. Caspase Activity Assay
2.10. Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick end Labeling (TUNEL) Assay
2.11. Statistical Analysis
3. Results
3.1. Without PrxIII, Mitochondrial H2O2 Levels Increase Following Diclofenac Treatment of HepG2 Cells
3.2. PrxIII Depletion Increases Diclofenac-Induced Mitochondrial Oxidative Injury in HepG2 Cells
3.3. PrxIII Depletion Exacerbates Mitochondrial Dysfunction Induced by Diclofenac in HepG2 Cells
3.4. PrxIII Depletion Promotes Apoptosis Induced by Diclofenac in HepG2 Cells
3.5. Mitochondrion-Targeted Catalase Expression Alleviates the Diclofenac-Induced Apoptosis Amplified in PrxIII-Depleted HepG2 Cells
3.6. Diclofenac-Induced Apoptosis of Primary PrxIII−/− Murine Hepatocytes Is Significantly Suppressed by Mitochondria-Specific Elimination of H2O2
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Schmeltzer, P.A.; Kosinski, A.S.; Kleiner, D.E.; Hoofnagle, J.H.; Stolz, A.; Fontana, R.J.; Russo, M.W.; Drug-Induced Liver Injury, N. Liver injury from nonsteroidal anti-inflammatory drugs in the United States. Liver Int. 2016, 36, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Masubuchi, Y.; Nakayama, S.; Horie, T. Role of mitochondrial permeability transition in diclofenac-induced hepatocyte injury in rats. Hepatology 2002, 35, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Lechon, M.J.; Ponsoda, X.; O’Connor, E.; Donato, T.; Castell, J.V.; Jover, R. Diclofenac induces apoptosis in hepatocytes by alteration of mitochondrial function and generation of ROS. Biochem. Pharmacol. 2003, 66, 2155–2167. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.H.; Lee, W.; Park, S.H.; Lee, K.Y.; Choi, Y.J.; Choi, S.; Kang, D.; Kim, S.; Chang, T.S.; Hong, S.S.; et al. Diclofenac impairs autophagic flux via oxidative stress and lysosomal dysfunction: Implications for hepatotoxicity. Redox Biol. 2020, 37, 101751. [Google Scholar] [CrossRef]
- Syed, M.; Skonberg, C.; Hansen, S.H. Mitochondrial toxicity of diclofenac and its metabolites via inhibition of oxidative phosphorylation (ATP synthesis) in rat liver mitochondria: Possible role in drug induced liver injury (DILI). Toxicol. Vitr. 2016, 31, 93–102. [Google Scholar] [CrossRef]
- Al-Attrache, H.; Sharanek, A.; Burban, A.; Burbank, M.; Gicquel, T.; Abdel-Razzak, Z.; Guguen-Guillouzo, C.; Morel, I.; Guillouzo, A. Differential sensitivity of metabolically competent and non-competent HepaRG cells to apoptosis induced by diclofenac combined or not with TNF-alpha. Toxicol. Lett. 2016, 258, 71–86. [Google Scholar] [CrossRef]
- Sandoval-Acuna, C.; Lopez-Alarcon, C.; Aliaga, M.E.; Speisky, H. Inhibition of mitochondrial complex I by various non-steroidal anti-inflammatory drugs and its protection by quercetin via a coenzyme Q-like action. Chem. Biol. Interact. 2012, 199, 18–28. [Google Scholar] [CrossRef]
- Ghosh, R.; Goswami, S.K.; Feitoza, L.; Hammock, B.; Gomes, A.V. Diclofenac induces proteasome and mitochondrial dysfunction in murine cardiomyocytes and hearts. Int. J. Cardiol. 2016, 223, 923–935. [Google Scholar] [CrossRef]
- Konstantinova, S.G.; Russanov, E.M. Studies on the nature of superoxide dismutase activity in sheep liver subcellular fractions. Acta Physiol. Pharmacol. Bulg. 1988, 14, 71–77. [Google Scholar]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS signaling in organismal homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef]
- Sena, L.A.; Chandel, N.S. Physiological Roles of Mitochondrial Reactive Oxygen Species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.G.; Winterbourn, C.C.; Hampton, M.B. Mitochondrial peroxiredoxin involvement in antioxidant defence and redox signalling. Biochem. J. 2010, 425, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.H.; Sung, S.H.; Lee, H.E.; Kang, H.T.; Lee, S.K.; Oh, S.Y.; Woo, H.A.; Kil, I.S.; Rhee, S.G. Peroxiredoxin III and sulfiredoxin together protect mice from pyrazole-induced oxidative liver injury. Antioxid. Redox Signal. 2012, 17, 1351–1361. [Google Scholar] [CrossRef] [PubMed]
- Sonn, S.K.; Song, E.J.; Seo, S.; Kim, Y.Y.; Um, J.H.; Yeo, F.J.; Lee, D.S.; Jeon, S.; Lee, M.N.; Jin, J.; et al. Peroxiredoxin 3 deficiency induces cardiac hypertrophy and dysfunction by impaired mitochondrial quality control. Redox Biol. 2022, 51, 102275. [Google Scholar] [CrossRef]
- Chang, T.S.; Cho, C.S.; Park, S.; Yu, S.; Kang, S.W.; Rhee, S.G. Peroxiredoxin III, a mitochondrion-specific peroxidase, regulates apoptotic signaling by mitochondria. J. Biol. Chem. 2004, 279, 41975–41984. [Google Scholar] [CrossRef]
- Baek, J.Y.; Han, S.H.; Sung, S.H.; Lee, H.E.; Kim, Y.M.; Noh, Y.H.; Bae, S.H.; Rhee, S.G.; Chang, T.S. Sulfiredoxin protein is critical for redox balance and survival of cells exposed to low steady-state levels of H2O2. J. Biol. Chem. 2012, 287, 81–89. [Google Scholar] [CrossRef]
- Rhee, S.G.; Kil, I.S. Mitochondrial H2O2 signaling is controlled by the concerted action of peroxiredoxin III and sulfiredoxin: Linking mitochondrial function to circadian rhythm. Free Radic. Biol. Med. 2016, 99, 120–127. [Google Scholar] [CrossRef]
- Kalyanaraman, B.; Darley-Usmar, V.; Davies, K.J.; Dennery, P.A.; Forman, H.J.; Grisham, M.B.; Mann, G.E.; Moore, K.; Roberts, L.J., 2nd; Ischiropoulos, H. Measuring reactive oxygen and nitrogen species with fluorescent probes: Challenges and limitations. Free Radic. Biol. Med. 2012, 52, 1065–1073. [Google Scholar] [CrossRef]
- Dickinson, B.C.; Lin, V.S.; Chang, C.J. Preparation and use of MitoPY1 for imaging hydrogen peroxide in mitochondria of live cells. Nat. Protoc. 2013, 8, 1249–1259. [Google Scholar] [CrossRef]
- Giorgio, M.; Trinei, M.; Migliaccio, E.; Pelicci, P.G. Hydrogen peroxide: A metabolic by-product or a common mediator of ageing signals? Nat. Rev. Mol. Cell Biol. 2007, 8, 722–728. [Google Scholar] [CrossRef]
- Nomura, K.; Imai, H.; Koumura, T.; Kobayashi, T.; Nakagawa, Y. Mitochondrial phospholipid hydroperoxide glutathione peroxidase inhibits the release of cytochrome c from mitochondria by suppressing the peroxidation of cardiolipin in hypoglycaemia-induced apoptosis. Biochem. J. 2000, 351, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Petit, J.M.; Maftah, A.; Ratinaud, M.H.; Julien, R. 10N-nonyl acridine orange interacts with cardiolipin and allows the quantification of this phospholipid in isolated mitochondria. Eur. J. Biochem. 1992, 209, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Scaduto, R.C., Jr.; Grotyohann, L.W. Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys. J. 1999, 76, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Cederbaum, A.I. Adenovirus-mediated overexpression of catalase in the cytosolic or mitochondrial compartment protects against cytochrome P450 2E1-dependent toxicity in HepG2 cells. J. Biol. Chem. 2001, 276, 4315–4321. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.A.; Isin, E.M.; Li, Y.; Weidolf, L.; Page, K.; Wilson, I.; Swallow, S.; Middleton, B.; Stahl, S.; Foster, A.J.; et al. In Vitro Approach to Assess the Potential for Risk of Idiosyncratic Adverse Reactions Caused by Candidate Drugs. Chem. Res. Toxicol. 2012, 25, 1616–1632. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; Visschers, R.G.J.; Duan, L.; Akakpo, J.Y.; Jaeschke, H. Mitochondrial dysfunction as a mechanism of drug-induced hepatotoxicity: Current understanding and future perspectives. J. Clin. Transl. Res. 2018, 4, 75–100. [Google Scholar] [CrossRef] [PubMed]
- Shah, I.; Barker, J.; Naughton, D.P.; Barton, S.J.; Ashraf, S.S. Determination of diclofenac concentrations in human plasma using a sensitive gas chromatography mass spectrometry method. Chem. Cent. J. 2016, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Willis, J.V.; Kendall, M.J.; Jack, D.B. A study of the Effect of Aspirin on the Pharmacokinetics of Oral and Intravenous Diclofenac Sodium. Eur. J. Clin. Pharmacol. 1980, 18, 415–418. [Google Scholar] [CrossRef]
- Lill, J.S.; O’Sullivan, T.; Bauer, L.A.; Horn, J.R.; Carithers, R., Jr.; Strandness, D.E.; Lau, H.; Chan, K.; Thakker, K. Pharmacokinetics of diclofenac sodium in chronic active hepatitis and alcoholic cirrhosis. J. Clin. Pharmacol. 2000, 40, 250–257. [Google Scholar] [CrossRef]
- Netter, P.; Lambert, H.; Larcan, A.; Godbillon, J.; Gosset, G. Diclofenac Sodium-Chlormezanone Poisoning. Eur. J. Clin. Pharmacol. 1984, 26, 535–536. [Google Scholar] [CrossRef]
- Hunter, L.J.; Wood, D.M.; Dargan, P.I. The patterns of toxicity and management of acute nonsteroidal anti-inflammatory drug (NSAID) overdose. Open Access Emerg. Med. 2011, 3, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Panchal, N.K.; Prince Sabina, E. Non-steroidal anti-inflammatory drugs (NSAIDs): A current insight into its molecular mechanism eliciting organ toxicities. Food Chem. Toxicol. 2023, 172, 113598. [Google Scholar] [CrossRef] [PubMed]
- Bort, R.; Ponsoda, X.; Jover, R.; Gomez-Lechon, M.J.; Castell, J.V. Diclofenac toxicity to hepatocytes: A role for drug metabolism in cell toxicity. J. Pharmacol. Exp. Ther. 1999, 288, 65–72. [Google Scholar] [PubMed]
- Giulivi, C.; Poderoso, J.J.; Boveris, A. Production of nitric oxide by mitochondria. J. Biol. Chem. 1998, 273, 11038–11043. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, E. Mitochondrial free radical production and cell signaling. Mol. Asp. Med. 2004, 25, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Ghafourifar, P.; Cadenas, E. Mitochondrial nitric oxide synthase. Trends Pharmacol. Sci. 2005, 26, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C. Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat. Rev. Mol. Cell Biol. 2022, 23, 499–515. [Google Scholar] [CrossRef]
- Imai, H.; Nakagawa, Y. Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radic. Biol. Med. 2003, 34, 145–169. [Google Scholar] [CrossRef]
- Kil, I.S.; Lee, S.K.; Ryu, K.W.; Woo, H.A.; Hu, M.C.; Bae, S.H.; Rhee, S.G. Feedback control of adrenal steroidogenesis via H2O2-dependent, reversible inactivation of peroxiredoxin III in mitochondria. Mol. Cell 2012, 46, 584–594. [Google Scholar] [CrossRef]
- Cardozo, G.; Mastrogiovanni, M.; Zeida, A.; Viera, N.; Radi, R.; Reyes, A.M.; Trujillo, M. Mitochondrial Peroxiredoxin 3 Is Rapidly Oxidized and Hyperoxidized by Fatty Acid Hydroperoxides. Antioxidants 2023, 12, 408. [Google Scholar] [CrossRef]
- De Armas, M.I.; Esteves, R.; Viera, N.; Reyes, A.M.; Mastrogiovanni, M.; Alegria, T.G.P.; Netto, L.E.S.; Tortora, V.; Radi, R.; Trujillo, M. Rapid peroxynitrite reduction by human peroxiredoxin 3: Implications for the fate of oxidants in mitochondria. Free Radic. Biol. Med. 2019, 130, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.; Lin, C.C.; Lett, C.; Karpinska, B.; Wright, M.H.; Foyer, C.H. Catalase: A critical node in the regulation of cell fate. Free Radic. Biol. Med. 2023, 199, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Masubuchi, Y.; Yamada, S.; Horie, T. Possible mechanism of hepatocyte injury induced by diphenylamine and its structurally related nonsteroidal anti-inflammatory drugs. J. Pharmacol. Exp. Ther. 2000, 292, 982–987. [Google Scholar] [PubMed]
- Demel, R.A.; Jordi, W.; Lambrechts, H.; van Damme, H.; Hovius, R.; de Kruijff, B. Differential interactions of apo- and holocytochrome c with acidic membrane lipids in model systems and the implications for their import into mitochondria. J. Biol. Chem. 1989, 264, 3988–3997. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Borisenko, G.G.; Tyurina, Y.Y.; Tyurin, V.A.; Jiang, J.; Potapovich, A.I.; Kini, V.; Amoscato, A.A.; Fujii, Y. Oxidative lipidomics of apoptosis: Redox catalytic interactions of cytochrome c with cardiolipin and phosphatidylserine. Free Radic. Biol. Med. 2004, 37, 1963–1985. [Google Scholar] [CrossRef]
- Orrenius, S.; Zhivotovsky, B. Cardiolipin oxidation sets cytochrome c free. Nat. Chem. Biol. 2005, 1, 188–189. [Google Scholar] [CrossRef]
- Shidoji, Y.; Hayashi, K.; Komura, S.; Ohishi, N.; Yagi, K. Loss of molecular interaction between cytochrome c and cardiolipin due to lipid peroxidation. Biochem. Biophys. Res. Commun. 1999, 264, 343–347. [Google Scholar] [CrossRef]
- Ott, M.; Robertson, J.D.; Gogvadze, V.; Zhivotovsky, B.; Orrenius, S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc. Natl. Acad. Sci. USA 2002, 99, 1259–1263. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.R.; Park, J.W.; Choi, Y.-J.; Sonn, S.K.; Oh, G.T.; Lee, B.-H.; Chang, T.-S. Mitochondrial H2O2 Is a Central Mediator of Diclofenac-Induced Hepatocellular Injury. Antioxidants 2024, 13, 17. https://doi.org/10.3390/antiox13010017
Kim SR, Park JW, Choi Y-J, Sonn SK, Oh GT, Lee B-H, Chang T-S. Mitochondrial H2O2 Is a Central Mediator of Diclofenac-Induced Hepatocellular Injury. Antioxidants. 2024; 13(1):17. https://doi.org/10.3390/antiox13010017
Chicago/Turabian StyleKim, Sin Ri, Ji Won Park, You-Jin Choi, Seong Keun Sonn, Goo Taeg Oh, Byung-Hoon Lee, and Tong-Shin Chang. 2024. "Mitochondrial H2O2 Is a Central Mediator of Diclofenac-Induced Hepatocellular Injury" Antioxidants 13, no. 1: 17. https://doi.org/10.3390/antiox13010017
APA StyleKim, S. R., Park, J. W., Choi, Y.-J., Sonn, S. K., Oh, G. T., Lee, B.-H., & Chang, T.-S. (2024). Mitochondrial H2O2 Is a Central Mediator of Diclofenac-Induced Hepatocellular Injury. Antioxidants, 13(1), 17. https://doi.org/10.3390/antiox13010017