1. Introduction
Pancreatic β-cells play a pivotal role in the management of glucose homeostasis by synthesizing and secreting insulin at appropriate rates. At low levels, oxidative stress is essential for normal β-cell function and has a role in different activities, such as the stimulation of basal insulin secretion [
1]. However, due to the relatively low expression of antioxidant enzymes (e.g., catalase and glutathione peroxidase) [
2], these cells are profoundly susceptible to oxidative damage upon sustained insult [
3]. In particular, reactive oxygen species (ROS) are key mediators for nucleic acids, lipids, and protein damage, resulting in undesirable biochemical and functional modifications [
4]. Sustained excessive iron and glucose exposure are key mediators of oxidative stress in pancreatic β-cells, ultimately leading to mitochondrial and cellular membrane damage and disruption [
5]. High amounts of free ferrous iron lead to ROS production via the Fenton and Haber–Weiss reactions, involving the production of hydroxyl radical (
•OH). These free radicals can react rapidly with biological molecules, causing significant damage to cellular components and dysfunctions including insulin resistance and eventual β-cell failure [
1]. In this context, recent evidence demonstrated that high iron exposure (100 µM) can result in a marked decrease in MIN6 insulin secretory capacity as well as cellular insulin content. From a mechanistic point of view, the perturbations in mitochondrial functionality related to iron-induced oxidative stress is associated with a reduction in MIN6 β-cell insulin secretion, indicating a fundamental role for iron overload in the onset and progression of T2DM [
6].
The potential efficacy of bioactive compounds of natural origin in the treatment of diabetes through the ability to control blood glucose levels to different degrees and through varying mechanisms of action has been well described in the scientific literature [
7]. Interestingly, immature fruits derived from fruit thinning, the manual elimination of fruitlets, have gained increasing attention for nutraceutical applications as food byproducts rich in bioactive compounds with potential health benefits [
8]. Specifically, recent findings suggested that thinned nectarines (TNs) (
Prunus persica L.) are promising for the management of diabetes due to their high content of polyphenols and abscisic acid (ABA) [
9,
10]. ABA is a naturally occurring plant hormone found at the immature stage in various fruits, including nectarines [
11]. Moreover, several studies have also demonstrated a role for ABA as an endogenous hormone exhibiting antidiabetic properties in humans by enhancing glucose uptake and improving insulin sensitivity [
12,
13]. Among plant-derived compounds, polyphenols have also demonstrated potential in diabetes control [
14]. These molecules exert antioxidant and anti-inflammatory properties that can help regulate glycemic levels and improve insulin resistance [
15]. Notably, TNs have been shown to contain high levels of polyphenols compared to their mature counterparts, making them an intriguing option for individuals with diabetes [
9,
16,
17]. Overall, the combined presence of abscisic acid and polyphenols in TNs suggests a synergistic effect in terms of their potential antidiabetic properties.
Although several natural compounds have shown promising effects, their application has often been limited by issues related to their relatively low stability, bioavailability, and efficacy at the site of action [
18]. In this context, nanotechnology-based formulations have proven to be beneficial in overcoming these limitations and enhancing the potential of bioactive molecules [
18,
19]. Polymeric micelles (PMs) are such nanoparticle delivery systems and have been used as drug carriers as they provide a suitable platform for the incorporation of active ingredients. In this regard, several studies successfully demonstrated the role of these vesicles as carriers for hydrophobic molecules [
20]. Among different polymeric micelles, the polyethylene glycol-grafted 1,2-distearoyl-sn-glycero-3-phosphatidylethanolamine (DSPE-PEG) stands out due to its ability to form micelles in aqueous environments rather than creating bilayered structures [
21]. Ascorbyl palmitate (AP), a lipophilic derivative of ascorbic acid, is commonly utilized in the cosmetic industry. Due to its hydrophobic nature, AP spontaneously combines with the amphiphilic derivative DSPE-PEG to form vesicular structures in an aqueous environment [
21,
22].
Based on these considerations, this study aimed to develop, for the first time, nanoformulations (NF) containing a pure standard of ABA or a thinned nectarine extract (TNE) at the same ABA concentration using DSPE-PEG-AP as the nanocarrier system. Moreover, the insulinotropic and antioxidant potential of these nanoformulated compounds and their unformulated forms were tested and compared in MIN-6 pancreatic cells incubated with a combination of different glucose (5.5 mM and 20 mM) and iron (100 µM) concentrations.
2. Materials and Methods
2.1. Reagents and Materials
All chemicals and reagents used for the HPLC-DAD analysis were either analytical-reagent or HPLC grade. The water was treated in a Milli-Q water purification system (Millipore, Bedford, MA, USA) before use. (±)-2-cis-4-trans-abscisic acid (ABA), 2′,7′-Dichlorofluorescein diacetate (DCFH-DA), ethanol, methanol, acetonitrile, and formic acid were all purchased from Sigma-Aldrich (Milano, Italy). Iron(II) sulfate heptahydrate (FeSO4) was used for all experiments involving the use of iron as a prooxidant agent. All chemicals for cell culture experiments were of cell-culture grade and purchased from Sigma-Aldrich (Dorset, UK) unless otherwise stated. Protease Inhibitor Cocktail (PIC, catalog no. P8340), dimethyl sulfoxide (DMSO), Thiazolyl, and Blue Tetrazolium Blue (MTT) were obtained from Sigma-Aldrich (Dorset, UK). Fisher Scientific (Loughborough, UK) supplied Minimum Essential Medium (MEM), Dulbecco’s modified Eagle’s medium (DMEM) Glutamax®, fetal bovine serum (FBS), and 100× antibiotic–antimycotic solution. The 6- and 12-well cell culture dishes, 96-well microtiter plates, and flasks were purchased from Nunc (Roskilde, Denmark). A mouse insulin ELISA kit was supplied by Mercodia (Cat No. 10-1247-01—Mercodia, D.B.A., Milan, Italy). DSPE-PEG was purchased from Lipoid (Steinhausen, Switzerland), and ascorbyl palmitate was from Sigma-Aldrich (Dorset, UK). Experimental reagents were prepared using ultrapure water (resistivity of 18.2 MΩ cm).
2.2. Preparation of Thinned Nectarine Extract
TNs were harvested in June 2022 at the orchards of “Giaccio Frutta” society (Vitulazio, Caserta, Italy, 41°10′ N–14°13′ E) about one month after full bloom in concomitance with the fruit thinning stage. The whole fruits (pulp, peel, and seed) were frozen at −20 °C, lyophilized, and ground to obtain a homogenous powder. With regards to the extraction procedure, a previous method reported by Schiano et al. was used with slight modifications [
23]. Briefly, 2.5 g of homogenized sample was suspended in 25 mL of a 100% ethanolic solution and thoroughly vortexed for 1 min. The suspension was then sonicated for 10 min using a VWR Ultrasonic cleaner bath USC300T (VWR International Limited, Lutterworth, UK) and stirred lightly for 40 min at 25°, 600 RPM speed. After centrifugation (10 min, 9000×
g, 25°), the supernatant was decanted, filtered with a 0.22 μm nylon filter (CellTreat, Shirley, MA, USA), and evaporated using a rotary evaporator (Hei-VAP Advantage Rotary Evaporator, Heidolph, Germany). The ABA content of the dried extract was evaluated as reported in the following section. For all experiments described below, the dried extract was dissolved with a defined volume of media (MEM) in order to obtain the equivalent ABA concentrations contained in free ABA and NF-ABA samples.
2.3. HPLC-DAD Analysis of Abscisic Acid
The chromatographic system utilized was a Jasco Extrema LC-4000 (Jasco Inc., Easton, MD, USA) equipped with the following modules: a vacuum degasser, an autoinjector, a quaternary pump, a column oven, and a diode array detector photodiode array detector (DAD). This setup was used for the chromatographic determination of ABA content in TNE based on a previous method, with slight modifications [
24]. The chosen column was a Kinetex
® C
18 column 100A (250 mm × 4.6 mm i.d., 5 μm) from Phenomenex (Torrance, CA, USA). The mobile phases consisted of 1% formic acid in water (A) and acetonitrile (B). The elution procedure started with a 3-min hold at 5% of solvent B, followed by a gradient to 75% (B) over 20 min and a 1 min maintenance; the column was then re-equilibrated to the initial conditions for another minute. Separation conditions included a 30 °C column temperature, 20 µL injection volume, and a 1 mL/min flow rate. Abscisic acid detection was monitored at 265 nm, with quantification based on a calibration curve covering 0.1–100 ppm concentrations and triplicate injections at each level.
2.4. Evaluation of the Physiochemical Properties of the Nanoformulations
2.4.1. Preparation of Nanoformulated Samples
The preparation of DSPE-PEG or DSPE-PEG ascorbyl palmitate nanocarriers was carried out by following the thin-film hydration method, as previously described [
21]. Briefly, DSPE-PEG alone or ascorbyl palmitate (AP) and DSPE-PEG were dissolved in methanol and then dried in a round-bottom flask using a rotary evaporator (Hei-VAPAdvantage Rotary Evaporator, Heidolph, Schwabach, Germany) at 60 °C to form a lipid film. This film was then purged with nitrogen gas to remove residual solvent and then hydrated with either an ABA or TNE solution (4 mg/10 mL or 52 mg/5 mL) or distilled water in the case of corresponding blank nanocarrier formulations. In order to remove any unloaded compound, the obtained solution was filtered with a sterile 0.22 μm filter. The nanocarriers were then stored in 10 mL vials at 4 °C until use. Preparations were coded: 1. DSPE-PEG-TNE, 2. DSPE-PEG-ABA, 3. DSPE-PEG-blank, 4. DSPE-PEG-AP-TNE, 5. DSPE-PEG-AP-ABA, and 6. DSPE-PEG-AP-blank. To allow the storage of samples for further analysis, samples were freeze-dried using a Virtis AdVantage 2.0 BenchTop freeze dryer (SP Industries, Bradford, UK).
2.4.2. Size and Surface Charge of the Nanoformulations
The particle size and surface charge of the NFs were determined using the Zetasizer Nano ZS (Malvern Instruments, Malvern, UK). For these measurements, the nanodispersions were added individually to the ZetaMaster electrophoresis cell to assess their size distribution via photon correlation spectroscopy as mean volume diameters (MVD) and polydispersity index (PDI), carrying out three measurements on each sample. The zeta potential of NF samples was determined similarly using the electrophoretic light-scattering technique (Zetasizer Nano ZS, Malvern Instruments, UK), measuring electrophoretic mobility in triplicate for all samples (
Table 1).
2.4.3. Nanoformulation Morphology
Transmission electron microscopy (TEM) was used to analyze the complex NF morphology, including the size and surface characteristics, using an FEI CM 120 BioTwin transmission electron microscope (Philips Electron Optics BV, Eindhoven, The Netherlands) at an acceleration voltage of 120.0 kV. Samples of approximately 40 μL of the nanoparticle dispersion were placed on a Formvar/carbon-coated copper grid, negatively stained with 1% uranyl acetate, and observed at magnifications of 13,500-, 17,500-, 46,000-, and 65,000-times magnification to capture digital images (
Figure S1).
2.4.4. Determination of Nanoformulation Loading and Encapsulation Efficiency
HPLC-DAD analysis was utilized to study the drug loading and encapsulation efficiency of NF, following the method previously described in
Section 2.3. The percentage of drug loading (DG) and encapsulation efficiency (EE) were calculated based on the following equations:
2.5. MIN6 Cell Culture
MIN6 β-cells from mice (originally from Prof. Jun-ichi Miyazaki, Osaka University, Osaka, Japan) were kindly provided by the Beta Cell Group (Department of Diabetes, King’s College London, London, UK) at passage 35. Cells stored at cryogenic temperatures (−196 °C) in liquid nitrogen were reconstituted rapidly by placing them in an incubator with an atmosphere of 95% air and 5% CO
2 at 37 °C for 2 min. After transferring the cells to a 15 mL centrifuge tube, 5 mL of fresh growth medium was added. The cell suspension was then centrifuged at 5000 RPM for 5 min. The resulting pellet was resuspended in a 75 cm
2 T-flask containing 10 mL of fresh growth media. MIN6 cells were cultured in a Dulbecco’s Modified Eagle Medium (DMEM)—GlutaMax
®, pH 7.4, supplemented with 1% antibiotic/antimycotic solution and 10% Fetal Bovine Serum (FBS), and maintained in a 95% air, 5% CO
2 atmosphere at 37 °C with constant humidity. Cells were seeded in 12-well or 6-well plates for all experimental procedures and reached confluence within five days. At this time, small clusters of cells were formed, indicating that the cells were assembled as physiologically relevant mature pseudoislets and were viable and functional (
Figure S2).
2.6. Determination of Cell Viability by MTT Assay
To assess any potential cytotoxicity of the testing samples (free ABA, free TNE, NF-ABA, and NF-TNE) on MIN6 beta cells, cells were incubated with each sample at three different ABA concentrations (0.001, 0.01, and 0.1 µM) or media (MEM) for 24, 48, and 72 h. After each incubation time, a volume of 20 µL of 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solubilized in sterile Dulbecco’s Phosphate-Buffered Saline (DPBS) was added to the wells at the concentration of 5 mg/mL [
19]. After incubation at 37 °C for 4 h, the formazan crystals formed were dissolved in 100 µL of DMSO, with the previous medium aspirated. The plates were ultimately placed on The MaxQ 4000 benchtop orbital shaker (Thermo Fisher Scientific, Loughborough, UK) at 75 rpm for 15 min to ensure the DMSO was mixed well. The absorbance was subsequently measured at 570 nm and compared to the control cells (MEM).
2.7. Cellular Antioxidant Activity (CAA) Assay
The cellular antioxidant activity (CAA) assay was performed to assess and compare the antioxidant power and cellular accessibility of the formulated and unformulated samples at varying concentrations. To this end, a pre-incubation with a cell-permeable 2′,7′-Dichlorofluorescein diacetate (DCFH-DA) fluorescence probe dye was performed. DCFH-DA is deacetylated by cellular esterase and turns into its fluorescent form DCFH upon oxidation, triggered by the addition of iron (100 µM) as a prooxidant agent. The cellular antioxidant activity was assessed based on the original assay developed by Wolfe and colleagues [
25]. MIN6 cells were first cultured in a 96-well black-walled cell culture plate until confluence. Cells were then washed with DPBS and treated with 200 µL of one of the testing samples at the same ABA concentration (0.05 µM) for 1 h at 37 °C. Subsequently, cells were washed with MEM and mixed with 200 µL of DCFH-DA (100 µM) and further incubated for 30 min at 37 °C. Following aspiration, each well was treated with 100 µL of iron at 100 µM. Cell fluorescence was read every 5 min for 1 h at 528 and 485 nm emission and excitation wavelengths, respectively, using a Fluostar Optima Fluorescence Plate Reader (BMG Labtech, Offenburg, Germany). The bioflavonoid Quercetin, ranging from 0 to 2000 μM, was used as a positive control. Based on the hypothesis of the assay, Quercetin and test antioxidant samples inhibit the development of DCF by inhibiting free radical production in a concentration-dependent manner. The CAA units were calculated using the following equations:
where RFU0 is the relative fluorescence value of time zero and RFUx is the relative fluorescence at each time point (e.g., RFU10 is the relative fluorescence value at minute 10). The results of the positive control Quercetin were also reported as IC
50, while the samples’ antioxidant values were expressed as both CAA units and Quercetin equivalents (QE).
2.8. MIN6 Cells Exposure to Glucose and Iron
Cells were seeded at 75 × 10
4 cells/cm
2 for the glucose-stimulated insulin secretion (GSIS) and the iron challenge assay. The day before the experiment, MIN6 cells were iron-depleted with a serum-free MEM media (glucose 5.5 mM, supplemented with 2 mM L-glutamine and 1% antibiotic/antimycotic) and incubated overnight at 37 °C with 5% CO
2. For the GSIS experiment, MIN6 cells were washed with DPBS and preincubated in a Krebs–Ringer Bicarbonate (KRB) buffer (10 mM HEPES, 119 mM NaCl, 1.19 mM KH
2PO
4, 1.19 mM MgSO
4·7H
2O, 4.74 mM KCl, 25 mM NaHCO
3, 2.54 mM CaCl·6H
2O, pH 7.4, and 1% BSA) containing 1.1 mM glucose (basal level) for 2.5 h. For the iron challenge assay, cells were incubated in a KRB buffer containing a high iron concentration (100 µM) for 2.5 h. Afterward, an aliquot of the supernatant was collected for T0 analysis and cells were then preincubated for 30 min with the different sample treatments (free ABA, free TNE, NF-ABA, NF-TNE). A negative control (represented by MEM) was also tested. Finally, media were aspirated from all the wells, followed by 2 h exposure to KRB solution at two glucose concentrations, 5.5 mM (physiological glucose concentration) and 20 mM (supraphysiological glucose concentration). Immediately after the exposure to glucose, the supernatants were collected at different time points for analysis (i.e., T5, T10, T30, T60 min), while cells were lysed at the end of the incubation time (60 min).
Table S1 reports the list of samples at different glucose and iron concentrations. All supernatants and cell lysates were aliquoted and stored at −20° C.
2.9. Quantification of Intracellular Total Iron
Intracellular total iron content was quantified using an optimized FerroZine™-based iron protocol developed by previous authors, with slight modifications [
26]. Briefly, 200 µL of 0.1 M HCl solution was added to tubes containing 200 µL of the sample, while 200 µL of 50 mM NaOH solution was used for tubes containing 200 µL of each standard of iron (from 0 to 120 µM). A blank solution contained 400 µL of 50 mM NaOH. Subsequently, a volume of 200 µL of the freshly prepared iron-releasing agent, consisting of an equal volume of 1.4 M HCl and 4.5% (
w/
v) KMnO
4 solution, was added to each tube to promote the iron release from proteins, including ferritin. Samples were incubated for 2 h in a 60 °C dry bath within a fume hood and then cooled to room temperature for 10 min. Afterward, a volume of 60 µL of the iron detection reagent (6.5 mM FerroZine™, 2.5 M ammonium acetate, and 1 M ascorbic acid dissolved in water) was mixed into each tube and samples were incubated at 25° for 60 min. Lastly, a volume of 200 µL from standard and sample tubes was placed in duplicate into a 96-well plate and the color development was spectrometrically monitored by measuring the absorbance at 550 nm using a microplate reader (Fluostar Optima Plate Reader (BMG Labtech, Offenburg, Germany)). Intracellular iron concentration was determined by normalizing the obtained results against the total protein content of each well evaluated by the BCA (bicinchoninic acid) assay.
2.10. Evaluation of Insulin Secretion
The measurement of insulin secretion is considered pivotal for the investigation of β-cell function. To this aim, supernatants were obtained from MIN6 cells exposed to different concentrations of glucose and iron at various time points (T0, T5, T10, T30, T60 min) to determine the temporal profile of insulin secretion under these conditions and were then analyzed by insulin ELISA assay. Briefly, a volume of 10 µL of standards and samples was transferred into a 96-well plate in duplicate followed by the incubation steps, as described by the manufacturer’s protocol (Cat No. 10-1247-01—Mercodia, D.B.A., Milan, Italy). Subsequently, the absorbance was measured at 450 nm using a microplate reader.
2.11. Statistical Analysis
For all the experiments, the average was calculated from at least four replicates per condition, with data expressed as the mean ± standard error of the mean (SEM) unless otherwise stated. The MTT results were analyzed using a one-way analysis of variance (ANOVA) followed by Dunnett’s post hoc test and a two-way ANOVA followed by Sidak’s post hoc test. Total iron quantification was examined using a two-way ANOVA test followed by Sidak’s post hoc test. CAA results were evaluated with a two-tailed Student’s t-test, while ELISA results were analyzed using a two-way ANOVA test followed by Tukey’s multiple comparisons post hoc test (PRISM software package, Version 8, Graphpad Software Inc., San Diego, CA, USA).
4. Discussion
This study aimed to develop, for the first time, nanoformulations containing pure ABA or a TNE at the same ABA concentration. Additionally, the antioxidant and insulinotropic potential of these nanoformulated samples and their unformulated forms were tested and compared in a pancreatic β-cell model. The first set of experiments involved the assessment of cell viability after treatment with testing samples at three different ABA concentrations (0.001, 0.01, and 0.1 µM) at 24, 48, and 72 h. The time points represent short-term to extended chronic exposure, while the concentrations were selected based on prior work [
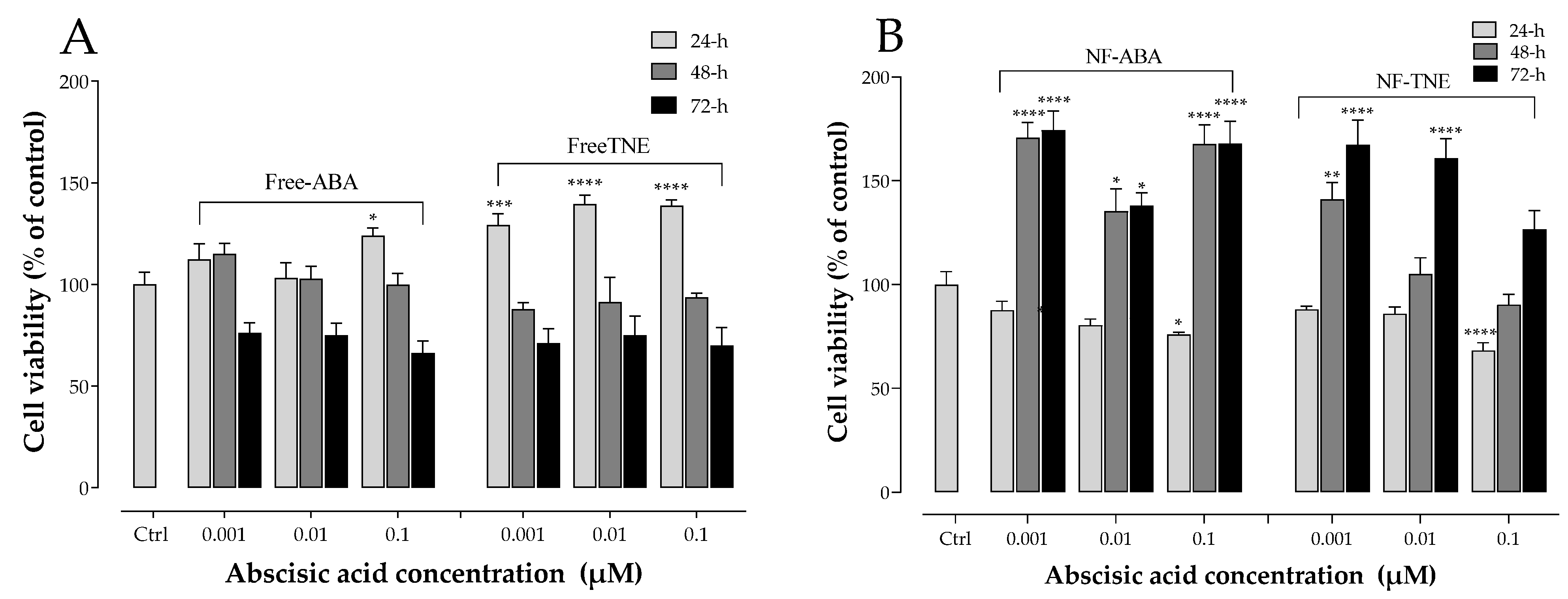
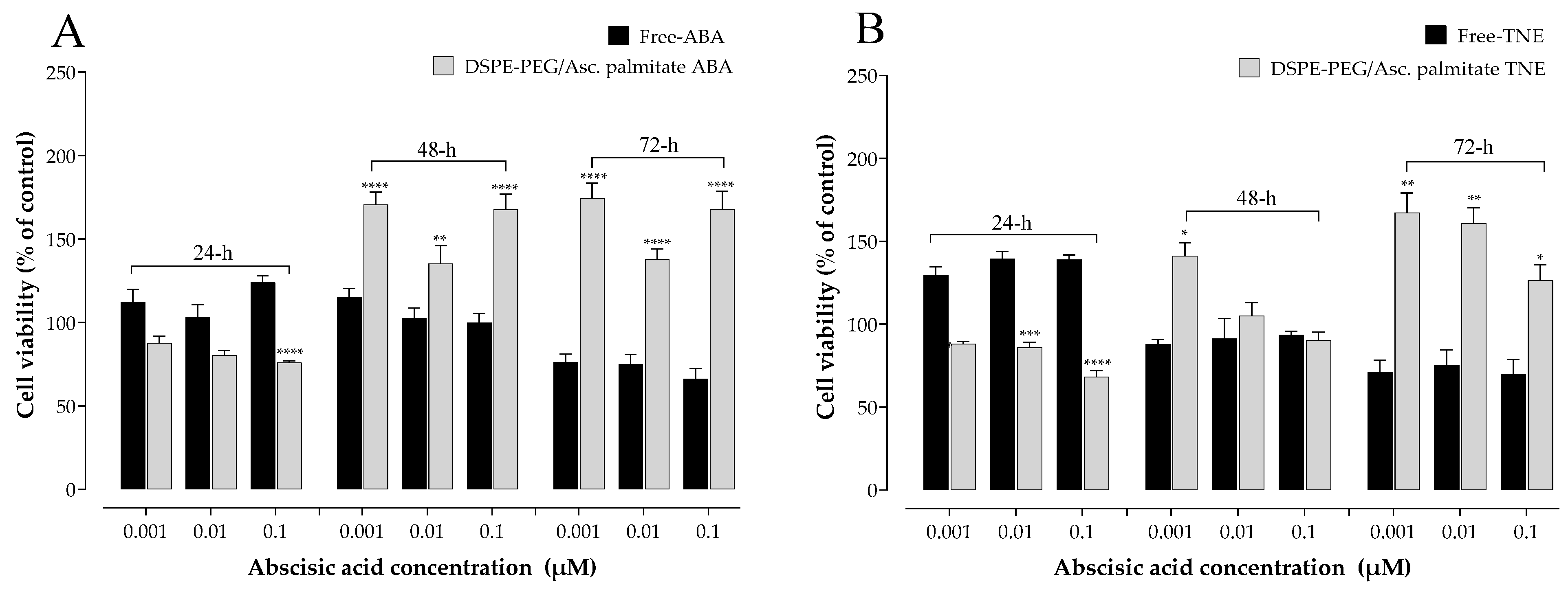
28]. In addition to assessing any potential cytotoxic effects of the samples, another objective of these experiments was to determine the optimum concentration range to be used in further cell exposure experiments involving NFs and free ABA/TNE. An interesting observation was that free TNE increased MIN6 cell viability at all concentrations tested at the initial 24 h time point (
Figure 1). One probable explanation may be that TNE contains a complex blend of bioactive compounds in addition to ABA, and it is possible that at the initial time point, these act in concert at the tested concentration range to evoke an acute proliferative effect on cell viability, which is then gradually reduced over time [
9]. Furthermore, as reported in the same Figure, a clear trend of increase in MIN6 cell survival was observed over the 72 h incubation period in both the NF-ABA and NF-TNE groups at almost all the concentrations tested, compared to the Ctrl group. Similarly, the increased cell viability observed in the NF samples was significant compared to the free samples at the same ABA concentrations (
Figure 2,
p < 0.05). One possible explanation could be that free compounds in their native, unformulated form evoke rapid effects on intracellular organelles, thereby compromising cell viability to some extent. In the case of formulated delivery systems of standards and extracts, the carrier system may facilitate a more gradual and temporal release of its payload into the intracellular environment upon cellular entry [
18]. In this way, the observed effects may be sustained and gradually mitigate any potential adverse effects that occur within the cells following sudden exposure to high levels of bioactive molecules. In addition, the specific composition of the NF may influence cell viability. In this regard, ascorbyl palmitate, a stable form of ascorbic acid containing palmitic acid was used as an excipient for NF preparation. The presence of the fatty acid palmitic acid may also present a nutrient for cells over the 72 h incubation, therefore promoting their eventual proliferation [
6]. In summary, the cytotoxicity results demonstrate the suitability of NFs for further cellular experiments and provide information about the concentrations of ABA that could be utilized in the experiments. Doubtless, the obtained results have also revealed intriguing trends that call for future more comprehensive studies to further investigate this phenomenon.
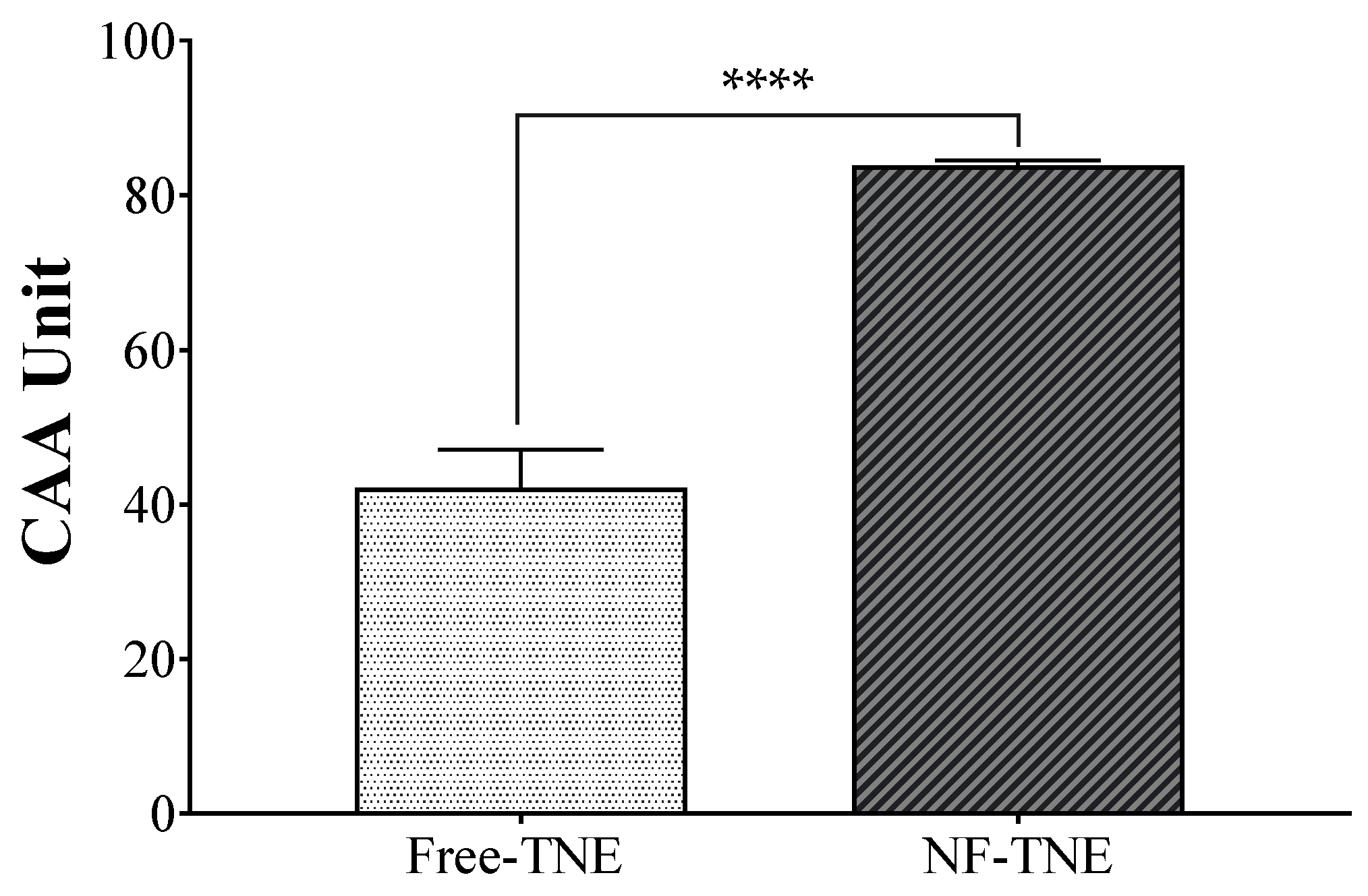
The antioxidant potential of pure ABA and TNE at the same ABA concentration was evaluated and compared in the MIN6 pancreatic line by performing the CAA assay. Since the cellular antioxidant activity of a bioactive compound depends on its cellular uptake, this assay also indicates the relative cellular uptake potential of active molecules from the free extract compared to NFs, as well as the differences between the formulations. Interestingly, neither of the ABA-based groups showed the ability to counteract oxidative stress in our assay, while the results from treatment with TNE demonstrated the ability of this vegetal extract to exert antioxidant properties when cells endure oxidative stress. However, NF-TNE showed significantly higher antioxidant activity compared to free TNE (
p < 0.001,
Figure 3). This result could be explained in part based on the rationale that the unformulated compounds had compromised antioxidant activity before and upon entry into the cellular environment. As explained earlier, many potent natural compounds, most notably curcumin, suffer from issues of stability and degradation that affect their potency [
18]. A formulation strategy such as the one used in this study not only preserves the stability and integrity of bioactive compounds but can also improve cellular delivery. In this regard, the DSPE-PEG-AP delivery system used in this study has previously been shown to significantly increase the cellular delivery of iron as the payload in human Caco-2 intestinal cells [
21]. Thus, the higher antioxidant activity may be attributed in part to the better preservation of antioxidant activity as well as enhanced cellular delivery, highlighting the beneficial potential of this approach. Furthermore, the components of the NF system retain their antioxidant activity, which may also have influenced cellular antioxidant behavior. As indeed reported in the scientific literature, the ascorbyl moiety of ascorbyl palmitate nanocarrier has been demonstrated to exert even much higher antioxidant activity than ascorbic acid itself [
22]. This observation further strengthens the beneficial potential of NF-TNE, as it contains different bioactive compounds that could act synergistically to exert significant cellular antioxidant activity.
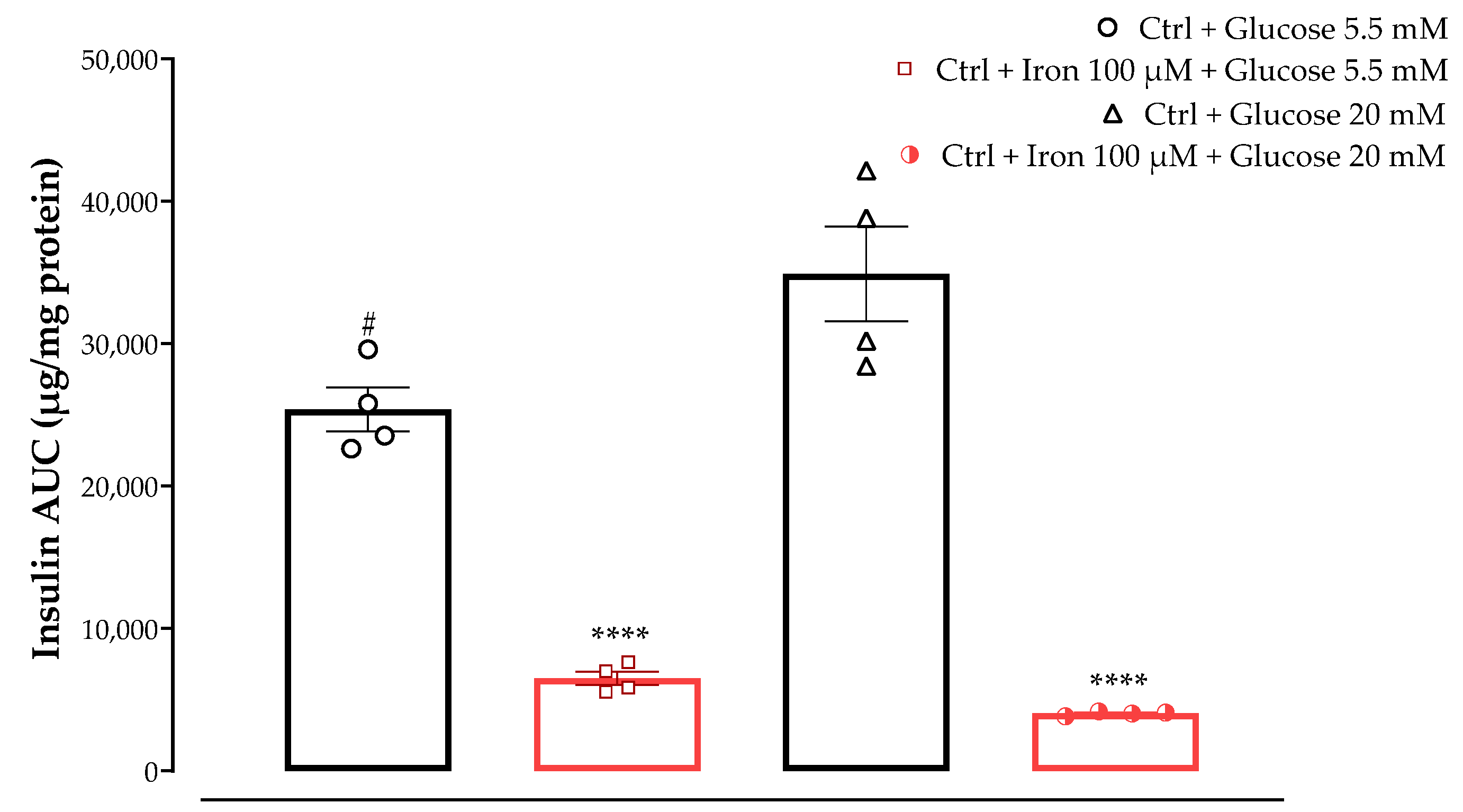
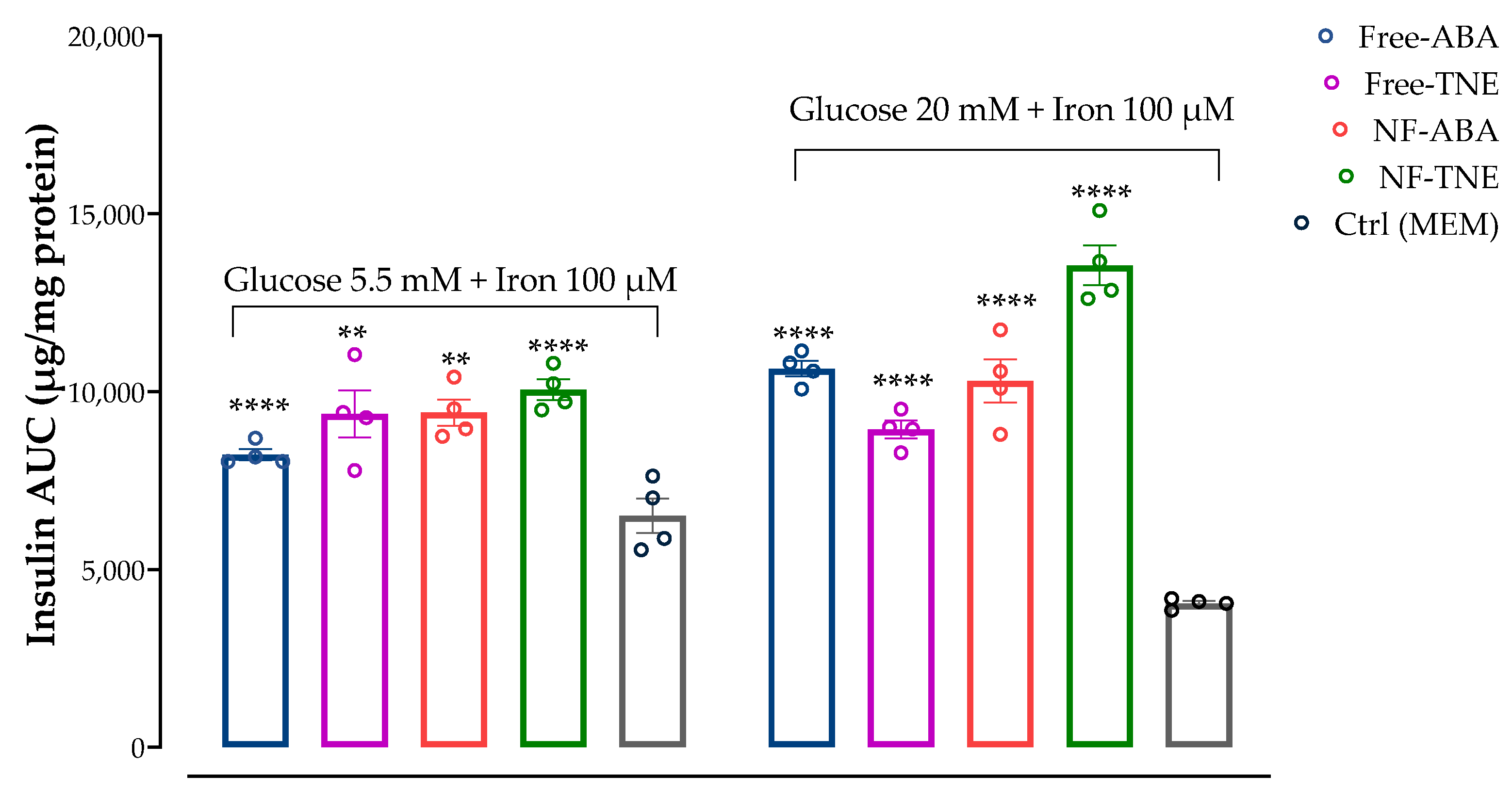
In the GSIS and iron-challenge assays, we aimed to evaluate the insulin response on MIN6 cells incubated with two different glucose concentrations, which were chosen to replicate physiological and supraphysiological glucose levels (5.5 and 20 mM respectively), w/wo preincubation with high iron levels (100 μM). The prooxidant role of excess free iron and its detrimental consequences have been demonstrated in various organ systems and disease states. Elevated iron levels have been shown to correlate with the pathogenesis of type-2 diabetes mellitus (T2DM) [
29], and previous studies on MIN6 cells have demonstrated 100 μM iron to cause cellular and mitochondrial dysfunction [
6]. Pre-treatment with one of the test samples (free ABA, free TNE, NF-ABA, and NF-TNE) or MEM (Ctrl) was then evaluated under the above-described conditions. The initial observation relates to the significant increase in insulin AUC of the Ctrl group incubated with 5.5 mM glucose compared with the same group incubated with higher glucose levels (
p < 0.05,
Figure 6). This is consistent with evidence from the literature on the insulin profile upon MIN6 cells’ exposure to the same glucose levels. In this regard, a progressive response starting at 5 mM glucose has been reported for this cell line, reaching its maximum at 25 mM and remaining at this level up to 50 mM [
30]. Moreover, a significant decrease in insulin secretion was observed for both Ctrl groups incubated with 5.5 and 20 mM glucose in combination with 100 µM iron, compared to the same groups without iron preincubation. The selection of iron dose for our experimental design was based on evidence obtained from a previous work performed on the same cellular line [
6]. The mentioned study compared the effects of two different iron concentrations, corresponding to 20 µM and 100 µM, on MIN6 β-cells. The study revealed that although both concentrations increased cellular iron accumulation, exposure to 100 µM iron led to a more pronounced increase in lipid peroxidation and a greater reduction in insulin secretion and cellular insulin content. By increasing mitochondrial iron transport, the overaccumulation of this element in β-cells may in fact be responsible for the dysfunction of the mitochondria and the impairment of insulin secretory machinery [
31,
32]. Moreover, the study reported a significant reduction in the expression of SNAP-25, a key protein in the core SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) complex responsible for insulin exocytosis, after cell incubation with high iron levels [
6]. Accordingly, as shown in
Figure 6, if we consider Ctrl cells exposed to both high glucose and iron levels, an even lower level of insulin AUC can be observed compared to cells incubated with 5.5 mM glucose and 100 µM iron, probably due to the detrimental combination of glucose and iron-induced toxicity to which these cells were exposed. Therefore, these results further support the validity of using these conditions as a model to investigate β-cell disruption and insulin secretory dysfunction.
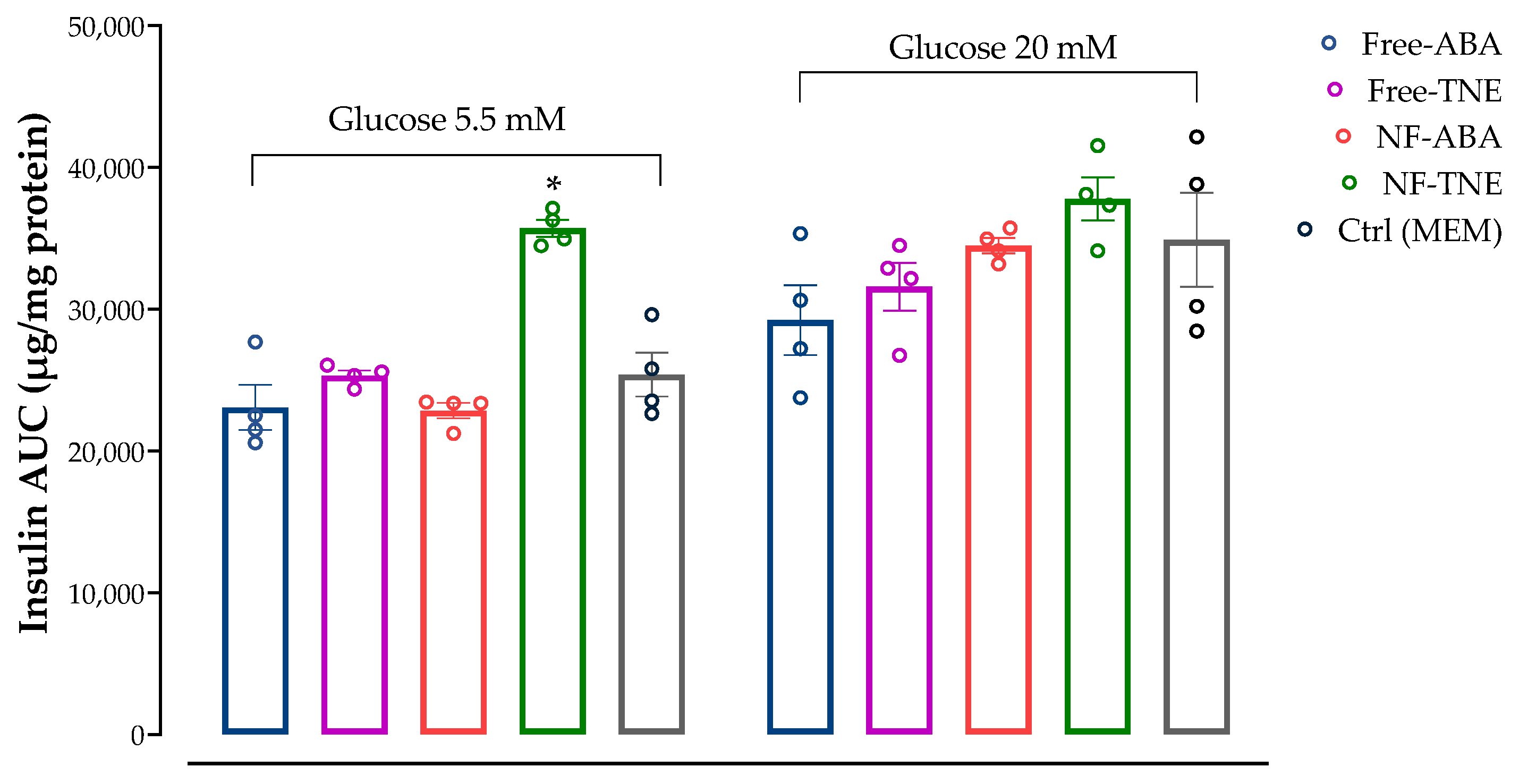
In the present work, we chose to evaluate and compare the effects of different samples on insulin secretion, using the same ABA concentration (0.05 µM) for all test treatments. This concentration was selected based on available evidence about the role of this bioactive molecule in pancreatic β-cell lines. Specifically, Bruzzone et al. reported the ability of ABA to significantly increase insulin secretion from RIN-m and INS-1 cells and murine and human pancreatic islets incubated with both low and high glucose levels (i.e., 5.5 and 16.7 mM glucose). This effect was observed after treatment with ABA at a concentration of 0.01 µM, although no statistical differences were observed with higher concentrations of the pure standard (from 0.01 to 10 µM ABA,
p = 0.6) [
28]. In our cellular model, treatment with ABA-containing samples did not show any significant increase in insulin response after incubation with physiological and supraphysiological glucose concentrations (
Figure 7). The only exception was represented by the treatment with free and NF-TNE samples, which resulted in a slight increase in insulin secretion compared with the Ctrl group. A completely different observation arises from results obtained in MIN6 cells incubated with both glucose and iron (
Figure 8). In this regard, significantly higher insulin AUC levels were observed for all test samples compared with the Ctrl group, with the greatest effect obtained after treatment with NF-TNE (+54% vs. Ctrl group incubated with 5.5 mM glucose + 100 μM iron; +235% vs. Ctrl group incubated with 20 mM glucose + 100 μM iron). As previously reported, the role of excessive iron accumulation in damaging essential biological components through the generation of ROS is well established [
3,
5,
33]. Therefore, ROS clearly have the ability to behave like a destructive agent that eventually leads to insulin resistance and β-cell failure [
34]. Notably, the impairment of insulin secretion due to iron-induced oxidative stress can lead to hyperglycemia and insulin resistance conditions, which are well-known features of T2DM. In this context, recent studies showed that iron excess negatively affected insulin activity [
5,
29,
35]. In these considerations, the presence of bioactive compounds with antioxidant potential, especially polyphenols, in the TNE-based sample has been shown to contribute to the protection of these cells by counteracting the cellular dysfunction induced by ROS in our model. Moreover, in both experiments involving exposure to high iron levels, NF samples were found to be more efficient in terms of insulin release compared to unformulated samples. These data are also supported by the results of our cell viability and antioxidant activity assays, where the NF samples generally demonstrated prominent positive effects. Therefore, this evidence further supports the advantages of using a formulation-based approach for the delivery of bioactive molecules [
18,
19,
21].
To better understand the different results obtained with the incubation with NF-ABA, NF-TNE, ABA, and TNE, it is important to consider both the effects of nanoformulations and the distinct nature of thinned nectarine extract compared to pure ABA. The composition of thinned nectarine extract (TNE) differs fundamentally from that of pure ABA [
9]. In this regard, TNE contains a complex mix of phytochemicals, including polyphenols, which may synergistically contribute to its biological activities [
9,
36]. This complexity can result in a variety of interactions with cellular components and signaling pathways, in contrast to the singular interaction of pure ABA. Moreover, nanoformulations significantly modify the properties of bioactive compounds, potentially enhancing their stability, solubility, and bioavailability [
37]. More specifically, nanoformulated compounds might demonstrate improved cellular delivery and efficacy of various bioactive substances, leading to more pronounced biological effects [
20,
38]. Nanoformulations might also change how active ingredients interact with cellular receptors or signal transduction pathways [
39]. For example, alterations in the molecular size or surface properties of nanoformulations could affect recognition by cellular receptors, thereby influencing signal transduction [
40]. Therefore, the differences between nanoformulated and non-nanoformulated ABA and TNE can be attributed to both the changes in the physicochemical properties of the nanoformulated samples and the complex nature of TNE compared to pure ABA. However, more specific mechanistic insights into signal transduction and receptor interactions would require further experimental studies.
The originality of the present project lies in the first-time evaluation and comparison of the TNE efficacy on pancreatic β-function at the same ABA concentrations of a pure standard and the first-time incorporation of these molecules into formulations using novel delivery systems. Specifically, the evaluation at the same concentration of the active ABA allowed us to distinguish the additional activity of TNE in relation to its content of bioactive compounds, especially polyphenols, which can contribute positively and synergistically to cellular antioxidant protection.

 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}