
Tetramethylpyrazine Analogue T-006 Protects Neuronal and Endothelial Cells Against Oxidative Stress via PI3K/AKT/mTOR and Nrf2 Signaling


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Cultures
2.3. Glutamate or OGD/R-Induced Oxidative Stress Models
2.4. Cell Viability Assay
2.5. Quantitative Analysis of Apoptotic Cells by Flow Cytometry
2.6. Reactive Oxygen Species (ROS) Detection
2.7. Measurement of TEER in Endothelial Monolayer
2.8. Immunofluorescence Staining of Tight Junction Protein
2.9. Western Blotting
2.10. Statistical Analysis
3. Results
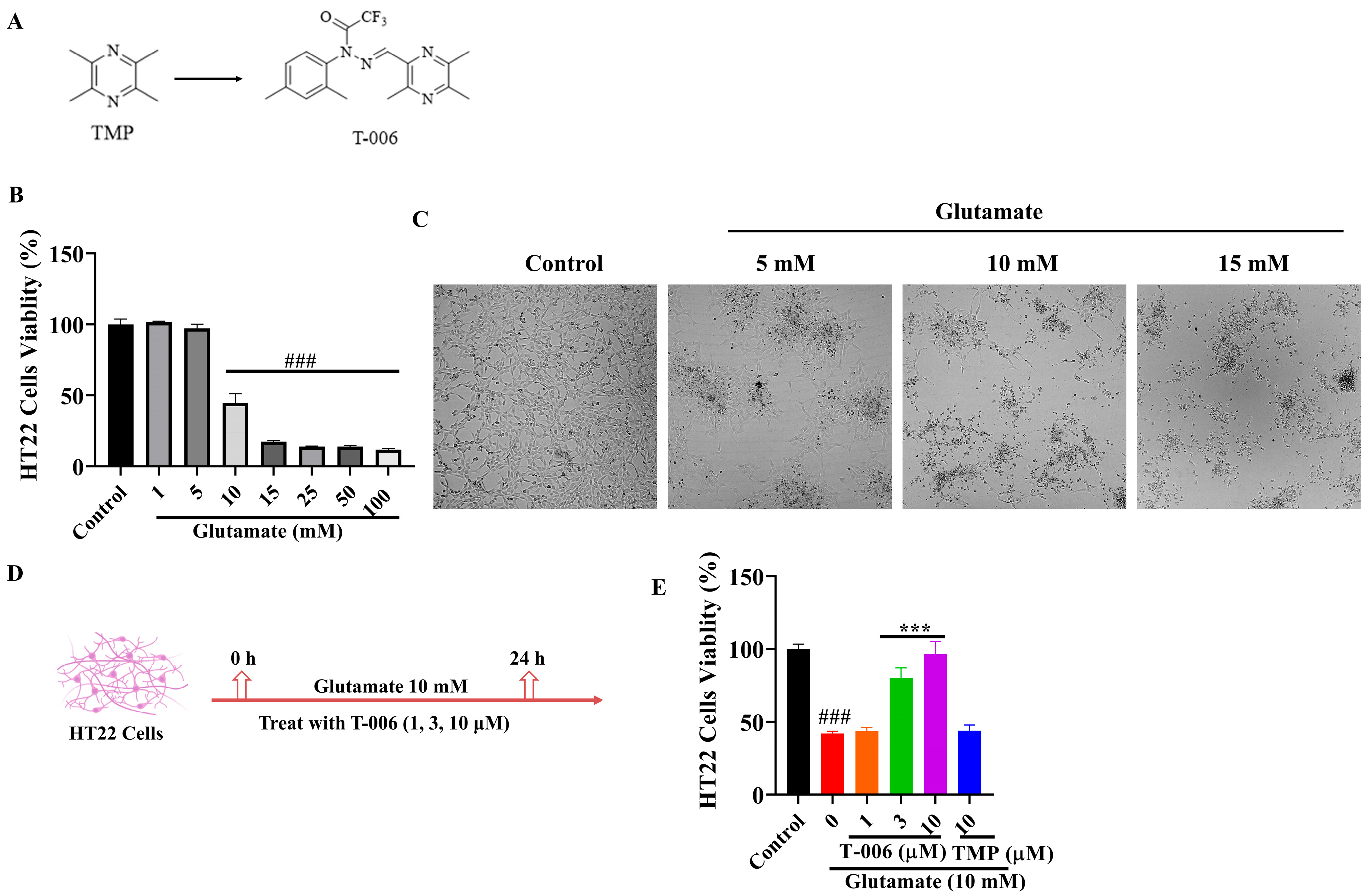
3.1. T-006 Protected HT22 Cells Against Glutamate-Induced Oxidative Cytotoxicity
3.2. T-006 Attenuated Glutamate-Induced ROS Production and Apoptosis in HT22 Cells
3.3. T-006 Enhanced mTOR Signaling and Suppressed Glutamate-Induced Autophagic Cell Death in HT22 Cells
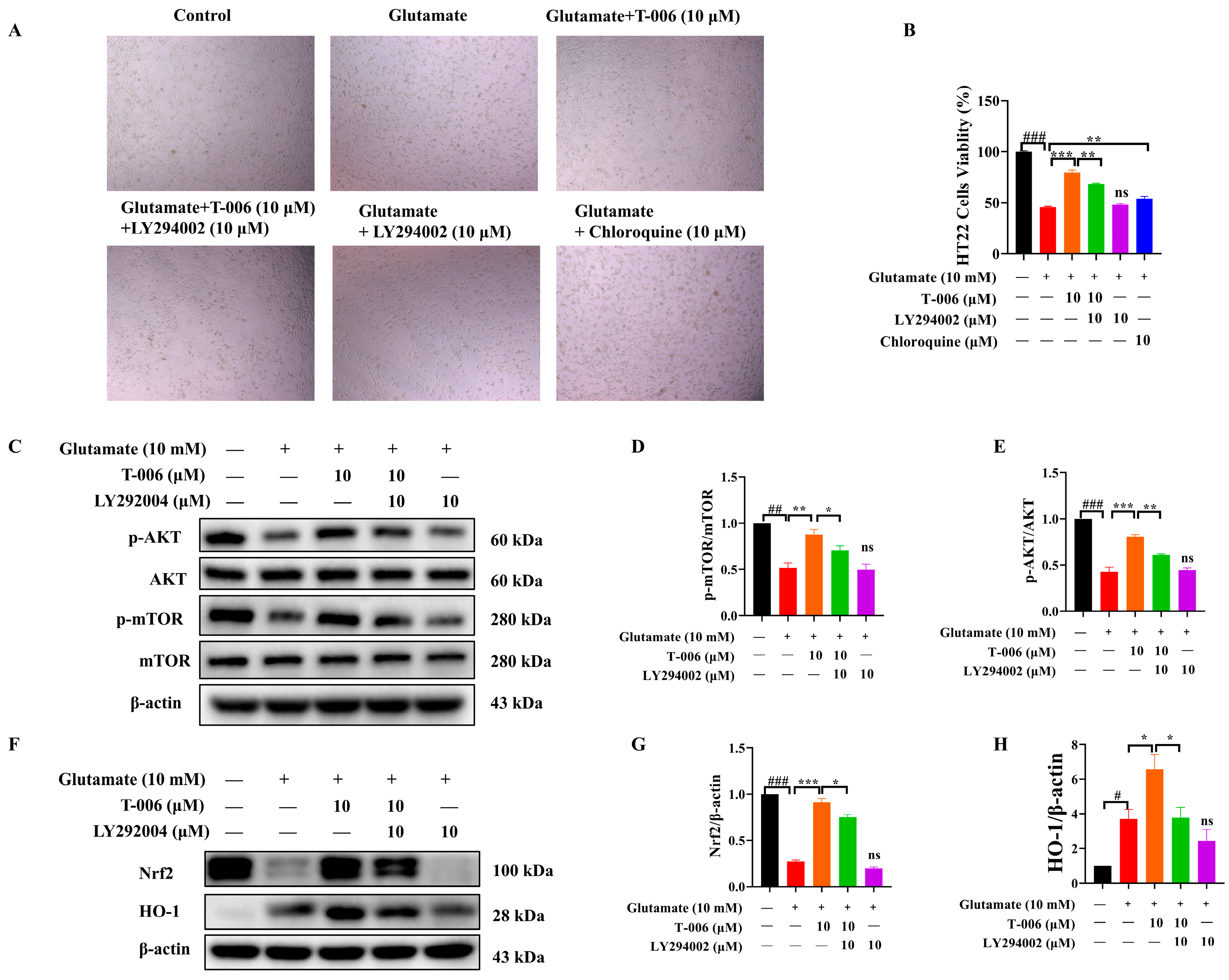
3.4. T-006 Protected HT22 Cells Against Glutamate-Induced Oxidative Toxicity via PI3K/AKT-Mediated mTOR and Nrf2/HO-1 Signaling Pathways
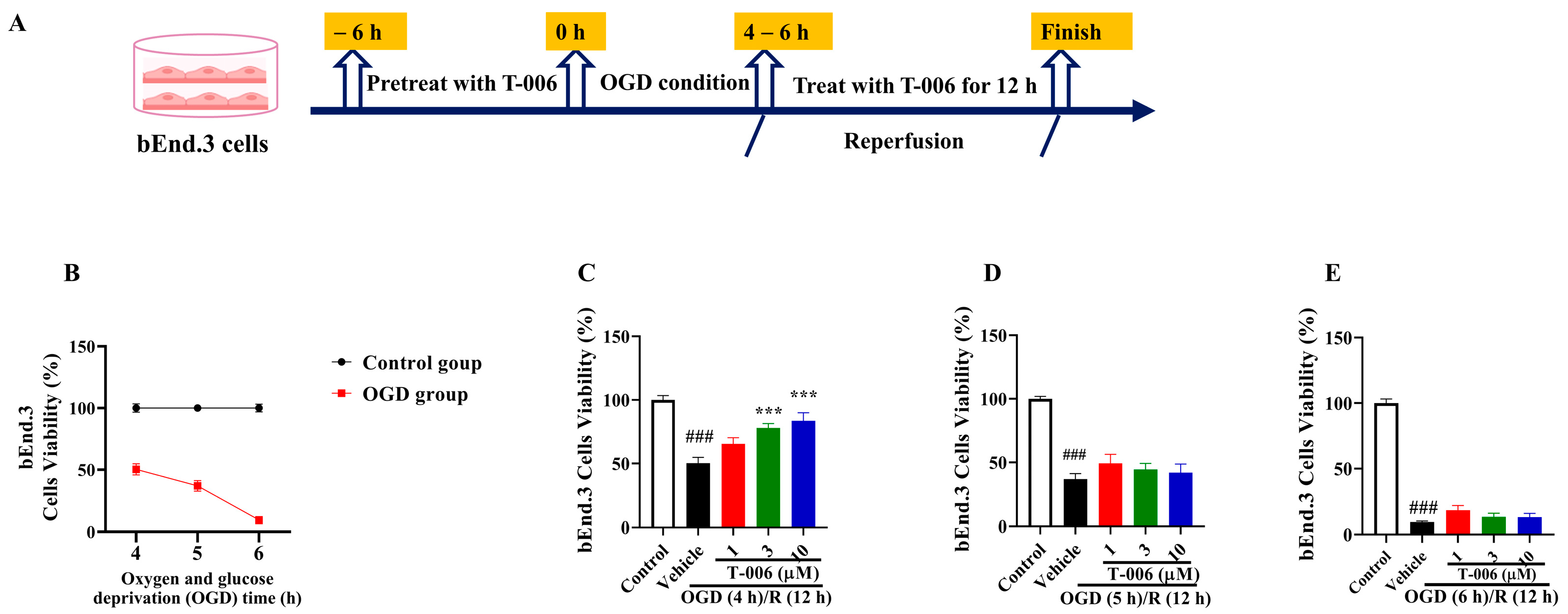
3.5. T-006 Protected bEnd.3 Cells Against OGD/R-Induced Oxidative Cytotoxicity
3.6. T-006 Attenuated Brain Endothelial Cell Dysfunction via AKT/Nrf2/HO-1 Pathway
4. Discussion
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Qin, C.; Yang, S.; Chu, Y.H.; Zhang, H.; Pang, X.W.; Chen, L.; Zhou, L.Q.; Chen, M.; Tian, D.S.; Wang, W. Signaling pathways involved in ischemic stroke: Molecular mechanisms and therapeutic interventions. Signal Transduct. Target. Ther. 2022, 7, 215. [Google Scholar] [CrossRef] [PubMed]
- Belov Kirdajova, D.; Kriska, J.; Tureckova, J.; Anderova, M. Ischemia-Triggered Glutamate Excitotoxicity from the Perspective of Glial Cells. Front. Cell. Neurosci. 2020, 14, 51. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Andjelkovic, A.V.; Zhu, L.; Yang, T.; Bennett, M.V.L.; Chen, J.; Keep, R.F.; Shi, Y. Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog. Neurobiol. 2018, 163–164, 144–171. [Google Scholar] [CrossRef] [PubMed]
- Meldrum, B.S. Glutamate as a neurotransmitter in the brain: Review of physiology and pathology. J. Nutr. 2000, 130, 1007s–1015s. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef]
- Wang, Y.; Qin, Z.H. Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosis Int. J. Program. Cell Death 2010, 15, 1382–1402. [Google Scholar] [CrossRef]
- Fukui, M.; Song, J.H.; Choi, J.; Choi, H.J.; Zhu, B.T. Mechanism of glutamate-induced neurotoxicity in HT22 mouse hippocampal cells. Eur. J. Pharmacol. 2009, 617, 1–11. [Google Scholar] [CrossRef]
- Kritis, A.A.; Stamoula, E.G.; Paniskaki, K.A.; Vavilis, T.D. Researching glutamate—Induced cytotoxicity in different cell lines: A comparative/collective analysis/study. Front. Cell. Neurosci. 2015, 9, 91. [Google Scholar] [CrossRef]
- Mehta, A.; Prabhakar, M.; Kumar, P.; Deshmukh, R.; Sharma, P.L. Excitotoxicity: Bridge to various triggers in neurodegenerative disorders. Eur. J. Pharmacol. 2013, 698, 6–18. [Google Scholar] [CrossRef]
- Verma, M.; Lizama, B.N.; Chu, C.T. Excitotoxicity, calcium and mitochondria: A triad in synaptic neurodegeneration. Transl. Neurodegener. 2022, 11, 3. [Google Scholar] [CrossRef]
- Chamorro, Á.; Dirnagl, U.; Urra, X.; Planas, A.M. Neuroprotection in acute stroke: Targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet. Neurol. 2016, 15, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Andjelkovic, A.V.; Xiang, J.; Stamatovic, S.M.; Hua, Y.; Xi, G.; Wang, M.M.; Keep, R.F. Endothelial Targets in Stroke: Translating Animal Models to Human. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 2240–2247. [Google Scholar] [CrossRef]
- Wong, B.W.; Marsch, E.; Treps, L.; Baes, M.; Carmeliet, P. Endothelial cell metabolism in health and disease: Impact of hypoxia. EMBO J. 2017, 36, 2187–2203. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Rosenberg, G.A. Blood-brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke 2011, 42, 3323–3328. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Guan, X.; Gao, C.L.; Ruan, W.; Zhao, S.; Kai, G.; Li, F.; Pang, T. Medioresinol as a novel PGC-1α activator prevents pyroptosis of endothelial cells in ischemic stroke through PPARα-GOT1 axis. Pharmacol. Res. 2021, 169, 105640. [Google Scholar] [CrossRef]
- Li, H.; Gao, A.; Feng, D.; Wang, Y.; Zhang, L.; Cui, Y.; Li, B.; Wang, Z.; Chen, G. Evaluation of the protective potential of brain microvascular endothelial cell autophagy on blood-brain barrier integrity during experimental cerebral ischemia-reperfusion injury. Transl. Stroke Res. 2014, 5, 618–626. [Google Scholar] [CrossRef]
- Gu, C.; Zhang, Q.; Li, Y.; Li, R.; Feng, J.; Chen, W.; Ahmed, W.; Soufiany, I.; Huang, S.; Long, J.; et al. The PI3K/AKT Pathway-The Potential Key Mechanisms of Traditional Chinese Medicine for Stroke. Front. Med. 2022, 9, 900809. [Google Scholar] [CrossRef]
- Heras-Sandoval, D.; Pérez-Rojas, J.M.; Hernández-Damián, J.; Pedraza-Chaverri, J. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell. Signal. 2014, 26, 2694–2701. [Google Scholar] [CrossRef]
- Zhang, R.; Xu, M.; Wang, Y.; Xie, F.; Zhang, G.; Qin, X. Nrf2-a Promising Therapeutic Target for Defensing Against Oxidative Stress in Stroke. Mol. Neurobiol. 2017, 54, 6006–6017. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
- Qiao, N.; An, Z.; Fu, Z.; Chen, X.; Tong, Q.; Zhang, Y.; Ren, H. Kinsenoside alleviates oxidative stress-induced blood-brain barrier dysfunction via promoting Nrf2/HO-1 pathway in ischemic stroke. Eur. J. Pharmacol. 2023, 949, 175717. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Y.; Xu, D.P.; Tan, G.L.; Cai, W.; Zhang, G.X.; Cui, W.; Wang, J.Z.; Long, C.; Sun, Y.W.; Yu, P.; et al. A Potent Multi-functional Neuroprotective Derivative of Tetramethylpyrazine. J. Mol. Neurosci. MN 2015, 56, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Chen, H.; Mak, S.; Hu, S.; Tsim, K.W.K.; Hu, Y.; Sun, Y.; Zhang, G.; Wang, Y.; Zhang, Z.; et al. Neuroprotection against glutamate-induced excitotoxicity and induction of neurite outgrowth by T-006, a novel multifunctional derivative of tetramethylpyrazine in neuronal cell models. Neurochem. Int. 2016, 99, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Cao, J.; Zha, L.; Wang, P.; Liu, Z.; Guo, B.; Zhang, G.; Sun, Y.; Zhang, Z.; Wang, Y. Neuroprotective and neurogenic effects of novel tetramethylpyrazine derivative T-006 in Parkinson’s disease models through activating the MEF2-PGC1α and BDNF/CREB pathways. Aging 2020, 12, 14897–14917. [Google Scholar] [CrossRef]
- Zhou, H.; Shao, M.; Guo, B.; Li, C.; Lu, Y.; Yang, X.; Li, S.; Li, H.; Zhu, Q.; Zhong, H.; et al. Tetramethylpyrazine Analogue T-006 Promotes the Clearance of Alpha-synuclein by Enhancing Proteasome Activity in Parkinson’s Disease Models. Neurother. J. Am. Soc. Exp. NeuroTher. 2019, 16, 1225–1236. [Google Scholar] [CrossRef]
- Zhou, H.; Shao, M.; Yang, X.; Li, C.; Cui, G.; Gao, C.; Di, L.; Zhong, H.; Wang, Y.; Zhang, Z.; et al. Tetramethylpyrazine Analogue T-006 Exerts Neuroprotective Effects against 6-Hydroxydopamine-Induced Parkinson’s Disease In Vitro and In Vivo. Oxidative Med. Cell. Longev. 2019, 2019, 8169125. [Google Scholar] [CrossRef]
- Zhang, G.; Wu, J.; Huang, C.; Cheng, J.; Su, Z.; Zhu, Z.; Yang, X.; Guo, B.; Wu, L.; Zhang, Z.; et al. The Tetramethylpyrazine Analogue T-006 Alleviates Cognitive Deficits by Inhibition of Tau Expression and Phosphorylation in Transgenic Mice Modeling Alzheimer’s Disease. J. Mol. Neurosci. MN 2021, 71, 1456–1466. [Google Scholar] [CrossRef]
- Wu, L.; Cao, J.; Li, N.; Chen, H.; Liu, W.; Zhang, G.; Liu, Y.; Wang, Y.; Sun, Y.; Zhang, Z. Novel neuroprotective tetramethylpyrazine analog T-006 promotes neurogenesis and neurological restoration in a rat model of stroke. Neuroreport 2019, 30, 658–663. [Google Scholar] [CrossRef]
- Huang, L.; Chen, Y.; Liu, R.; Li, B.; Fei, X.; Li, X.; Liu, G.; Li, Y.; Xu, B.; Fang, W. P-Glycoprotein Aggravates Blood Brain Barrier Dysfunction in Experimental Ischemic Stroke by Inhibiting Endothelial Autophagy. Aging Dis. 2022, 13, 1546–1561. [Google Scholar] [CrossRef]
- Shen, Z.; Xiang, M.; Chen, C.; Ding, F.; Wang, Y.; Shang, C.; Xin, L.; Zhang, Y.; Cui, X. Glutamate excitotoxicity: Potential therapeutic target for ischemic stroke. Biomed. Pharmacother. 2022, 151, 113125. [Google Scholar] [CrossRef]
- Murphy, T.H.; Miyamoto, M.; Sastre, A.; Schnaar, R.L.; Coyle, J.T. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 1989, 2, 1547–1558. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Schubert, D.; Maher, P. Oxytosis: A novel form of programmed cell death. Curr. Top. Med. Chem. 2001, 1, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Tobaben, S.; Grohm, J.; Seiler, A.; Conrad, M.; Plesnila, N.; Culmsee, C. Bid-mediated mitochondrial damage is a key mechanism in glutamate-induced oxidative stress and AIF-dependent cell death in immortalized HT-22 hippocampal neurons. Cell Death Differ. 2011, 18, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Choi, J.; Ryu, J.; Park, S.G.; Cho, S.; Park, B.C.; Lee, D.H. Activation of autophagy during glutamate-induced HT22 cell death. Biochem. Biophys. Res. Commun. 2009, 388, 339–344. [Google Scholar] [CrossRef]
- Jiang, P.; Mizushima, N. LC3- and p62-based biochemical methods for the analysis of autophagy progression in mammalian cells. Methods 2015, 75, 13–18. [Google Scholar] [CrossRef]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef]
- Liu, X.; Xiao, X.; Han, X.; Yao, L.; Lan, W. A New Therapeutic Trend: Natural Medicine for Ameliorating Ischemic Stroke via PI3K/Akt Signaling Pathway. Molecules 2022, 27, 7963. [Google Scholar] [CrossRef]
- Pasquier, B. Autophagy inhibitors. Cell. Mol. Life Sci. CMLS 2016, 73, 985–1001. [Google Scholar] [CrossRef]
- Xu, W.; Zheng, H.; Fu, Y.; Gu, Y.; Zou, H.; Yuan, Y.; Gu, J.; Liu, Z.; Bian, J. Role of PI3K/Akt-Mediated Nrf2/HO-1 Signaling Pathway in Resveratrol Alleviation of Zearalenone-Induced Oxidative Stress and Apoptosis in TM4 Cells. Toxins 2022, 14, 733. [Google Scholar] [CrossRef]
- Lee, D.; Choi, H.G.; Hwang, J.H.; Shim, S.H.; Kang, K.S. Neuroprotective Effect of Tricyclic Pyridine Alkaloids from Fusarium lateritium SSF2, against Glutamate-Induced Oxidative Stress and Apoptosis in the HT22 Hippocampal Neuronal Cell Line. Antioxidants 2020, 9, 1115. [Google Scholar] [CrossRef]
- Song, J.H.; Lee, H.J.; Kang, K.S. Procyanidin C1 Activates the Nrf2/HO-1 Signaling Pathway to Prevent Glutamate-Induced Apoptotic HT22 Cell Death. Int. J. Mol. Sci. 2019, 20, 142. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Ren, B.; Gao, Y. Tight junction proteins related to blood-brain barrier and their regulatory signaling pathways in ischemic stroke. Biomed. Pharmacother. 2023, 165, 115272. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, X.; Xiong, X.; Zhu, H.; Chen, R.; Zhang, S.; Chen, G.; Jian, Z. Nrf2 Regulates Oxidative Stress and Its Role in Cerebral Ischemic Stroke. Antioxidants 2022, 11, 2377. [Google Scholar] [CrossRef]
- Lin, J.; Wang, Q.; Zhou, S.; Xu, S.; Yao, K. Tetramethylpyrazine: A review on its mechanisms and functions. Biomed. Pharmacother. 2022, 150, 113005. [Google Scholar] [CrossRef]
- Qi, M.; Su, X.; Li, Z.; Huang, H.; Wang, J.; Lin, N.; Kong, X. Bibliometric analysis of research progress on tetramethylpyrazine and its effects on ischemia-reperfusion injury. Pharmacol. Ther. 2024, 259, 108656. [Google Scholar] [CrossRef]
- Prasansuklab, A.; Sukjamnong, S.; Theerasri, A.; Hu, V.W.; Sarachana, T.; Tencomnao, T. Transcriptomic analysis of glutamate-induced HT22 neurotoxicity as a model for screening anti-Alzheimer’s drugs. Sci. Rep. 2023, 13, 7225. [Google Scholar] [CrossRef]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative Stress in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef]
- He, M.; Liu, J.; Cheng, S.; Xing, Y.; Suo, W.Z. Differentiation renders susceptibility to excitotoxicity in HT22 neurons. Neural Regen. Res. 2013, 8, 1297–1306. [Google Scholar] [CrossRef]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Cao, Y.; Li, Q.; Liu, L.; Wu, H.; Huang, F.; Wang, C.; Lan, Y.; Zheng, F.; Xing, F.; Zhou, Q.; et al. Modafinil protects hippocampal neurons by suppressing excessive autophagy and apoptosis in mice with sleep deprivation. Br. J. Pharmacol. 2019, 176, 1282–1297. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef] [PubMed]
- Bendavit, G.; Aboulkassim, T.; Hilmi, K.; Shah, S.; Batist, G. Nrf2 Transcription Factor Can Directly Regulate mTOR: Linking cytoprotective gene expression to a major metabolic regulator that generates redox activity. J. Biol. Chem. 2016, 291, 25476–25488. [Google Scholar] [CrossRef] [PubMed]
- Woo, Y.; Lee, H.J.; Jung, Y.M.; Jung, Y.J. mTOR-Mediated Antioxidant Activation in Solid Tumor Radioresistance. J. Oncol. 2019, 2019, 5956867. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, X.; Chen, X.; Wei, Y. Neuronal injuries in cerebral infarction and ischemic stroke: From mechanisms to treatment (Review). Int. J. Mol. Med. 2022, 49, 15. [Google Scholar] [CrossRef] [PubMed]
- Abdullahi, W.; Tripathi, D.; Ronaldson, P.T. Blood-brain barrier dysfunction in ischemic stroke: Targeting tight junctions and transporters for vascular protection. Am. J. Physiol.-Cell Physiol. 2018, 315, C343–C356. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, G.; Liang, Z.; Wang, Y.; Zhang, Z.; Hoi, P.-M. Tetramethylpyrazine Analogue T-006 Protects Neuronal and Endothelial Cells Against Oxidative Stress via PI3K/AKT/mTOR and Nrf2 Signaling. Antioxidants 2024, 13, 1272. https://doi.org/10.3390/antiox13101272
Zhang G, Liang Z, Wang Y, Zhang Z, Hoi P-M. Tetramethylpyrazine Analogue T-006 Protects Neuronal and Endothelial Cells Against Oxidative Stress via PI3K/AKT/mTOR and Nrf2 Signaling. Antioxidants. 2024; 13(10):1272. https://doi.org/10.3390/antiox13101272
Chicago/Turabian StyleZhang, Guiliang, Zirong Liang, Yuqiang Wang, Zaijun Zhang, and Pui-Man Hoi. 2024. "Tetramethylpyrazine Analogue T-006 Protects Neuronal and Endothelial Cells Against Oxidative Stress via PI3K/AKT/mTOR and Nrf2 Signaling" Antioxidants 13, no. 10: 1272. https://doi.org/10.3390/antiox13101272
APA StyleZhang, G., Liang, Z., Wang, Y., Zhang, Z., & Hoi, P.-M. (2024). Tetramethylpyrazine Analogue T-006 Protects Neuronal and Endothelial Cells Against Oxidative Stress via PI3K/AKT/mTOR and Nrf2 Signaling. Antioxidants, 13(10), 1272. https://doi.org/10.3390/antiox13101272