
Scavenging of Alkylperoxyl Radicals by Addition to Ascorbate: An Alternative Mechanism to Electron Transfer



Abstract
:1. Introduction
2. Materials and Methods
3. Results
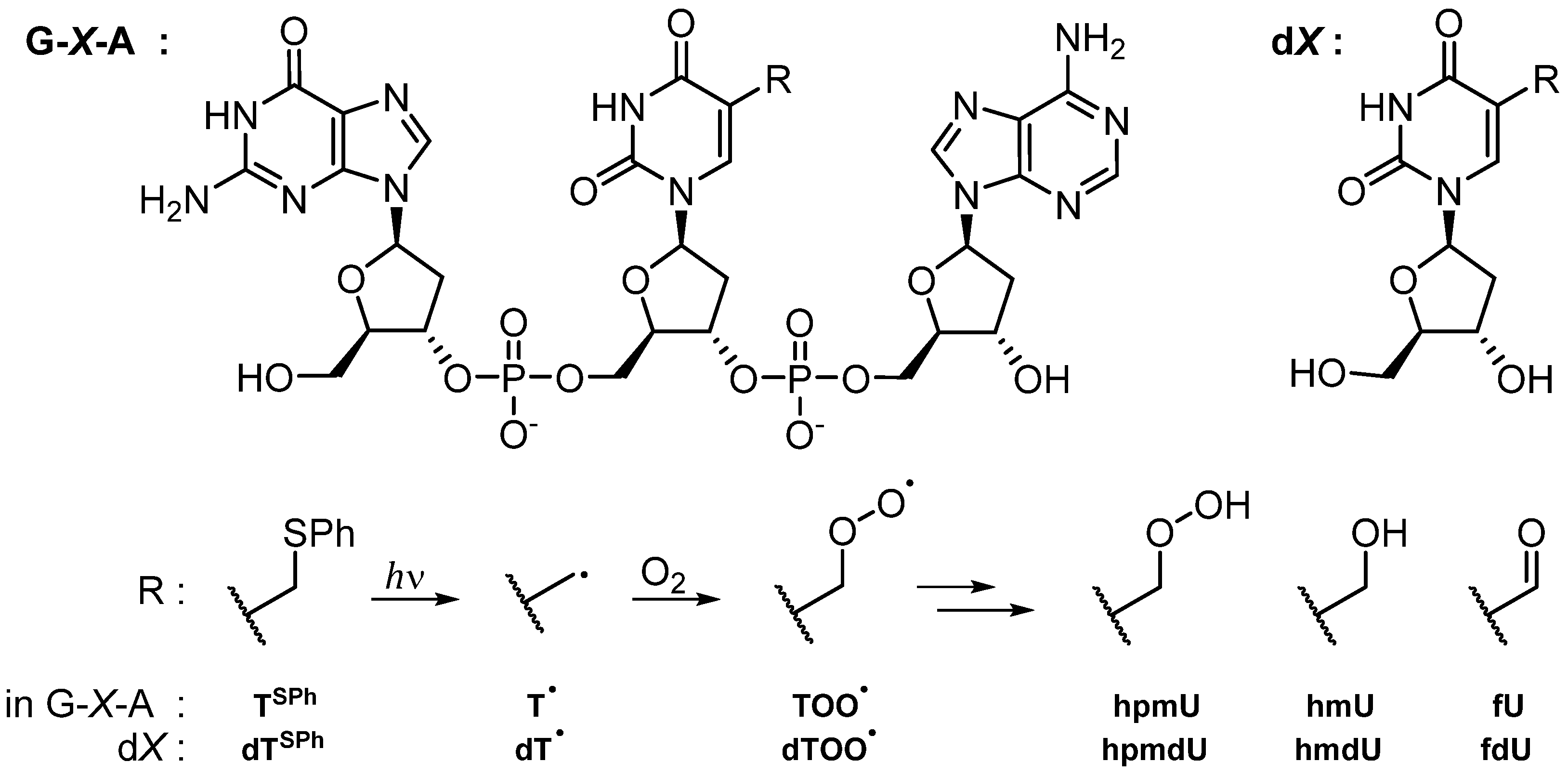
3.1. Fate of 5-(Uracilyl)Methyl Peroxyl Radicals in the Presence of Ascorbate
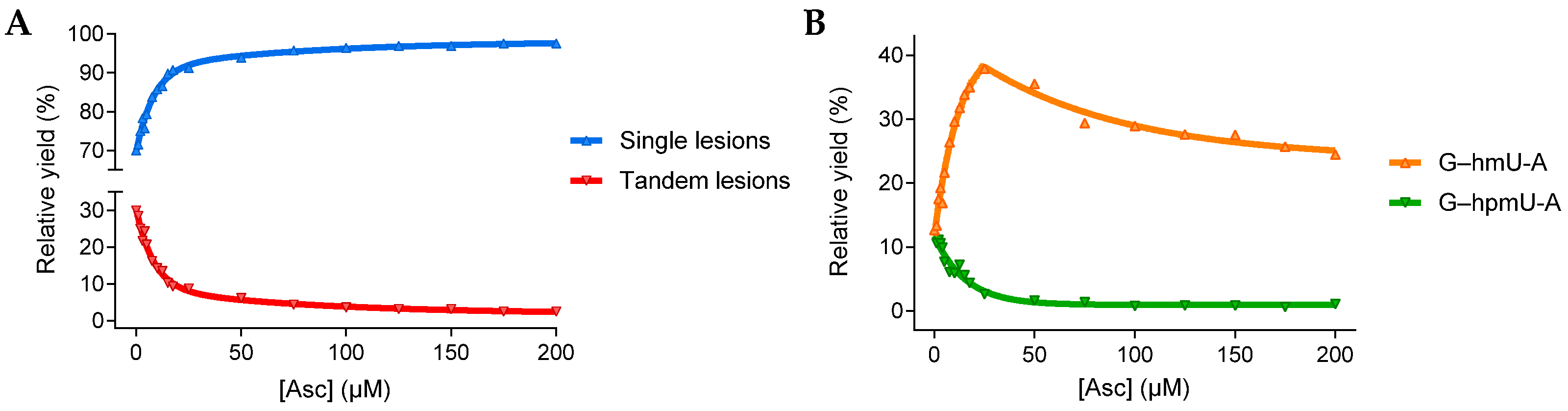
3.1.1. Single and Tandem Lesions Formation in a Trinucleotide Model
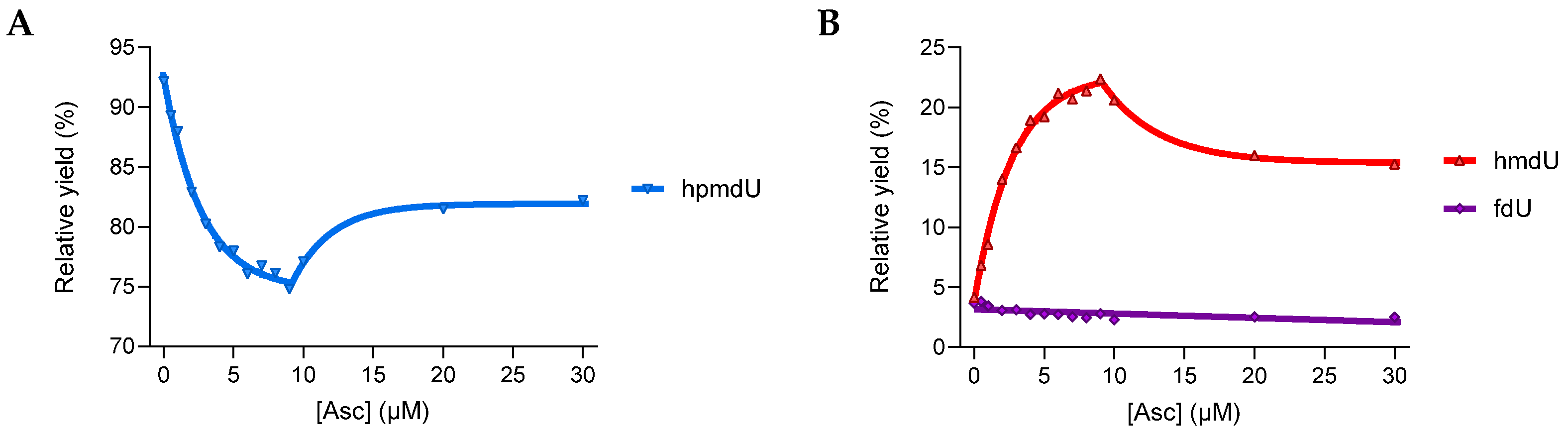
3.1.2. Reactivity in a 2′-Deoxyribonucleoside Model
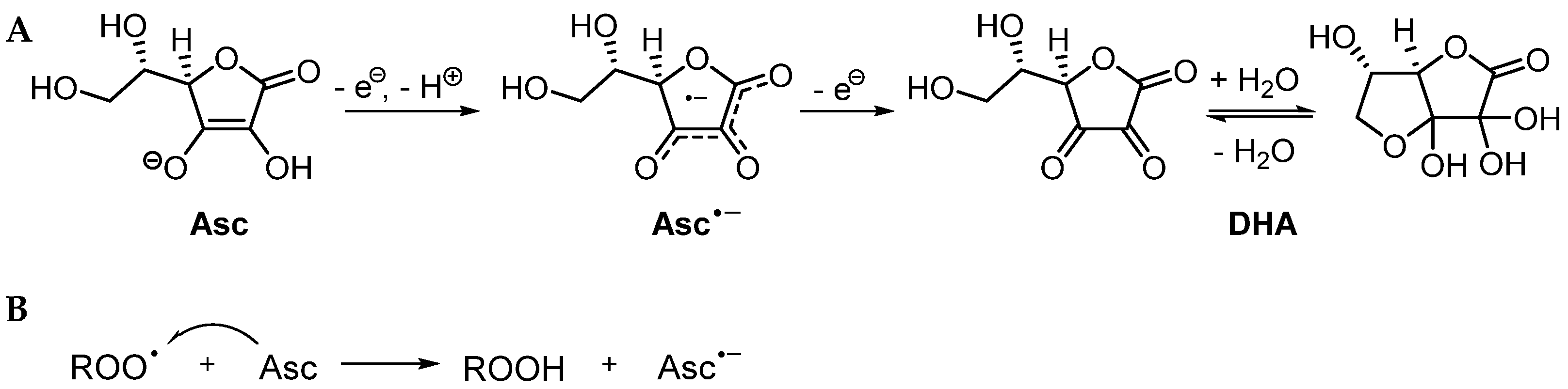
3.2. Mechanistic Studies
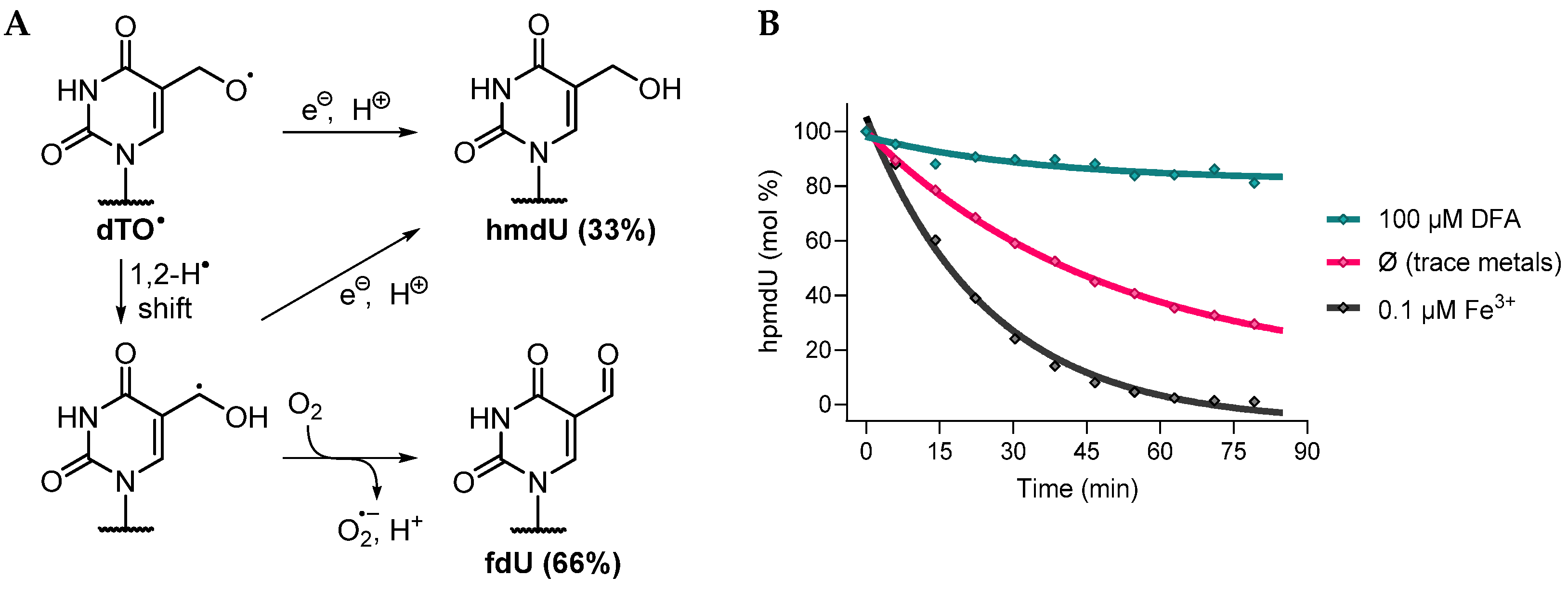
3.2.1. Possible Mechanism #1: Electron Transfers Involving Intermediate Alkoxyl Radical
- Pathway 1



3.2.2. Possible Mechanism #2: Peroxyl Radical Addition
- Pathway 2



3.2.3. Possible Mechanism #3: Peroxyl Radical Addition Followed by Epoxide Formation
- Pathway 3


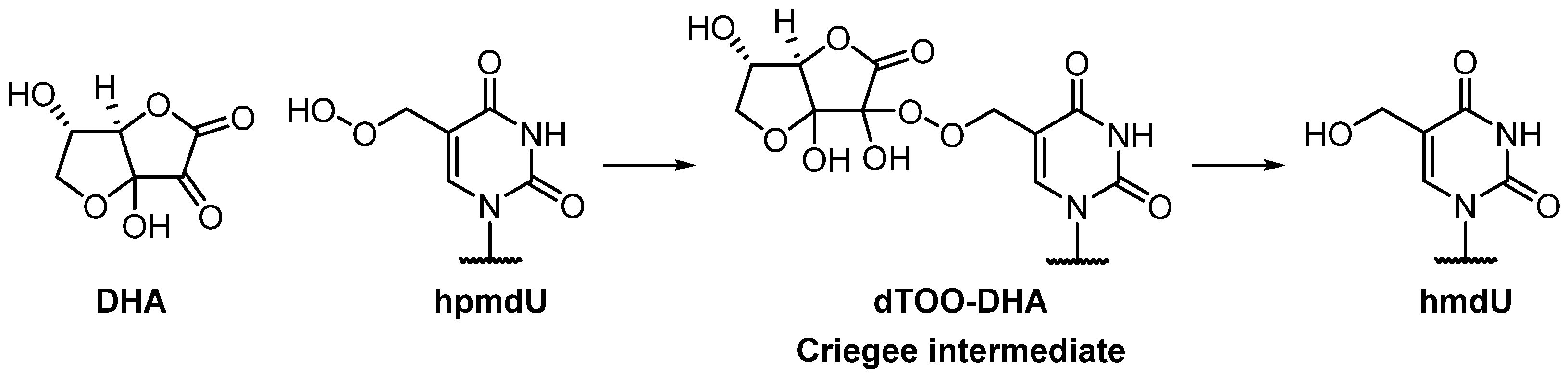
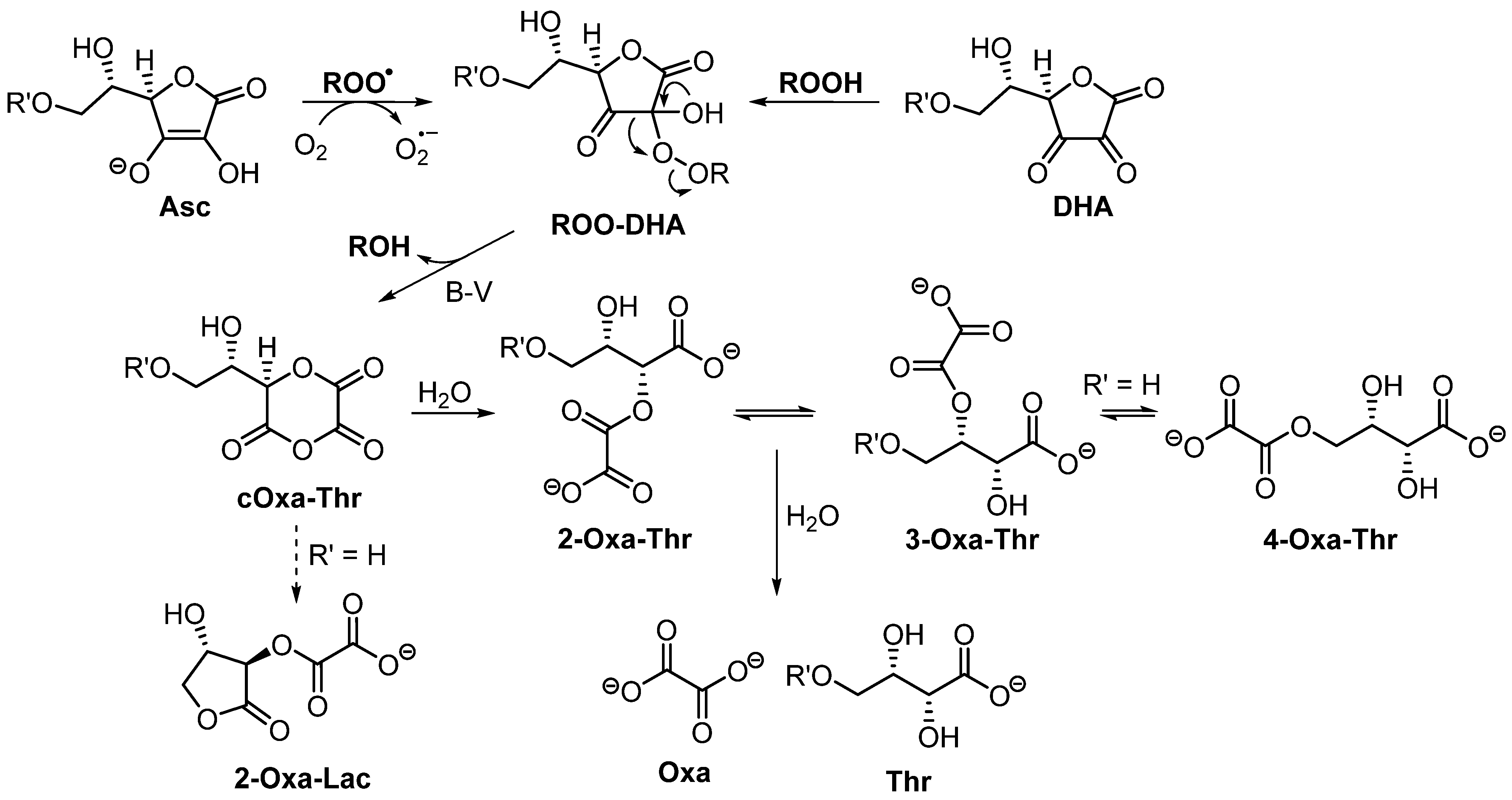
3.3. Product Characterization
3.4. Oxidation of Asc-PA by Peroxyl Radicals Derived from Azo Compounds
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Neta, P.; Huie, R.E.; Ross, A.B. Rate Constants for Reactions of Peroxyl Radicals in Fluid Solutions. J. Phys. Chem. Ref. Data 1990, 19, 413–513. [Google Scholar] [CrossRef]
- Valgimigli, L. Lipid Peroxidation and Antioxidant Protection. Biomolecules 2023, 13, 1291. [Google Scholar] [CrossRef] [PubMed]
- Greene, L.E.; Lincoln, R.; Cosa, G. Rate of Lipid Peroxyl Radical Production during Cellular Homeostasis Unraveled via Fluorescence Imaging. J. Am. Chem. Soc. 2017, 139, 15801–15811. [Google Scholar] [CrossRef] [PubMed]
- Isildar, M.; Schuchmann, M.N.; Schulte-Frohlinde, D.; von Sonntag, C. Oxygen Uptake in the Radiolysis of Aqueous Solutions of Nucleic Acids and Their Constituents. Int. J. Radiat. Biol. Relat. Stud. Physics, Chem. Med. 1982, 41, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, F.; Auvré, F.; Radicella, J.P.; Ravanat, J.L. HO• Radicals Induce an Unexpected High Proportion of Tandem Base Lesions Refractory to Repair by DNA Glycosylases. Proc. Natl. Acad. Sci. USA 2010, 107, 5528–5533. [Google Scholar] [CrossRef]
- Davies, M.J. Protein Oxidation and Peroxidation. Biochem. J. 2016, 473, 805–825. [Google Scholar] [CrossRef]
- Gebicki, J.M.; Nauser, T. Initiation and Prevention of Biological Damage by Radiation-Generated Protein Radicals. Int. J. Mol. Sci. 2021, 23, 396. [Google Scholar] [CrossRef]
- Robert, G.; Wagner, J.R.; Cadet, J. Oxidatively Generated Tandem DNA Modifications by Pyrimidinyl and 2-Deoxyribosyl Peroxyl Radicals. Free Radic. Biol. Med. 2023, 196, 22–36. [Google Scholar] [CrossRef]
- Robert, G.; Wagner, J.R. Tandem Lesions Arising from 5-(Uracilyl)Methyl Peroxyl Radical Addition to Guanine: Product Analysis and Mechanistic Studies. Chem. Res. Toxicol. 2020, 33, 565–575. [Google Scholar] [CrossRef]
- Frelon, S.; Douki, T.; Ravanat, J.L.; Pouget, J.P.; Tornabene, C.; Cadet, J. High-Performance Liquid Chromatography - Tandem Mass Spectrometry Measurement of Radiation-Induced Base Damage to Isolated and Cellular DNA. Chem. Res. Toxicol. 2000, 13, 1002–1010. [Google Scholar] [CrossRef]
- Samson-Thibault, F.; Madugundu, G.S.; Gao, S.; Cadet, J.; Wagner, J.R. Profiling Cytosine Oxidation in DNA by LC-MS/MS. Chem. Res. Toxicol. 2012, 25, 1902–1911. [Google Scholar] [CrossRef] [PubMed]
- Madugundu, G.S.; Cadet, J.; Wagner, J.R. Hydroxyl-Radical-Induced Oxidation of 5-Methylcytosine in Isolated and Cellular DNA. Nucleic Acids Res. 2014, 42, 7450–7460. [Google Scholar] [CrossRef]
- Chen, J.; Guo, L.; Zhang, L.; Wu, H.; Yang, J.; Liu, H.; Wang, X.; Hu, X.; Gu, T.; Zhou, Z.; et al. Vitamin C Modulates TET1 Function during Somatic Cell Reprogramming. Nat. Genet. 2013, 45, 1504–1509. [Google Scholar] [CrossRef]
- Young, J.I.; Züchner, S.; Wang, G. Regulation of the Epigenome by Vitamin C. Annu. Rev. Nutr. 2015, 35, 545–564. [Google Scholar] [CrossRef] [PubMed]
- Fujii, J.; Osaki, T.; Bo, T. Ascorbate Is a Primary Antioxidant in Mammals. Molecules 2022, 27, 6187. [Google Scholar] [CrossRef]
- Laroff, G.P.; Fessenden, R.W.; Schuler, R.H. The Electron Spin Resonance Spectra of Radical Intermediates in the Oxidation of Ascorbic Acid and Related Substances. J. Am. Chem. Soc. 1972, 94, 9062–9073. [Google Scholar] [CrossRef]
- Davis, H.F.; McManus, H.J.; Fessenden, R.W. An ESR Study of Free-Radical Protonation Equilibria in Strongly Acid Media. J. Phys. Chem. 1986, 90, 6400–6404. [Google Scholar] [CrossRef]
- Lagercrantz, C. Free Radicals in the Auto-Oxidation of Ascorbic Acid. Acta Chem. Scand. 1964, 18, 562. [Google Scholar] [CrossRef]
- Iyanagi, T.; Yamazaki, I.; Anan, K.F. One-Electron Oxidation-Reduction Properties of Ascorbic Acid. Biochim. Biophys. Acta (BBA)—Bioenerg. 1985, 806, 255–261. [Google Scholar] [CrossRef]
- Pelizzetti, E.; Mentasti, E.; Pramauro, E. Outer-Sphere Oxidation of Ascorbic Acid. Inorg. Chem. 1978, 17, 1181–1186. [Google Scholar] [CrossRef]
- Williams, N.H.; Yandell, J.K. Outer-Sphere Electron-Transfer Reactions of Ascorbate Anions. Aust. J. Chem. 1982, 35, 1133–1144. [Google Scholar] [CrossRef]
- Bielski, B.H.J. Chemistry of Ascorbic Acid Radicals. In Ascorbic Acid: Chemistry, Metabolism, and Uses; Seib, P.A., Tolbert, B.M., Eds.; American Chemical Society: Washington, DC, USA, 1982; pp. 81–100. [Google Scholar]
- Buettner, G.R.; Jurkiewicz, B.A. Catalytic Metals, Ascorbate and Free Radicals: Combinations to Avoid. Radiat. Res. 1996, 145, 532–541. [Google Scholar] [CrossRef]
- Njus, D.; Kelley, P.M.; Tu, Y.J.; Schlegel, H.B. Ascorbic Acid: The Chemistry Underlying Its Antioxidant Properties. Free Radic. Biol. Med. 2020, 159, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Buettner, G.R. The Pecking Order of Free Radicals and Antioxidants: Lipid Peroxidation, α-Tocopherol, and Ascorbate. Arch. Biochem. Biophys. 1993, 300, 535–543. [Google Scholar] [CrossRef]
- Nadezhdin, A.D.; Dunford, H.B. The Oxidation of Ascorbic Acid and Hydroquinone by Perhydroxyl Radicals. A Flash Photolysis Study. Can. J. Chem. 1979, 57, 3017–3022. [Google Scholar] [CrossRef]
- Packer, J.E.; Willson, R.L.; Bahnemann, D.; Asmus, K.-D. Electron Transfer Reactions of Halogenated Aliphatic Peroxyl Radicals: Measurement of Absolute Rate Constants by Pulse Radiolysis. J. Chem. Soc. Perkin Trans. 2 1980, 296. [Google Scholar] [CrossRef]
- Valgimigli, L.; Pratt, D.A. Antioxidants in Chemistry and Biology. In Encyclopedia of Radicals in Chemistry, Biology and Materials; John Wiley & Sons: Hoboken, NJ, USA, 2012; ISBN 9780470971253. [Google Scholar]
- Haidasz, E.A.; Van Kessel, A.T.M.; Pratt, D.A. A Continuous Visible Light Spectrophotometric Approach to Accurately Determine the Reactivity of Radical-Trapping Antioxidants. J. Org. Chem. 2016, 81, 737–744. [Google Scholar] [CrossRef]
- Domazou, A.S.; Zelenay, V.; Koppenol, W.H.; Gebicki, J.M. Efficient Depletion of Ascorbate by Amino Acid and Protein Radicals under Oxidative Stress. Free Radic. Biol. Med. 2012, 53, 1565–1573. [Google Scholar] [CrossRef]
- Domazou, A.S.; Gebicki, J.M.; Nauser, T.; Koppenol, W.H. Repair of Protein Radicals by Antioxidants. Isr. J. Chem. 2014, 54, 254–264. [Google Scholar] [CrossRef]
- Nauser, T.; Gebicki, J.M. Physiological Concentrations of Ascorbate Cannot Prevent the Potentially Damaging Reactions of Protein Radicals in Humans. Chem. Res. Toxicol. 2017, 30, 1702–1710. [Google Scholar] [CrossRef]
- Packer, J.E.; Slater, T.F.; Wilson, R.L. Direct Observation of a Free Radical Interaction between Vitamin E and Vitamin C. Nature 1979, 278, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Buettner, G.R. In the Absence of Catalytic Metals Ascorbate Does Not Autoxidize at PH 7: Ascorbate as a Test for Catalytic Metals. J. Biochem. Biophys. Methods 1988, 16, 27–40. [Google Scholar] [CrossRef]
- Boatright, W.L. Oxygen Dependency of One-Electron Reactions Generating Ascorbate Radicals and Hydrogen Peroxide from Ascorbic Acid. Food Chem. 2016, 196, 1361–1367. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Griffiths, P.T.; Campbell, S.J.; Utinger, B.; Kalberer, M.; Paulson, S.E. Ascorbate Oxidation by Iron, Copper and Reactive Oxygen Species: Review, Model Development, and Derivation of Key Rate Constants. Sci. Rep. 2021, 11, 7417. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Oe, T.; Blair, I.A. Vitamin C-Induced Decomposition of Lipid Hydroperoxides to Endogenous Genotoxins. Science 2001, 292, 2083–2086. [Google Scholar] [CrossRef]
- Chen, Q.; Espey, M.G.; Krishna, M.C.; Mitchell, J.B.; Corpe, C.P.; Buettner, G.R.; Shacter, E.; Levine, M. Pharmacologic Ascorbic Acid Concentrations Selectively Kill Cancer Cells: Action as a pro-Drug to Deliver Hydrogen Peroxide to Tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 13604–13609. [Google Scholar] [CrossRef]
- Verrax, J.; Calderon, P.B. Pharmacologic Concentrations of Ascorbate Are Achieved by Parenteral Administration and Exhibit Antitumoral Effects. Free Radic. Biol. Med. 2009, 47, 32–40. [Google Scholar] [CrossRef]
- Halliwell, B. Artefacts with Ascorbate and Other Redox-Active Compounds in Cell Culture: Epigenetic Modifications, and Cell Killing Due to Hydrogen Peroxide Generation in Cell Culture Media. Free Radic. Res. 2018, 52, 907–909. [Google Scholar] [CrossRef]
- Vissers, M.C.M.; Das, A.B. Potential Mechanisms of Action for Vitamin C in Cancer: Reviewing the Evidence. Front. Physiol. 2018, 9, 809. [Google Scholar] [CrossRef]
- Clément, M.-V.; Ramalingam, J.; Long, L.H.; Halliwell, B. The In Vitro Cytotoxicity of Ascorbate Depends on the Culture Medium Used to Perform the Assay and Involves Hydrogen Peroxide. Antioxid. Redox Signal. 2001, 3, 157–163. [Google Scholar] [CrossRef]
- Halliwell, B.; Clement, M.V.; Ramalingam, J.; Long, L.H. Hydrogen Peroxide. Ubiquitous in Cell Culture and In Vivo? IUBMB Life 2000, 50, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.; Frei, B. Does Vitamin C Act as a Pro-oxidant under Physiological Conditions? FASEB J. 1999, 13, 1007–1024. [Google Scholar] [CrossRef] [PubMed]
- Møller, P.; Loft, S. Interventions with Antioxidants and Nutrients in Relation to Oxidative DNA Damage and Repair. Mutat. Res. Mol. Mech. Mutagen. 2004, 551, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Lee, J.-H.; Krishna, M.C.; Shacter, E.; Choyke, P.L.; Pooput, C.; Kirk, K.L.; Buettner, G.R.; et al. Ascorbate in Pharmacologic Concentrations Selectively Generates Ascorbate Radical and Hydrogen Peroxide in Extracellular Fluid in Vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 8749–8754. [Google Scholar] [CrossRef]
- Böttger, F.; Vallés-Martí, A.; Cahn, L.; Jimenez, C.R. High-Dose Intravenous Vitamin C, a Promising Multi-Targeting Agent in the Treatment of Cancer. J. Exp. Clin. Cancer Res. 2021, 40, 343. [Google Scholar] [CrossRef]
- Wagner, J.R.; Van Lier, J.E.; Decarroz, C.; Berger, M.; Cadet, J. Photodynamic Methods for Oxy Radical-Induced DNA Damage. In Methods in Enzymology. Oxygen Radicals in Biological Systems; Packer, L., Glazer, A.N., Eds.; Academic Press Inc.: London, UK, 1990; Volume 186, pp. 502–511. [Google Scholar]
- Wagner, J.R.; van Lier, J.E.; Berger, M.; Cadet, J. Thymidine Hydroperoxides: Structural Assignment, Conformational Features, and Thermal Decomposition in Water. J. Am. Chem. Soc. 1994, 116, 2235–2242. [Google Scholar] [CrossRef]
- Cousins, R.C.; Seib, P.A.; Hoseney, R.C.; Deyoe, C.W.; Liang, Y.T.; Lillard, D.W. Synthesis of 6-fatty Acid Esters of L-ascorbic Acid. J. Am. Oil Chem. Soc. 1977, 54, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Washko, P.W.; Wang, Y.; Levine, M. Ascorbic Acid Recycling in Human Neutrophils. J. Biol. Chem. 1993, 268, 15531–15535. [Google Scholar] [CrossRef]
- Renz, M.; Meunier, B. 100 Years of Baeyer–Villiger Oxidations. Eur. J. Org. Chem. 1999, 1999, 737–750. [Google Scholar] [CrossRef]
- Jackson, K.G.A.; Jones, J.K.N. Synthesis of 3-Hexuloses. Can. J. Chem. 1969, 47, 2498–2501. [Google Scholar] [CrossRef]
- Gilbert, B.C.; Holmes, R.G.G.; Laue, H.A.H.; Norman, R.O.C. Electron Spin Resonance Studies. Part L. Reactions of Alkoxyl Radicals Generated from Alkyl Hydroperoxides and Titanium(III) Ion in Aqueous Solution. J. Chem. Soc. Perkin Trans. 2 1976, 9, 1047. [Google Scholar] [CrossRef]
- Elford, P.E.; Roberts, B.P. EPR Studies of the Formation and Transformation of Isomeric Radicals [C3H5O]˙. Rearrangement of the Allyloxyl Radical in Non-Aqueous Solution Involving a Formal 1,2-Hydrogen-Atom Shift Promoted by Alcohols. J. Chem. Soc. Perkin Trans. 2 1996, 5, 2247–2256. [Google Scholar] [CrossRef]
- Ingold, K.U.; Paul, T.; Young, M.J.; Doiron, L. Invention of the First Azo Compound To Serve as a Superoxide Thermal Source under Physiological Conditions: Concept, Synthesis, and Chemical Properties. J. Am. Chem. Soc. 1997, 119, 12364–12365. [Google Scholar] [CrossRef]
- Konya, K.G.; Paul, T.; Lin, S.; Lusztyk, J.; Ingold, K.U. Laser Flash Photolysis Studies on the First Superoxide Thermal Source. First Direct Measurements of the Rates of Solvent-Assisted 1,2-Hydrogen Atom Shifts and a Proposed New Mechanism for This Unusual Rearrangement. J. Am. Chem. Soc. 2000, 122, 7518–7527. [Google Scholar] [CrossRef]
- Görner, H. Oxygen Uptake and Involvement of Superoxide Radicals upon Photolysis of Ketones in Air-saturated Aqueous Alcohol, Formate, Amine or Ascorbic Acid Solutions. Photochem. Photobiol. 2006, 82, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Ramos, A.; Zgierski, M.Z. Theoretical Study of the Rate Constants and Kinetic Isotope Effects of the 1,2-Hydrogen-Atom Shift of Methoxyl and Benzyloxyl Radicals Assisted by Water. J. Phys. Chem. A 2002, 106, 10578–10583. [Google Scholar] [CrossRef]
- Litwinienko, G.; Beckwith, A.L.J.; Ingold, K.U. The Frequently Overlooked Importance of Solvent in Free Radical Syntheses. Chem. Soc. Rev. 2011, 40, 2157–2163. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, D.; Liu, S.; Ge, Y.; Lan, Y.; Chen, Y. Visible-Light-Induced Alkoxyl Radicals Enable α-C(Sp3)-H Bond Allylation. iScience 2020, 23, 100755. [Google Scholar] [CrossRef]
- Saraev, D.D.; Wu, Z.; Kim, H.-Y.H.; Porter, N.A.; Pratt, D.A. Intramolecular H-Atom Transfers in Alkoxyl Radical Intermediates Underlie the Apparent Oxidation of Lipid Hydroperoxides by Fe(II). ACS Chem. Biol. 2023, 18, 2073–2081. [Google Scholar] [CrossRef]
- Do, Q.; Lee, D.D.; Dinh, A.N.; Seguin, R.P.; Zhang, R.; Xu, L. Development and Application of a Peroxyl Radical Clock Approach for Measuring Both Hydrogen-Atom Transfer and Peroxyl Radical Addition Rate Constants. J. Org. Chem. 2021, 86, 153–168. [Google Scholar] [CrossRef]
- Zielinski, Z.A.M.; Pratt, D.A. H-Atom Abstraction vs Addition: Accounting for the Diverse Product Distribution in the Autoxidation of Cholesterol and Its Esters. J. Am. Chem. Soc. 2019, 141, 3037–3051. [Google Scholar] [CrossRef] [PubMed]
- Rabani, J.; Klug-Roth, D.; Henglein, A. Pulse Radiolytic Investigations of OHCH₂O₂ Radicals. J. Phys. Chem. 1974, 78, 2089–2093. [Google Scholar] [CrossRef]
- Ilan, Y.; Rabani, J.; Henglein, A. Pulse Radiolytic Investigations of Peroxy Radicals Produced from 2-Propanol and Methanol. J. Phys. Chem. 1976, 80, 1558–1562. [Google Scholar] [CrossRef]
- Bothe, E.; Behrens, G.; Schulte-Frohlinde, D. Mechanism of the First Order Decay of 2-Hydroxy-Propyl-2-Peroxyl Radicals and of O₂˙¯ Formation in Aqueous Solution. Zeitschrift fur Naturforsch.—Sect. B J. Chem. Sci. 1977, 32, 886–889. [Google Scholar] [CrossRef]
- Bothe, E.; Schulte-Frohlinde, D.; von Sonntag, C. Radiation Chemistry of Carbohydrates. Part 16. Kinetics of HO₂˙ Elimination from Peroxyl Radicals Derived from Glucose and Polyhydric Alcohols. J. Chem. Soc. Perkin Trans. 2 1978, 5, 416. [Google Scholar] [CrossRef]
- Bothe, E.; Schuchmann, M.N.; Schulte-Frohlinde, D.; Sonntag, C. von HO₂˙ Elimination from α-Hydroxyalkylperoxyl Radicals in Aqueous Solution. Photochem. Photobiol. 1978, 28, 639–643. [Google Scholar] [CrossRef]
- Schuchmann, M.N.; Schuchmann, H.; von Sonntag, C. Oxidation of Hydroxymalonic Acid by OH Radicals in the Presence and in the Absence of Molecular Oxygen. A Pulse-Radiolysis and Product Study. J. Phys. Chem. 1995, 99, 9122–9129. [Google Scholar] [CrossRef]
- Bothe, E.; Schuchmann, M.N.; Schulte-Frohlinde, D.; Sonntag, C. von Hydroxyl Radical-Induced Oxidation of Ethanol in Oxygenated Aqueous Solutions. A Pulse Radiolysis and Product Study. Zeitschrift für Naturforsch. B 1983, 38, 212–219. [Google Scholar] [CrossRef]
- Yaremenko, I.A.; Vil’, V.A.; Demchuk, D.V.; Terent’ev, A.O. Rearrangements of Organic Peroxides and Related Processes. Beilstein J. Org. Chem. 2016, 12, 1647–1748. [Google Scholar] [CrossRef]
- Cioffi, N.; Losito, I.; Terzano, R.; Zambonin, C.G. An Electrospray Ionization Ion Trap Mass Spectrometric (ESI-MS-MSn) Study of Dehydroascorbic Acid Hydrolysis at Neutral PH. Analyst 2000, 125, 2244–2248. [Google Scholar] [CrossRef]
- Bora, P.; Bora, B.; Bora, U. Recent Developments in Synthesis of Catechols by Dakin Oxidation. New J. Chem. 2021, 45, 17077–17084. [Google Scholar] [CrossRef]
- ten Brink, G.-J.; Arends, I.W.C.E.; Sheldon, R.A. The Baeyer−Villiger Reaction: New Developments toward Greener Procedures. Chem. Rev. 2004, 104, 4105–4124. [Google Scholar] [CrossRef] [PubMed]
- Mayo, F.R. The Oxidation of Unsaturated Compounds. VIII. The Oxidation of Aliphatic Unsaturated Compounds. J. Am. Chem. Soc. 1958, 80, 2497–2500. [Google Scholar] [CrossRef]
- Mayo, F.R.; Miller, A.A.; Russell, G.A. The Oxidation of Unsaturated Compounds. IX. The Effects of Structure on the Rates and Products of Oxidation of Unsaturated Compounds. J. Am. Chem. Soc. 1958, 80, 2500–2507. [Google Scholar] [CrossRef]
- Bloodworth, A.J.; Courtneidge, J.L.; Davies, A.G. Rate Constants for the Formation of Oxiranes by γ-Scission in Secondary β-t-Butylperoxyalkyl Radicals. J. Chem. Soc. Perkin Trans. 2 1984, 3, 523–527. [Google Scholar] [CrossRef]
- Yin, H.; Xu, L.; Porter, N.A. Free Radical Lipid Peroxidation: Mechanisms and Analysis. Chem. Rev. 2011, 111, 5944–5972. [Google Scholar] [CrossRef]
- Kennedy, T.A.; Liebler, D.C. Peroxyl Radical Oxidation of β-Carotene: Formation of β-Carotene Epoxides. Chem. Res. Toxicol. 1991, 4, 290–295. [Google Scholar] [CrossRef]
- Wei, C.C.; De Bernardo, S.; Tengi, J.P.; Borgese, J.; Weigele, M. Synthesis of Chiral β-Lactams Using L-Ascorbic Acid. J. Org. Chem. 1985, 50, 3462–3467. [Google Scholar] [CrossRef]
- Kirino, Y.; Kwan, T. Free Radicals Formed during the Oxidation of L-Ascorbic Acid or Hydroxytetronic Acid with Hydrogen Peroxide and Titanium (III) Ions. Chem. Pharm. Bull. 1971, 19, 718–721. [Google Scholar] [CrossRef]
- Kirino, Y.; Schuler, R.H. Electron Spin Resonance Spectra of Radicals Related to the Intermediates in the Oxidation of Ascorbic Acid. Radical Produced from γ-Methyl-α-Hydroxytetronic Acid. J. Am. Chem. Soc. 1973, 95, 6926–6928. [Google Scholar] [CrossRef]
- Schuler, M.A.; Bhatia, K.; Schuler, R.H. Radiation Chemical Studies on Systems Related to Ascorbic Acid. The Radiolysis of Aqueous Solutions of α-Bromotetronic Acid. J. Phys. Chem. 1974, 78, 1063–1074. [Google Scholar] [CrossRef]
- Fessenden, R.W.W.; Verma, N.C.C. A Time-Resolved Electron Spin Resonance Study of the Oxidation of Ascorbic Acid by Hydroxyl Radical. Biophys. J. 1978, 24, 93–101. [Google Scholar] [CrossRef]
- Cabelli, D.E.; Comstock, D.A.; Bielski, B.H.J. Free Radical Mechanisms for the Oxidation of Substituted Ascorbates. A Pulse Radiolysis Study of L-Ascorbic Acid-2-Sulfate. Radiat. Res. 1983, 95, 530–540. [Google Scholar] [CrossRef]
- Cabelli, D.E.; Bielski, B.H.J. Kinetics and Mechanism for the Oxidation of Ascorbic Acid/Ascorbate by HO₂˙/O₂˙¯ Radicals. A Pulse Radiolysis and Stopped-Flow Photolysis Study. J. Phys. Chem. 1983, 87, 1809–1812. [Google Scholar] [CrossRef]
- Isbell, H.S.; Frush, H.L. Oxidation of L-Ascorbic Acid by Hydrogen Peroxide: Preparation of L-Threonic Acid. Carbohydr. Res. 1979, 72, 301–304. [Google Scholar] [CrossRef]
- Deutsch, J.C. Ascorbic Acid Oxidation by Hydrogen Peroxide. Anal. Biochem. 1998, 255, 1–7. [Google Scholar] [CrossRef]
- Green, M.A.; Fry, S.C. Vitamin C Degradation in Plant Cells via Enzymatic Hydrolysis of 4-O-Oxalyl-l-Threonate. Nature 2005, 433, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Parsons, H.T.; Fry, S.C. Oxidation of Dehydroascorbic Acid and 2,3-Diketogulonate under Plant Apoplastic Conditions. Phytochemistry 2012, 75, 41–49. [Google Scholar] [CrossRef]
- Barclay, L.R.C.; Locke, S.J.; MacNeil, J.M. The Autoxidation of Unsaturated Lipids in Micelles. Synergism of Inhibitors Vitamins C and E. Can. J. Chem. 1983, 61, 1288–1290. [Google Scholar] [CrossRef]
- Doba, T.; Burton, G.W.; Ingold, K.U. Antioxidant and Co-Antioxidant Activity of Vitamin C. The Effect of Vitamin C, Either Alone or in the Presence of Vitamin E or a Water-Soluble Vitamin E Analogue, upon the Peroxidation of Aqueous Multilamellar Phospholipid Liposomes. Biochim. Biophys. Acta (BBA)/Lipids Lipid Metab. 1985, 835, 298–303. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd | Calcd for | Calcd | Found | Δ ppm |
|---|---|---|---|---|
| Asc-PA | C14H14O7 | 293.0667 | 293.0675 | 2.7 |
| DHA-PA | C14H12O5 | 291.0510 | 291.0515 | 1.7 |
| cOxa-Thr-PA | C14H12O8 | 307.0459 | 307.0458 | 0.3 |
| Oxa-Thr-PA | C14H14O9 | 325.0565 | 325.0567 | 0.6 |
| Thr-PA | C12H14O6 | 253.0718 | 253.0720 | 0.8 |
| 1H-NMR | Asc-PA (Ace) | cOxa-Thr-PA (Ace) | Oxa-Thr-PA (Ace) | Oxa-Thr-PA (Major/Minor) (D2O) | Thr-PA (DMSO) |
| H4 | 4.72 (d) | 5.66 (d) | 5.30 (d) | 5.23 (d)/4.56 | 3.56 (dd) |
| H5 | 4.24–4.29 (m) | 5.37 (ddd) | 4.52 (dd) | 4.49 (m)/5.59 | 3.81 (m) |
| H6a | 4.12–4.21 (m) | 4.84 (dd) | 4.24 (dd) | 4.30 (m)/4.45 | 3.88 (m) |
| H6b | 4.12–4.21 (m) | 4.64 (dd) | 4.31 (dd) | 4.23 (m)/4.43 | 4.03 (dd) |
| PhCH2 | 3.68 (s) | 3.65 (s) | 3.69 (s) | 3.77 (s) | 3.66 (s) |
| Ar-H | 7.19–7.30 (m) | 7.22–7.34 (m) | 7.22–7.34 (m) | 7.28–7.44 (m) | 7.24–7.35 (m) |
| 13C-NMR | Asc-PA (Ace) | cOxa-Thr-PA (Ace) | Oxa-Thr-PA (Ace) | Oxa-Thr-PA (Major/Minor) (D2O) | Thr-PA (DMSO) |
| C1 | 170.3 | 152.5 | 158.4 | 161.3/161.2 | --- |
| C2 | 120.0 | 153.2 | 158.6 | 161.7/161.2 | --- |
| C3 | 151.1 | 169.2 | 168.1 | 170.7/174.0 | 174.6 |
| C4 | 76.1 | 76.0 | 75.3 | 74.2/73.4 | 70.1 |
| C5 | 67.5 | 76.0 | 69.1 | 67.9/69.2 | 69.6 |
| C6 | 65.8 | 64.0 | 64.9 | 64.3/62.9 | 66.3 |
| PhCH2 | 41.3 | 41.0 | 41.3 | 40.3/40.5 | 40.2 |
| PhCH2CO | 171.7 | 171.1 | 171.6 | 174.2/174.0 | 171.3 |
| Ar-C | 127.7, 129.2, 130.2, 135.4 | 127.9, 129.3, 130.3, 134.7 | 127.7, 129.2, 130.2, 134.3 | 127.5, 128.9, 129.4, 133.8 | 126.7, 128.3, 129.4, 134.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robert, G.; Wagner, J.R. Scavenging of Alkylperoxyl Radicals by Addition to Ascorbate: An Alternative Mechanism to Electron Transfer. Antioxidants 2024, 13, 1194. https://doi.org/10.3390/antiox13101194
Robert G, Wagner JR. Scavenging of Alkylperoxyl Radicals by Addition to Ascorbate: An Alternative Mechanism to Electron Transfer. Antioxidants. 2024; 13(10):1194. https://doi.org/10.3390/antiox13101194
Chicago/Turabian StyleRobert, Gabriel, and J. Richard Wagner. 2024. "Scavenging of Alkylperoxyl Radicals by Addition to Ascorbate: An Alternative Mechanism to Electron Transfer" Antioxidants 13, no. 10: 1194. https://doi.org/10.3390/antiox13101194