
Neuroinflammation in Age-Related Neurodegenerative Diseases: Role of Mitochondrial Oxidative Stress
 , , , ,
, , , ,  ,
,  , ,
, ,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. The Immune System in the Brain and Its Role in Neurodegenerative Disorders
The Contribution of the Innate–Adaptive Immunity to Neurodegeneration
The Role of Microglia in the Neurodegenerative Process
2. Mitochondrial Control of the Inflammatory Response
The mtDNA as a Danger Signal That Activates the cGAS–STING Signaling Pathway
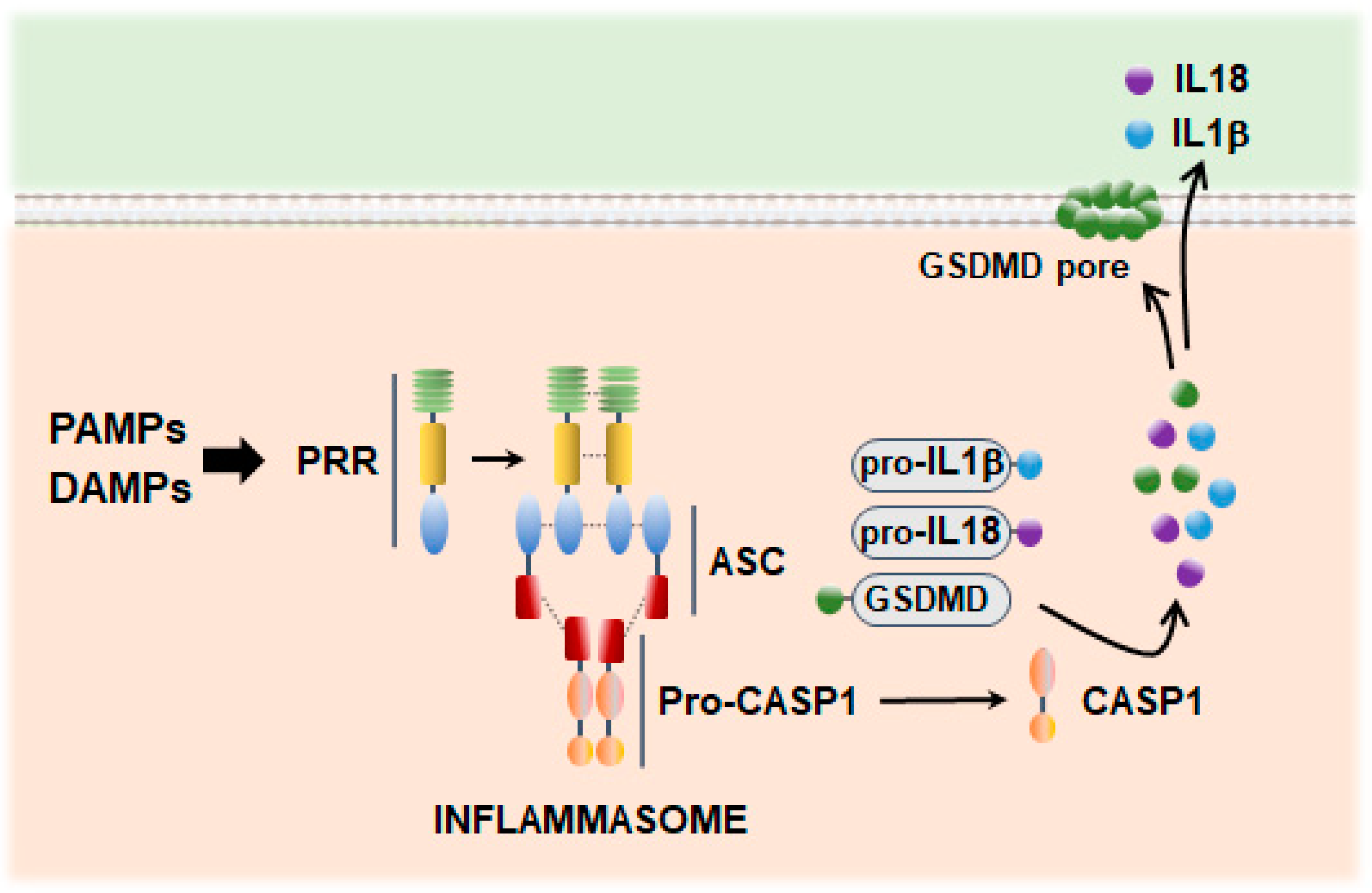
3. Inflammasome Signaling in Neuroinflammation
3.1. Inflammasome Scaffold Proteins
3.2. Dysfunctional Mitochondria and Mitochondrial ROS as Drivers of Inflammasome Activation
3.3. GSDM-Mediated Inflammatory Cell Death: Pyroptosis
3.4. Inflammasome-Mediated Neuroinflammation in Alzheimer’s and Parkinson’s Disease
4. Role of Immunogenic Cell Death as a Contributor to Neurodegeneration
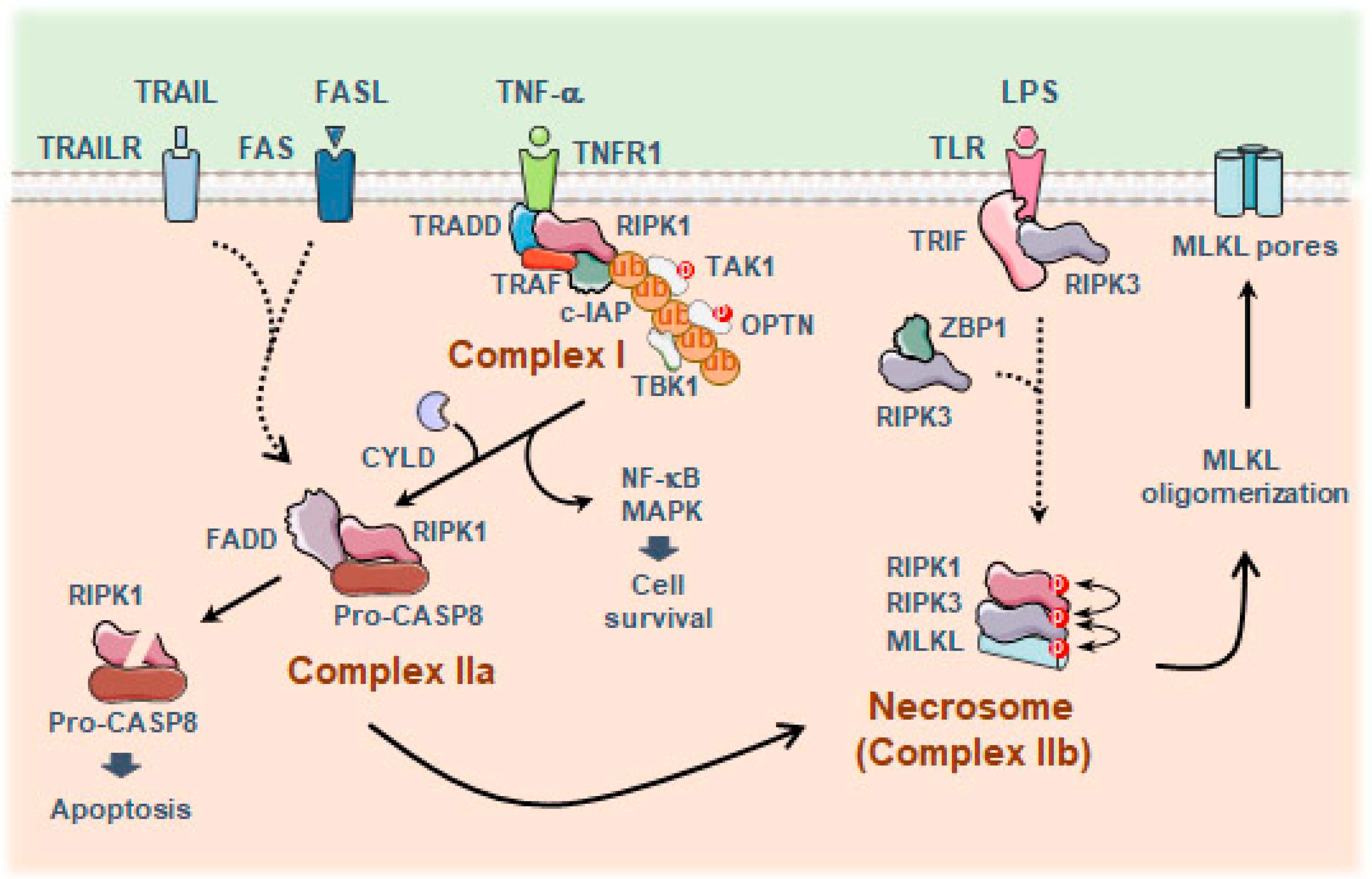
4.1. Necroptosis
Necroptosis and Aging-Related Neurodegenerative Diseases
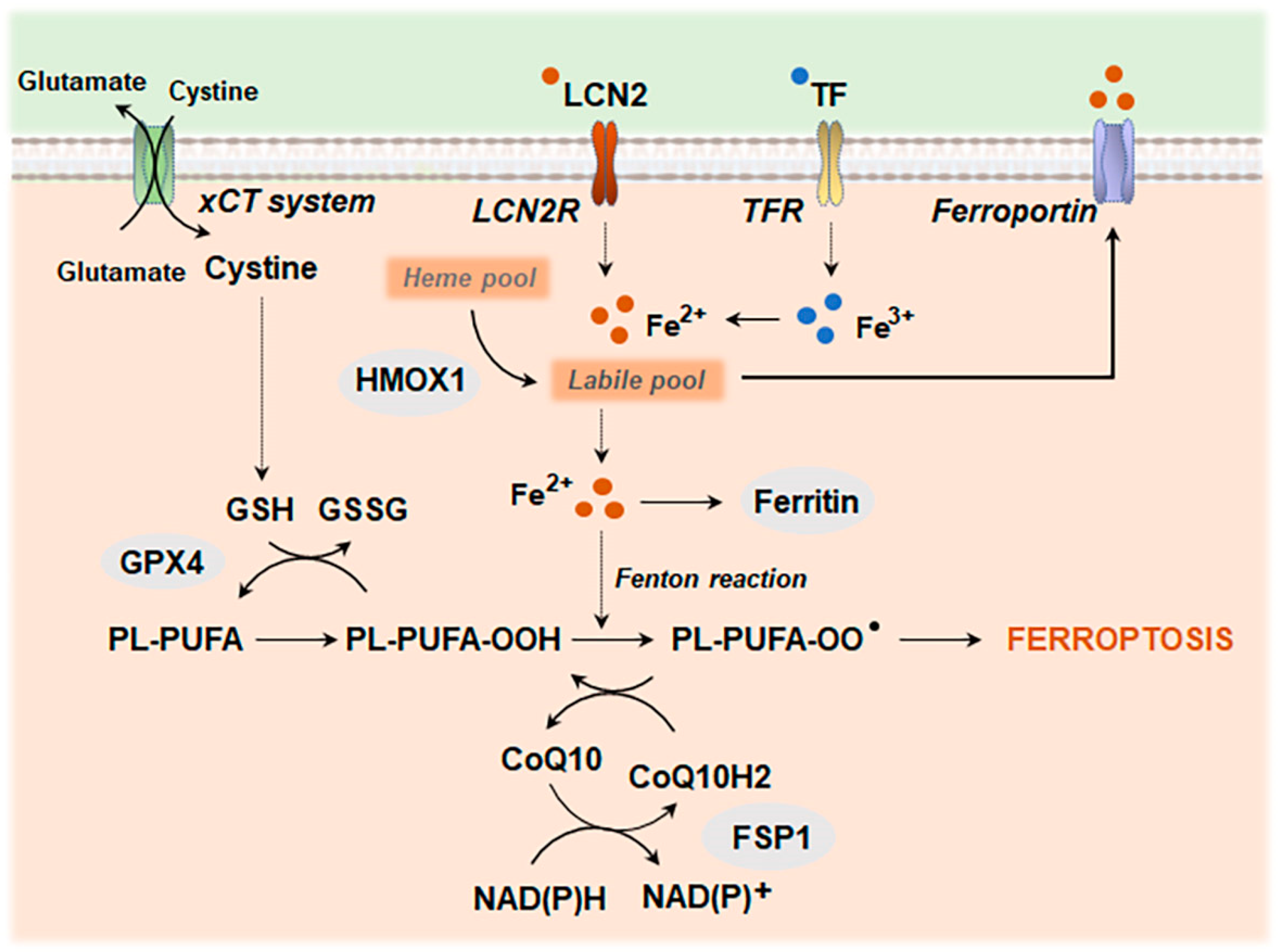
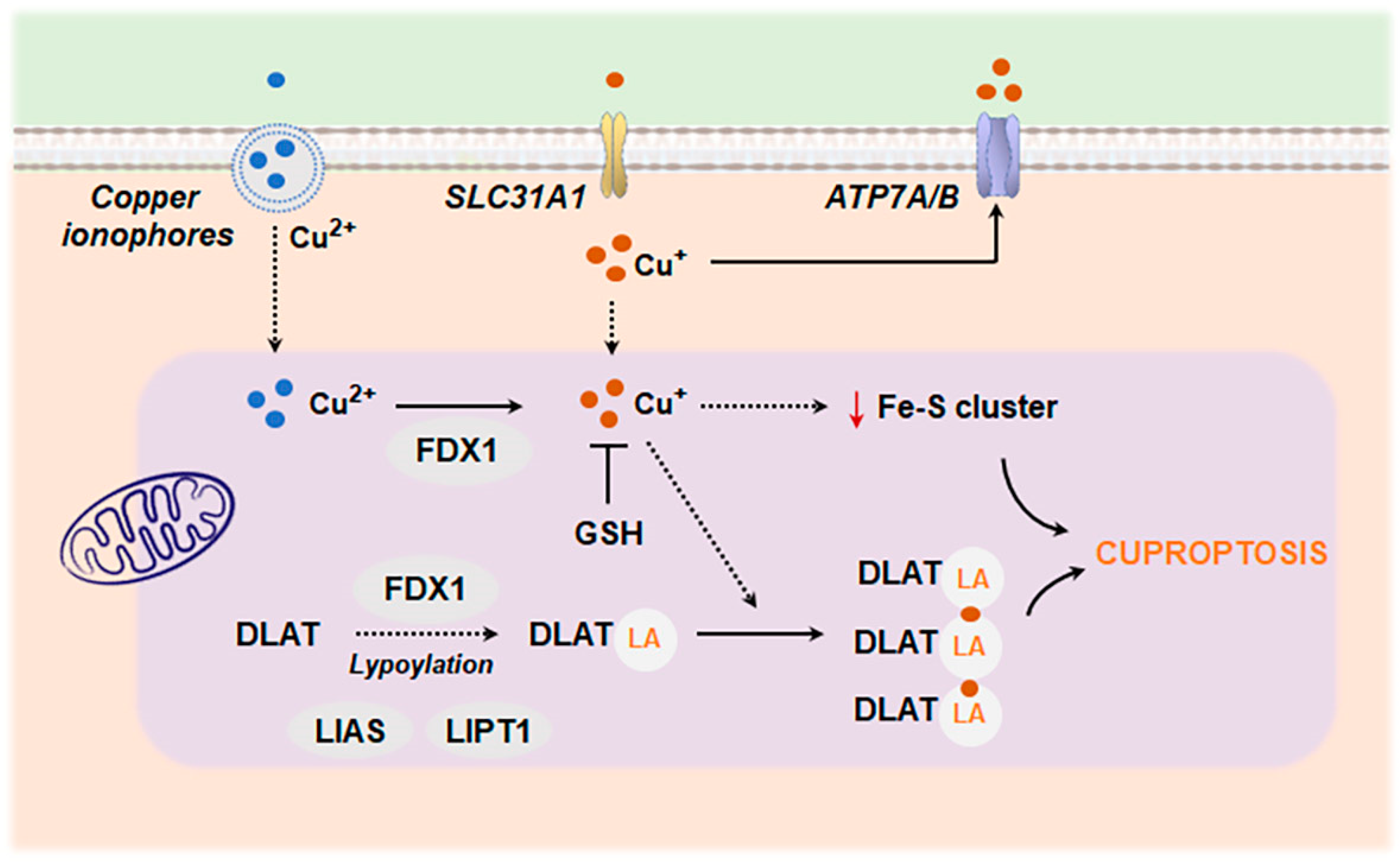
4.2. Metal Ion-Induced Cell Death: Ferroptosis and Cuproptosis
4.2.1. Ferroptosis
4.2.2. Cuproptosis
5. Concluding Remarks and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Greenhalgh, A.D.; David, S.; Bennett, F.C. Immune Cell Regulation of Glia during CNS Injury and Disease. Nat. Rev. Neurosci. 2020, 21, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Priller, J. The Role of Peripheral Immune Cells in the CNS in Steady State and Disease. Nat. Neurosci. 2017, 20, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Greenhalgh, A.D.; Zarruk, J.G.; Healy, L.M.; Baskar Jesudasan, S.J.; Jhelum, P.; Salmon, C.K.; Formanek, A.; Russo, M.V.; Antel, J.P.; McGavern, D.B.; et al. Peripherally Derived Macrophages Modulate Microglial Function to Reduce Inflammation after CNS Injury. PLoS Biol. 2018, 16, e2005264. [Google Scholar] [CrossRef] [PubMed]
- Netzahualcoyotzi, C.; Santillán-Cigales, J.J.; Adalid-Peralta, L.V.; Velasco, I. Infiltration of Immune Cells to the Brain and Its Relation to the Pathogenesis of Alzheimer’s and Parkinson’s Diseases. J. Neurochem. 2024, 168, 2316–2334. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, R.; Chen, H.; Jin, C.; Jin, Z.; Lu, J.; Xu, L.; Lu, Y.; Zhang, J.; Shi, L. Aged Microglia Promote Peripheral T Cell Infiltration by Reprogramming the Microenvironment of Neurogenic Niches. Immun. Ageing 2022, 19, 34. [Google Scholar] [CrossRef]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-Brain Barrier Breakdown in the Aging Human Hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef]
- Wyss-Coray, T. Ageing, Neurodegeneration and Brain Rejuvenation. Nature 2016, 539, 180–186. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Bourdenx, M.; Koulakiotis, N.S.; Sanoudou, D.; Bezard, E.; Dehay, B.; Tsarbopoulos, A. Protein Aggregation and Neurodegeneration in Prototypical Neurodegenerative Diseases: Examples of Amyloidopathies, Tauopathies and Synucleinopathies. Prog. Neurobiol. 2017, 155, 171–193. [Google Scholar] [CrossRef]
- Efthymiou, A.G.; Goate, A.M. Late Onset Alzheimer’s Disease Genetics Implicates Microglial Pathways in Disease Risk. Mol. Neurodegener. 2017, 12, 43. [Google Scholar] [CrossRef]
- Bellenguez, C.; Küçükali, F.; Jansen, I.E.; Kleineidam, L.; Moreno-Grau, S.; Amin, N.; Naj, A.C.; Campos-Martin, R.; Grenier-Boley, B.; Andrade, V.; et al. New Insights into the Genetic Etiology of Alzheimer’s Disease and Related Dementias. Nat. Genet. 2022, 54, 412–436. [Google Scholar] [CrossRef] [PubMed]
- Gorlé, N.; Vandenbroucke, R.E. Interferons: A Molecular Switch between Damage and Repair in Ageing and Alzheimer’s Disease. Mech. Ageing Dev. 2019, 183, 111148. [Google Scholar] [CrossRef] [PubMed]
- Marsh, S.E.; Abud, E.M.; Lakatos, A.; Karimzadeh, A.; Yeung, S.T.; Davtyan, H.; Fote, G.M.; Lau, L.; Weinger, J.G.; Lane, T.E.; et al. The Adaptive Immune System Restrains Alzheimer’s Disease Pathogenesis by Modulating Microglial Function. Proc. Natl. Acad. Sci. USA 2016, 113, E1316–E1325. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Theodore, S.; Standaert, D.G. Fcγ Receptors Are Required for NF-κB Signaling, Microglial Activation and Dopaminergic Neurodegeneration in an AAV-Synuclein Mouse Model of Parkinson’s Disease. Mol. Neurodegener. 2010, 5, 42. [Google Scholar] [CrossRef] [PubMed]
- Teeling, J.L.; Carare, R.O.; Glennie, M.J.; Perry, V.H. Intracerebral Immune Complex Formation Induces Inflammation in the Brain That Depends on Fc Receptor Interaction. Acta Neuropathol. 2012, 124, 479–490. [Google Scholar] [CrossRef]
- Kim, K.; Wang, X.; Ragonnaud, E.; Bodogai, M.; Illouz, T.; DeLuca, M.; McDevitt, R.A.; Gusev, F.; Okun, E.; Rogaev, E.; et al. Therapeutic B-Cell Depletion Reverses Progression of Alzheimer’s Disease. Nat. Commun. 2021, 12, 2185. [Google Scholar] [CrossRef]
- Batchu, S. Prefrontal Cortex Transcriptomic Deconvolution Implicates Monocyte Infiltration in Parkinson’s Disease. Neurodegener. Dis. 2020, 20, 110–112. [Google Scholar] [CrossRef]
- Muñoz-Castro, C.; Mejias-Ortega, M.; Sanchez-Mejias, E.; Navarro, V.; Trujillo-Estrada, L.; Jimenez, S.; Garcia-Leon, J.A.; Fernandez-Valenzuela, J.J.; Sanchez-Mico, M.V.; Romero-Molina, C.; et al. Monocyte-Derived Cells Invade Brain Parenchyma and Amyloid Plaques in Human Alzheimer’s Disease Hippocampus. Acta Neuropathol. Commun. 2023, 11, 31. [Google Scholar] [CrossRef]
- Koronyo, Y.; Salumbides, B.C.; Sheyn, J.; Pelissier, L.; Li, S.; Ljubimov, V.; Moyseyev, M.; Daley, D.; Fuchs, D.-T.; Pham, M.; et al. Therapeutic Effects of Glatiramer Acetate and Grafted CD115+ Monocytes in a Mouse Model of Alzheimer’s Disease. Brain 2015, 138, 2399–2422. [Google Scholar] [CrossRef]
- Harms, A.S.; Thome, A.D.; Yan, Z.; Schonhoff, A.M.; Williams, G.P.; Li, X.; Liu, Y.; Qin, H.; Benveniste, E.N.; Standaert, D.G. Peripheral Monocyte Entry Is Required for Alpha-Synuclein Induced Inflammation and Neurodegeneration in a Model of Parkinson Disease. Exp. Neurol. 2018, 300, 179–187. [Google Scholar] [CrossRef]
- Subbarayan, M.S.; Hudson, C.; Moss, L.D.; Nash, K.R.; Bickford, P.C. T Cell Infiltration and Upregulation of MHCII in Microglia Leads to Accelerated Neuronal Loss in an α-Synuclein Rat Model of Parkinson’s Disease. J. Neuroinflammation 2020, 17, 242. [Google Scholar] [CrossRef] [PubMed]
- Laurent, C.; Dorothée, G.; Hunot, S.; Martin, E.; Monnet, Y.; Duchamp, M.; Dong, Y.; Légeron, F.-P.; Leboucher, A.; Burnouf, S.; et al. Hippocampal T Cell Infiltration Promotes Neuroinflammation and Cognitive Decline in a Mouse Model of Tauopathy. Brain 2017, 140, 184–200. [Google Scholar] [CrossRef] [PubMed]
- Zenaro, E.; Pietronigro, E.; Della Bianca, V.; Piacentino, G.; Marongiu, L.; Budui, S.; Turano, E.; Rossi, B.; Angiari, S.; Dusi, S.; et al. Neutrophils Promote Alzheimer’s Disease-like Pathology and Cognitive Decline via LFA-1 Integrin. Nat. Med. 2015, 21, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Firulyova, M.; Manis, M.; Herz, J.; Smirnov, I.; Aladyeva, E.; Wang, C.; Bao, X.; Finn, M.B.; Hu, H.; et al. Microglia-Mediated T Cell Infiltration Drives Neurodegeneration in Tauopathy. Nature 2023, 615, 668–677. [Google Scholar] [CrossRef]
- Lindestam Arlehamn, C.S.; Dhanwani, R.; Pham, J.; Kuan, R.; Frazier, A.; Rezende Dutra, J.; Phillips, E.; Mallal, S.; Roederer, M.; Marder, K.S.; et al. α-Synuclein-Specific T Cell Reactivity Is Associated with Preclinical and Early Parkinson’s Disease. Nat. Commun. 2020, 11, 1875. [Google Scholar] [CrossRef]
- Gate, D.; Tapp, E.; Leventhal, O.; Shahid, M.; Nonninger, T.J.; Yang, A.C.; Strempfl, K.; Unger, M.S.; Fehlmann, T.; Oh, H.; et al. CD4+ T Cells Contribute to Neurodegeneration in Lewy Body Dementia. Science 2021, 374, 868–874. [Google Scholar] [CrossRef]
- Gate, D.; Saligrama, N.; Leventhal, O.; Yang, A.C.; Unger, M.S.; Middeldorp, J.; Chen, K.; Lehallier, B.; Channappa, D.; De Los Santos, M.B.; et al. Clonally Expanded CD8 T Cells Patrol the Cerebrospinal Fluid in Alzheimer’s Disease. Nature 2020, 577, 399–404. [Google Scholar] [CrossRef]
- Piehl, N.; van Olst, L.; Ramakrishnan, A.; Teregulova, V.; Simonton, B.; Zhang, Z.; Tapp, E.; Channappa, D.; Oh, H.; Losada, P.M.; et al. Cerebrospinal Fluid Immune Dysregulation During Healthy Brain Aging and Cognitive Impairment. Cell 2022, 185, 5028–5039.e13. [Google Scholar] [CrossRef]
- Tan, Y.-L.; Yuan, Y.; Tian, L. Microglial Regional Heterogeneity and Its Role in the Brain. Mol. Psychiatry 2020, 25, 351–367. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R.; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.-B. Microglia Promote Learning-Dependent Synapse Formation through Brain-Derived Neurotrophic Factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef] [PubMed]
- Sierra, A.; Encinas, J.M.; Deudero, J.J.P.; Chancey, J.H.; Enikolopov, G.; Overstreet-Wadiche, L.S.; Tsirka, S.E.; Maletic-Savatic, M. Microglia Shape Adult Hippocampal Neurogenesis Through Apoptosis-Coupled Phagocytosis. Cell Stem Cell 2010, 7, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma In Vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. A Polarizing Question: Do M1 and M2 Microglia Exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Gerrits, E.; Brouwer, N.; Kooistra, S.M.; Woodbury, M.E.; Vermeiren, Y.; Lambourne, M.; Mulder, J.; Kummer, M.; Möller, T.; Biber, K.; et al. Distinct Amyloid-β and Tau-Associated Microglia Profiles in Alzheimer’s Disease. Acta Neuropathol. 2021, 141, 681–696. [Google Scholar] [CrossRef]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef]
- Uriarte Huarte, O.; Kyriakis, D.; Heurtaux, T.; Pires-Afonso, Y.; Grzyb, K.; Halder, R.; Buttini, M.; Skupin, A.; Mittelbronn, M.; Michelucci, A. Single-Cell Transcriptomics and In Situ Morphological Analyses Reveal Microglia Heterogeneity Across the Nigrostriatal Pathway. Front. Immunol. 2021, 12, 639613. [Google Scholar] [CrossRef]
- Singh, N.; Das, B.; Zhou, J.; Hu, X.; Yan, R. Targeted BACE-1 Inhibition in Microglia Enhances Amyloid Clearance and Improved Cognitive Performance. Sci. Adv. 2022, 8, eabo3610. [Google Scholar] [CrossRef]
- Heckmann, B.L.; Teubner, B.J.W.; Tummers, B.; Boada-Romero, E.; Harris, L.; Yang, M.; Guy, C.S.; Zakharenko, S.S.; Green, D.R. LC3-Associated Endocytosis Facilitates β-Amyloid Clearance and Mitigates Neurodegeneration in Murine Alzheimer’s Disease. Cell 2019, 178, 536–551.e14. [Google Scholar] [CrossRef] [PubMed]
- McAlpine, C.S.; Park, J.; Griciuc, A.; Kim, E.; Choi, S.H.; Iwamoto, Y.; Kiss, M.G.; Christie, K.A.; Vinegoni, C.; Poller, W.C.; et al. Astrocytic Interleukin-3 Programs Microglia and Limits Alzheimer’s Disease. Nature 2021, 595, 701–706. [Google Scholar] [CrossRef] [PubMed]
- d’Errico, P.; Ziegler-Waldkirch, S.; Aires, V.; Hoffmann, P.; Mezö, C.; Erny, D.; Monasor, L.S.; Liebscher, S.; Ravi, V.M.; Joseph, K.; et al. Microglia Contribute to the Propagation of Aβ into Unaffected Brain Tissue. Nat. Neurosci. 2022, 25, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Propson, N.E.; Du, S.; Xiong, W.; Zheng, H. Autophagy Deficiency Modulates Microglial Lipid Homeostasis and Aggravates Tau Pathology and Spreading. Proc. Natl. Acad. Sci. USA 2021, 118, e2023418118. [Google Scholar] [CrossRef]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-Derived ASC Specks Cross-Seed Amyloid-β in Alzheimer’s Disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef]
- Huang, Y.; Happonen, K.E.; Burrola, P.G.; O’Connor, C.; Hah, N.; Huang, L.; Nimmerjahn, A.; Lemke, G. Microglia Use TAM Receptors to Detect and Engulf Amyloid β Plaques. Nat. Immunol. 2021, 22, 586–594. [Google Scholar] [CrossRef]
- Choi, I.; Zhang, Y.; Seegobin, S.P.; Pruvost, M.; Wang, Q.; Purtell, K.; Zhang, B.; Yue, Z. Microglia Clear Neuron-Released α-Synuclein via Selective Autophagy and Prevent Neurodegeneration. Nat. Commun. 2020, 11, 1386. [Google Scholar] [CrossRef]
- Joshi, P.; Turola, E.; Ruiz, A.; Bergami, A.; Libera, D.D.; Benussi, L.; Giussani, P.; Magnani, G.; Comi, G.; Legname, G.; et al. Microglia Convert Aggregated Amyloid-β into Neurotoxic Forms through the Shedding of Microvesicles. Cell Death Differ. 2014, 21, 582–593. [Google Scholar] [CrossRef]
- Clayton, K.; Delpech, J.C.; Herron, S.; Iwahara, N.; Ericsson, M.; Saito, T.; Saido, T.C.; Ikezu, S.; Ikezu, T. Plaque Associated Microglia Hyper-Secrete Extracellular Vesicles and Accelerate Tau Propagation in a Humanized APP Mouse Model. Mol. Neurodegener. 2021, 16, 18. [Google Scholar] [CrossRef]
- Xia, Y.; Zhang, G.; Han, C.; Ma, K.; Guo, X.; Wan, F.; Kou, L.; Yin, S.; Liu, L.; Huang, J.; et al. Microglia as Modulators of Exosomal Alpha-Synuclein Transmission. Cell Death Dis. 2019, 10, 174. [Google Scholar] [CrossRef]
- Ruan, Z.; Delpech, J.-C.; Venkatesan Kalavai, S.; Van Enoo, A.A.; Hu, J.; Ikezu, S.; Ikezu, T. P2RX7 Inhibitor Suppresses Exosome Secretion and Disease Phenotype in P301S Tau Transgenic Mice. Mol. Neurodegener. 2020, 15, 47. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.B.; Butler, M.S.; Rowe, D.B.; O’Neill, L.A.; et al. Inflammasome Inhibition Prevents α-Synuclein Pathology and Dopaminergic Neurodegeneration in Mice. Sci. Transl. Med. 2018, 10, eaah4066. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Ng, W.L.; Goh, S.Y.; Gulam, M.Y.; Wang, L.-F.; Tan, E.-K.; Ahn, M.; Chao, Y.-X. Targeting the Inflammasome in Parkinson’s Disease. Front. Aging Neurosci. 2022, 14, 957705. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 Is Activated in Alzheimer’s Disease and Contributes to Pathology in APP/PS1 Mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-Sensing Receptors in Sterile Inflammation and Inflammatory Diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Peruzzotti-Jametti, L.; Willis, C.M.; Krzak, G.; Hamel, R.; Pirvan, L.; Ionescu, R.-B.; Reisz, J.A.; Prag, H.A.; Garcia-Segura, M.E.; Wu, V.; et al. Mitochondrial Complex I Activity in Microglia Sustains Neuroinflammation. Nature 2024, 628, 195–203. [Google Scholar] [CrossRef]
- Suomalainen, A.; Nunnari, J. Mitochondria at the Crossroads of Health and Disease. Cell 2024, 187, 2601–2627. [Google Scholar] [CrossRef]
- Murphy, M.P.; O’Neill, L.A.J. A Break in Mitochondrial Endosymbiosis as a Basis for Inflammatory Diseases. Nature 2024, 626, 271–279. [Google Scholar] [CrossRef]
- Patergnani, S.; Bouhamida, E.; Leo, S.; Pinton, P.; Rimessi, A. Mitochondrial Oxidative Stress and “Mito-Inflammation”: Actors in the Diseases. Biomedicines 2021, 9, 216. [Google Scholar] [CrossRef]
- Park, J.; Min, J.-S.; Kim, B.; Chae, U.-B.; Yun, J.W.; Choi, M.-S.; Kong, I.-K.; Chang, K.-T.; Lee, D.-S. Mitochondrial ROS Govern the LPS-Induced pro-Inflammatory Response in Microglia Cells by Regulating MAPK and NF-κB Pathways. Neurosci. Lett. 2015, 584, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Kim, S.H.; Hamasaki, N. Mitochondrial Transcription Factor A (TFAM): Roles in Maintenance of mtDNA and Cellular Functions. Mitochondrion 2007, 7, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Alam, K.; Moinuddin; Jabeen, S. Immunogenicity of Mitochondrial DNA Modified by Hydroxyl Radical. Cell. Immunol. 2007, 247, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Xian, H.; Watari, K.; Sanchez-Lopez, E.; Offenberger, J.; Onyuru, J.; Sampath, H.; Ying, W.; Hoffman, H.M.; Shadel, G.S.; Karin, M. Oxidized DNA Fragments Exit Mitochondria via mPTP- and VDAC-Dependent Channels to Activate NLRP3 Inflammasome and Interferon Signaling. Immunity 2022, 55, 1370–1385.e8. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA Stress Primes the Antiviral Innate Immune Response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef]
- McArthur, K.; Whitehead, L.W.; Heddleston, J.M.; Li, L.; Padman, B.S.; Oorschot, V.; Geoghegan, N.D.; Chappaz, S.; Davidson, S.; San Chin, H.; et al. BAK/BAX Macropores Facilitate Mitochondrial Herniation and mtDNA Efflux during Apoptosis. Science 2018, 359, eaao6047. [Google Scholar] [CrossRef]
- Kim, J.; Gupta, R.; Blanco, L.P.; Yang, S.; Shteinfer-Kuzmine, A.; Wang, K.; Zhu, J.; Yoon, H.E.; Wang, X.; Kerkhofs, M.; et al. VDAC Oligomers Form Mitochondrial Pores to Release mtDNA Fragments and Promote Lupus-like Disease. Science 2019, 366, 1531–1536. [Google Scholar] [CrossRef]
- Newman, L.E.; Weiser Novak, S.; Rojas, G.R.; Tadepalle, N.; Schiavon, C.R.; Grotjahn, D.A.; Towers, C.G.; Tremblay, M.-È.; Donnelly, M.P.; Ghosh, S.; et al. Mitochondrial DNA Replication Stress Triggers a Pro-Inflammatory Endosomal Pathway of Nucleoid Disposal. Nat. Cell Biol. 2024, 26, 194–206. [Google Scholar] [CrossRef]
- Lu, T.; Zhang, Z.; Bi, Z.; Lan, T.; Zeng, H.; Liu, Y.; Mo, F.; Yang, J.; Chen, S.; He, X.; et al. TFAM Deficiency in Dendritic Cells Leads to Mitochondrial Dysfunction and Enhanced Antitumor Immunity through cGAS-STING Pathway. J. Immunother. Cancer 2023, 11, e005430. [Google Scholar] [CrossRef]
- Lepelley, A.; Della Mina, E.; Van Nieuwenhove, E.; Waumans, L.; Fraitag, S.; Rice, G.I.; Dhir, A.; Frémond, M.-L.; Rodero, M.P.; Seabra, L.; et al. Enhanced cGAS-STING-Dependent Interferon Signaling Associated with Mutations in ATAD3A. J. Exp. Med. 2021, 218, e20201560. [Google Scholar] [CrossRef] [PubMed]
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS-STING Pathway as a Therapeutic Target in Inflammatory Diseases. Nat. Rev. Immunol. 2021, 21, 548–569. [Google Scholar] [CrossRef] [PubMed]
- Stetson, D.B.; Ko, J.S.; Heidmann, T.; Medzhitov, R. Trex1 Prevents Cell-Intrinsic Initiation of Autoimmunity. Cell 2008, 134, 587–598. [Google Scholar] [CrossRef]
- Gehrke, N.; Mertens, C.; Zillinger, T.; Wenzel, J.; Bald, T.; Zahn, S.; Tüting, T.; Hartmann, G.; Barchet, W. Oxidative Damage of DNA Confers Resistance to Cytosolic Nuclease TREX1 Degradation and Potentiates STING-Dependent Immune Sensing. Immunity 2013, 39, 482–495. [Google Scholar] [CrossRef]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 Mitigate STING-Induced Inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef]
- Gulen, M.F.; Samson, N.; Keller, A.; Schwabenland, M.; Liu, C.; Glück, S.; Thacker, V.V.; Favre, L.; Mangeat, B.; Kroese, L.J.; et al. cGAS-STING Drives Ageing-Related Inflammation and Neurodegeneration. Nature 2023, 620, 374–380. [Google Scholar] [CrossRef]
- Jiménez-Loygorri, J.I.; Villarejo-Zori, B.; Viedma-Poyatos, Á.; Zapata-Muñoz, J.; Benítez-Fernández, R.; Frutos-Lisón, M.D.; Tomás-Barberán, F.A.; Espín, J.C.; Area-Gómez, E.; Gomez-Duran, A.; et al. Mitophagy Curtails Cytosolic mtDNA-Dependent Activation of cGAS/STING Inflammation during Aging. Nat. Commun. 2024, 15, 830. [Google Scholar] [CrossRef]
- Larrick, J.W.; Mendelsohn, A.R. Modulation of cGAS-STING Pathway by Nicotinamide Riboside in Alzheimer’s Disease. Rejuvenation Res. 2021, 24, 397–402. [Google Scholar] [CrossRef]
- Xie, X.; Ma, G.; Li, X.; Zhao, J.; Zhao, Z.; Zeng, J. Activation of Innate Immune cGAS-STING Pathway Contributes to Alzheimer’s Pathogenesis in 5×FAD Mice. Nat. Aging 2023, 3, 202–212. [Google Scholar] [CrossRef]
- Hinkle, J.T.; Patel, J.; Panicker, N.; Karuppagounder, S.S.; Biswas, D.; Belingon, B.; Chen, R.; Brahmachari, S.; Pletnikova, O.; Troncoso, J.C.; et al. STING Mediates Neurodegeneration and Neuroinflammation in Nigrostriatal α-Synucleinopathy. Proc. Natl. Acad. Sci. USA 2022, 119, e2118819119. [Google Scholar] [CrossRef]
- Ma, C.; Liu, Y.; Li, S.; Ma, C.; Huang, J.; Wen, S.; Yang, S.; Wang, B. Microglial cGAS Drives Neuroinflammation in the MPTP Mouse Models of Parkinson’s Disease. CNS Neurosci. Ther. 2023, 29, 2018–2035. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of proIL-Beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Tan, M.-S.; Tan, L.; Jiang, T.; Zhu, X.-C.; Wang, H.-F.; Jia, C.-D.; Yu, J.-T. Amyloid-β Induces NLRP1-Dependent Neuronal Pyroptosis in Models of Alzheimer’s Disease. Cell Death Dis. 2014, 5, e1382. [Google Scholar] [CrossRef]
- Lu, A.; Magupalli, V.G.; Ruan, J.; Yin, Q.; Atianand, M.K.; Vos, M.R.; Schröder, G.F.; Fitzgerald, K.A.; Wu, H.; Egelman, E.H. Unified Polymerization Mechanism for the Assembly of ASC-Dependent Inflammasomes. Cell 2014, 156, 1193–1206. [Google Scholar] [CrossRef]
- Walker, N.P.; Talanian, R.V.; Brady, K.D.; Dang, L.C.; Bump, N.J.; Ferenz, C.R.; Franklin, S.; Ghayur, T.; Hackett, M.C.; Hammill, L.D. Crystal Structure of the Cysteine Protease Interleukin-1 Beta-Converting Enzyme: A (P20/P10)2 Homodimer. Cell 1994, 78, 343–352. [Google Scholar] [CrossRef]
- Kuida, K.; Lippke, J.A.; Ku, G.; Harding, M.W.; Livingston, D.J.; Su, M.S.; Flavell, R.A. Altered Cytokine Export and Apoptosis in Mice Deficient in Interleukin-1 Beta Converting Enzyme. Science 1995, 267, 2000–2003. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Kalai, M.; Saelens, X.; Declercq, W.; Vandenabeele, P. Caspase-1 Activates Nuclear Factor of the Kappa-Enhancer in B Cells Independently of Its Enzymatic Activity. J. Biol. Chem. 2004, 279, 24785–24793. [Google Scholar] [CrossRef]
- Miggin, S.M.; Pålsson-McDermott, E.; Dunne, A.; Jefferies, C.; Pinteaux, E.; Banahan, K.; Murphy, C.; Moynagh, P.; Yamamoto, M.; Akira, S.; et al. NF-kappaB Activation by the Toll-IL-1 Receptor Domain Protein MyD88 Adapter-like Is Regulated by Caspase-1. Proc. Natl. Acad. Sci. USA 2007, 104, 3372–3377. [Google Scholar] [CrossRef]
- Boucher, D.; Monteleone, M.; Coll, R.C.; Chen, K.W.; Ross, C.M.; Teo, J.L.; Gomez, G.A.; Holley, C.L.; Bierschenk, D.; Stacey, K.J.; et al. Caspase-1 Self-Cleavage Is an Intrinsic Mechanism to Terminate Inflammasome Activity. J. Exp. Med. 2018, 215, 827–840. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, Q. Uncoupled Pyroptosis and IL-1β Secretion Downstream of Inflammasome Signaling. Front. Immunol. 2023, 14, 1128358. [Google Scholar] [CrossRef]
- Carty, M.; Kearney, J.; Shanahan, K.A.; Hams, E.; Sugisawa, R.; Connolly, D.; Doran, C.G.; Muñoz-Wolf, N.; Gürtler, C.; Fitzgerald, K.A.; et al. Cell Survival and Cytokine Release after Inflammasome Activation Is Regulated by the Toll-IL-1R Protein SARM. Immunity 2019, 50, 1412–1424.e6. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.; Wu, H. Structural Mechanisms of Inflammasome Assembly. FEBS J. 2015, 282, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Janowski, A.M.; Sutterwala, F.S. Atypical Inflammasomes. Methods Mol. Biol. 2016, 1417, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Ciążyńska, M.; Bednarski, I.A.; Wódz, K.; Narbutt, J.; Lesiak, A. NLRP1 and NLRP3 Inflammasomes as a New Approach to Skin Carcinogenesis. Oncol. Lett. 2020, 19, 1649–1656. [Google Scholar] [CrossRef]
- McKee, C.M.; Coll, R.C. NLRP3 Inflammasome Priming: A Riddle Wrapped in a Mystery inside an Enigma. J. Leukoc. Biol. 2020, 108, 937–952. [Google Scholar] [CrossRef]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting Edge: NF-kappaB Activating Pattern Recognition and Cytokine Receptors License NLRP3 Inflammasome Activation by Regulating NLRP3 Expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Shi, H.; Wang, Y.; Li, X.; Zhan, X.; Tang, M.; Fina, M.; Su, L.; Pratt, D.; Bu, C.H.; Hildebrand, S.; et al. NLRP3 Activation and Mitosis Are Mutually Exclusive Events Coordinated by NEK7, a New Inflammasome Component. Nat. Immunol. 2016, 17, 250–258. [Google Scholar] [CrossRef]
- Zhang, Y.; Luo, L.; Xu, X.; Wu, J.; Wang, F.; Lu, Y.; Zhang, N.; Ding, Y.; Lu, B.; Zhao, K. Acetylation Is Required for Full Activation of the NLRP3 Inflammasome. Nat. Commun. 2023, 14, 8396. [Google Scholar] [CrossRef]
- Niu, T.; De Rosny, C.; Chautard, S.; Rey, A.; Patoli, D.; Groslambert, M.; Cosson, C.; Lagrange, B.; Zhang, Z.; Visvikis, O.; et al. NLRP3 Phosphorylation in Its LRR Domain Critically Regulates Inflammasome Assembly. Nat. Commun. 2021, 12, 5862. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K+ Efflux Is the Common Trigger of NLRP3 Inflammasome Activation by Bacterial Toxins and Particulate Matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [PubMed]
- Katsnelson, M.A.; Lozada-Soto, K.M.; Russo, H.M.; Miller, B.A.; Dubyak, G.R. NLRP3 Inflammasome Signaling Is Activated by Low-Level Lysosome Disruption but Inhibited by Extensive Lysosome Disruption: Roles for K+ Efflux and Ca2+ Influx. Am. J. Physiol. Cell Physiol. 2016, 311, C83–C100. [Google Scholar] [CrossRef] [PubMed]
- Groß, C.J.; Mishra, R.; Schneider, K.S.; Médard, G.; Wettmarshausen, J.; Dittlein, D.C.; Shi, H.; Gorka, O.; Koenig, P.-A.; Fromm, S.; et al. K+ Efflux-Independent NLRP3 Inflammasome Activation by Small Molecules Targeting Mitochondria. Immunity 2016, 45, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-J.; Yoon, J.-H.; Ryu, J.-H. Mitophagy: A Balance Regulator of NLRP3 Inflammasome Activation. BMB Rep. 2016, 49, 529–535. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A Role for Mitochondria in NLRP3 Inflammasome Activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-κB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef]
- Maurya, S.R.; Mahalakshmi, R. VDAC-2: Mitochondrial Outer Membrane Regulator Masquerading as a Channel? FEBS J. 2016, 283, 1831–1836. [Google Scholar] [CrossRef]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical Role for Calcium Mobilization in Activation of the NLRP3 Inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-Interacting Protein Links Oxidative Stress to Inflammasome Activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Choi, E.-H.; Park, S.-J. TXNIP: A Key Protein in the Cellular Stress Response Pathway and a Potential Therapeutic Target. Exp. Mol. Med. 2023, 55, 1348–1356. [Google Scholar] [CrossRef]
- Hou, Y.; Wang, Y.; He, Q.; Li, L.; Xie, H.; Zhao, Y.; Zhao, J. Nrf2 Inhibits NLRP3 Inflammasome Activation Through Regulating Trx1/TXNIP Complex in Cerebral Ischemia Reperfusion Injury. Behav. Brain Res. 2018, 336, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Elliott, E.I.; Miller, A.N.; Banoth, B.; Iyer, S.S.; Stotland, A.; Weiss, J.P.; Gottlieb, R.A.; Sutterwala, F.S.; Cassel, S.L. Cutting Edge: Mitochondrial Assembly of the NLRP3 Inflammasome Complex Is Initiated at Priming. J. Immunol. 2018, 200, 3047–3052. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial Cardiolipin Is Required for Nlrp3 Inflammasome Activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Miao, R.; Jiang, C.; Chang, W.Y.; Zhang, H.; An, J.; Ho, F.; Chen, P.; Zhang, H.; Junqueira, C.; Amgalan, D.; et al. Gasdermin D Permeabilization of Mitochondrial Inner and Outer Membranes Accelerates and Enhances Pyroptosis. Immunity 2023, 56, 2523–2541.e8. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy Proteins Regulate Innate Immune Responses by Inhibiting the Release of Mitochondrial DNA Mediated by the NALP3 Inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.-J.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New Mitochondrial DNA Synthesis Enables NLRP3 Inflammasome Activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef]
- Dang, E.V.; McDonald, J.G.; Russell, D.W.; Cyster, J.G. Oxysterol Restraint of Cholesterol Synthesis Prevents AIM2 Inflammasome Activation. Cell 2017, 171, 1057–1071.e11. [Google Scholar] [CrossRef]
- Cookson, B.T.; Brennan, M.A. Pro-Inflammatory Programmed Cell Death. Trends Microbiol. 2001, 9, 113–114. [Google Scholar] [CrossRef]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef]
- He, W.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.-H.; Zhong, C.-Q.; Han, J. Gasdermin D Is an Executor of Pyroptosis and Required for Interleukin-1β Secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- DiPeso, L.; Ji, D.X.; Vance, R.E.; Price, J.V. Cell Death and Cell Lysis Are Separable Events During Pyroptosis. Cell Death Discov. 2017, 3, 17070. [Google Scholar] [CrossRef] [PubMed]
- Devant, P.; Boršić, E.; Ngwa, E.M.; Xiao, H.; Chouchani, E.T.; Thiagarajah, J.R.; Hafner-Bratkovič, I.; Evavold, C.L.; Kagan, J.C. Gasdermin D Pore-Forming Activity Is Redox-Sensitive. Cell Rep. 2023, 42, 112008. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, P.; Chen, Q.; Huang, Z.; Zou, D.; Zhang, J.; Gao, X.; Lin, Z. Mitochondrial ROS Promote Macrophage Pyroptosis by Inducing GSDMD Oxidation. J. Mol. Cell Biol. 2019, 11, 1069–1082. [Google Scholar] [CrossRef] [PubMed]
- Evavold, C.L.; Hafner-Bratkovič, I.; Devant, P.; D’Andrea, J.M.; Ngwa, E.M.; Boršić, E.; Doench, J.G.; LaFleur, M.W.; Sharpe, A.H.; Thiagarajah, J.R.; et al. Control of Gasdermin D Oligomerization and Pyroptosis by the Ragulator-Rag-mTORC1 Pathway. Cell 2021, 184, 4495–4511.e19. [Google Scholar] [CrossRef]
- Du, G.; Healy, L.B.; David, L.; Walker, C.; El-Baba, T.J.; Lutomski, C.A.; Goh, B.; Gu, B.; Pi, X.; Devant, P.; et al. ROS-Dependent S-Palmitoylation Activates Cleaved and Intact Gasdermin D. Nature 2024, 630, 437–446. [Google Scholar] [CrossRef]
- Rogers, C.; Erkes, D.A.; Nardone, A.; Aplin, A.E.; Fernandes-Alnemri, T.; Alnemri, E.S. Gasdermin Pores Permeabilize Mitochondria to Augment Caspase-3 Activation during Apoptosis and Inflammasome Activation. Nat. Commun. 2019, 10, 1689. [Google Scholar] [CrossRef]
- Miao, N.; Wang, Z.; Wang, Q.; Xie, H.; Yang, N.; Wang, Y.; Wang, J.; Kang, H.; Bai, W.; Wang, Y.; et al. Oxidized Mitochondrial DNA Induces Gasdermin D Oligomerization in Systemic Lupus Erythematosus. Nat. Commun. 2023, 14, 872. [Google Scholar] [CrossRef]
- Shen, H.; Han, C.; Yang, Y.; Guo, L.; Sheng, Y.; Wang, J.; Li, W.; Zhai, L.; Wang, G.; Guan, Q. Pyroptosis Executive Protein GSDMD as a Biomarker for Diagnosis and Identification of Alzheimer’s Disease. Brain Behav. 2021, 11, e02063. [Google Scholar] [CrossRef]
- Li, S.; Wu, Y.; Yang, D.; Wu, C.; Ma, C.; Liu, X.; Moynagh, P.N.; Wang, B.; Hu, G.; Yang, S. Gasdermin D in Peripheral Myeloid Cells Drives Neuroinflammation in Experimental Autoimmune Encephalomyelitis. J. Exp. Med. 2019, 216, 2562–2581. [Google Scholar] [CrossRef]
- Humphries, F.; Shmuel-Galia, L.; Ketelut-Carneiro, N.; Li, S.; Wang, B.; Nemmara, V.V.; Wilson, R.; Jiang, Z.; Khalighinejad, F.; Muneeruddin, K.; et al. Succination Inactivates Gasdermin D and Blocks Pyroptosis. Science 2020, 369, 1633–1637. [Google Scholar] [CrossRef]
- Zhang, D.; Qian, J.; Zhang, P.; Li, H.; Shen, H.; Li, X.; Chen, G. Gasdermin D Serves as a Key Executioner of Pyroptosis in Experimental Cerebral Ischemia and Reperfusion Model Both In Vivo and In Vitro. J. Neurosci. Res. 2019, 97, 645–660. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, X.; Qiao, S.; Yang, D.; Li, Z.; Xu, J.; Li, W.; Su, L.; Liu, W. Occludin Degradation Makes Brain Microvascular Endothelial Cells More Vulnerable to Reperfusion Injury In Vitro. J. Neurochem. 2021, 156, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Zhong, W.; Gu, Y.; Li, Y. Emerging Mechanisms and Targeted Therapy of Pyroptosis in Central Nervous System Trauma. Front. Cell Dev. Biol. 2022, 10, 832114. [Google Scholar] [CrossRef] [PubMed]
- Freeman, L.C.; Ting, J.P.-Y. The Pathogenic Role of the Inflammasome in Neurodegenerative Diseases. J. Neurochem. 2016, 136, 29–38. [Google Scholar] [CrossRef]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 Inflammasome Is Involved in the Innate Immune Response to Amyloid-Beta. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef]
- Lučiūnaitė, A.; McManus, R.M.; Jankunec, M.; Rácz, I.; Dansokho, C.; Dalgėdienė, I.; Schwartz, S.; Brosseron, F.; Heneka, M.T. Soluble Aβ Oligomers and Protofibrils Induce NLRP3 Inflammasome Activation in Microglia. J. Neurochem. 2020, 155, 650–661. [Google Scholar] [CrossRef]
- Stancu, I.-C.; Cremers, N.; Vanrusselt, H.; Couturier, J.; Vanoosthuyse, A.; Kessels, S.; Lodder, C.; Brône, B.; Huaux, F.; Octave, J.-N.; et al. Aggregated Tau Activates NLRP3-ASC Inflammasome Exacerbating Exogenously Seeded and Non-Exogenously Seeded Tau Pathology In Vivo. Acta Neuropathol. 2019, 137, 599–617. [Google Scholar] [CrossRef]
- Friker, L.L.; Scheiblich, H.; Hochheiser, I.V.; Brinkschulte, R.; Riedel, D.; Latz, E.; Geyer, M.; Heneka, M.T. β-Amyloid Clustering around ASC Fibrils Boosts Its Toxicity in Microglia. Cell Rep. 2020, 30, 3743–3754.e6. [Google Scholar] [CrossRef]
- Scheiblich, H.; Bousset, L.; Schwartz, S.; Griep, A.; Latz, E.; Melki, R.; Heneka, M.T. Microglial NLRP3 Inflammasome Activation upon TLR2 and TLR5 Ligation by Distinct α-Synuclein Assemblies. J. Immunol. 2021, 207, 2143–2154. [Google Scholar] [CrossRef]
- Codolo, G.; Plotegher, N.; Pozzobon, T.; Brucale, M.; Tessari, I.; Bubacco, L.; de Bernard, M. Triggering of Inflammasome by Aggregated α-Synuclein, an Inflammatory Response in Synucleinopathies. PLoS ONE 2013, 8, e55375. [Google Scholar] [CrossRef]
- von Herrmann, K.M.; Salas, L.A.; Martinez, E.M.; Young, A.L.; Howard, J.M.; Feldman, M.S.; Christensen, B.C.; Wilkins, O.M.; Lee, S.L.; Hickey, W.F.; et al. NLRP3 Expression in Mesencephalic Neurons and Characterization of a Rare NLRP3 Polymorphism Associated with Decreased Risk of Parkinson’s Disease. npj Park. Dis. 2018, 4, 24. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Pan, Y.-T.; Zhang, Z.-Y.; Yang, H.; Yu, S.-Y.; Zheng, Y.; Ma, J.-H.; Wang, X.-M. Systemic Activation of NLRP3 Inflammasome and Plasma α-Synuclein Levels Are Correlated with Motor Severity and Progression in Parkinson’s Disease. J. Neuroinflammation 2020, 17, 11. [Google Scholar] [CrossRef] [PubMed]
- Anderson, F.L.; von Herrmann, K.M.; Andrew, A.S.; Kuras, Y.I.; Young, A.L.; Scherzer, C.R.; Hickey, W.F.; Lee, S.L.; Havrda, M.C. Plasma-Borne Indicators of Inflammasome Activity in Parkinson’s Disease Patients. npj Park. Dis. 2021, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, H.; Yang, L.; Dong, X.; Han, Y.; Su, Y.; Li, W.; Li, W. Inhibition of NLRP1 Inflammasome Improves Autophagy Dysfunction and Aβ Disposition in APP/PS1 Mice. Behav. Brain Funct. 2023, 19, 7. [Google Scholar] [CrossRef]
- Huang, S.; Chen, Z.; Fan, B.; Chen, Y.; Zhou, L.; Jiang, B.; Long, H.; Zhong, W.; Li, X.; Li, Y. A Selective NLRP3 Inflammasome Inhibitor Attenuates Behavioral Deficits and Neuroinflammation in a Mouse Model of Parkinson’s Disease. J. Neuroimmunol. 2021, 354, 577543. [Google Scholar] [CrossRef]
- Wang, B.; Ma, Y.; Li, S.; Yao, H.; Gu, M.; Liu, Y.; Xue, Y.; Ding, J.; Ma, C.; Yang, S.; et al. GSDMD in Peripheral Myeloid Cells Regulates Microglial Immune Training and Neuroinflammation in Parkinson’s Disease. Acta Pharm. Sin. B 2023, 13, 2663–2679. [Google Scholar] [CrossRef]
- Yan, Y.; Jiang, W.; Liu, L.; Wang, X.; Ding, C.; Tian, Z.; Zhou, R. Dopamine Controls Systemic Inflammation through Inhibition of NLRP3 Inflammasome. Cell 2015, 160, 62–73. [Google Scholar] [CrossRef]
- Qiao, C.; Zhang, L.-X.; Sun, X.-Y.; Ding, J.-H.; Lu, M.; Hu, G. Caspase-1 Deficiency Alleviates Dopaminergic Neuronal Death via Inhibiting Caspase-7/AIF Pathway in MPTP/p Mouse Model of Parkinson’s Disease. Mol. Neurobiol. 2017, 54, 4292–4302. [Google Scholar] [CrossRef]
- Martinez, E.M.; Young, A.L.; Patankar, Y.R.; Berwin, B.L.; Wang, L.; von Herrmann, K.M.; Weier, J.M.; Havrda, M.C. Editor’s Highlight: Nlrp3 Is Required for Inflammatory Changes and Nigral Cell Loss Resulting from Chronic Intragastric Rotenone Exposure in Mice. Toxicol. Sci. 2017, 159, 64–75. [Google Scholar] [CrossRef]
- Mao, Z.; Liu, C.; Ji, S.; Yang, Q.; Ye, H.; Han, H.; Xue, Z. The NLRP3 Inflammasome Is Involved in the Pathogenesis of Parkinson’s Disease in Rats. Neurochem. Res. 2017, 42, 1104–1115. [Google Scholar] [CrossRef]
- Sarkar, S.; Malovic, E.; Harishchandra, D.S.; Ghaisas, S.; Panicker, N.; Charli, A.; Palanisamy, B.N.; Rokad, D.; Jin, H.; Anantharam, V.; et al. Mitochondrial Impairment in Microglia Amplifies NLRP3 Inflammasome Proinflammatory Signaling in Cell Culture and Animal Models of Parkinson’s Disease. npj Park. Dis. 2017, 3, 30. [Google Scholar] [CrossRef] [PubMed]
- de Dios, C.; Abadin, X.; Roca-Agujetas, V.; Jimenez-Martinez, M.; Morales, A.; Trullas, R.; Mari, M.; Colell, A. Inflammasome Activation under High Cholesterol Load Triggers a Protective Microglial Phenotype While Promoting Neuronal Pyroptosis. Transl. Neurodegener. 2023, 12, 10. [Google Scholar] [CrossRef] [PubMed]
- Barbero-Camps, E.; Fernández, A.; Martínez, L.; Fernández-Checa, J.C.; Colell, A. APP/PS1 Mice Overexpressing SREBP-2 Exhibit Combined Aβ Accumulation and Tau Pathology Underlying Alzheimer’s Disease. Hum. Mol. Genet. 2013, 22, 3460–3476. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Weinlich, R.; Oberst, A.; Beere, H.M.; Green, D.R. Necroptosis in Development, Inflammation and Disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 127–136. [Google Scholar] [CrossRef]
- Petrie, E.J.; Czabotar, P.E.; Murphy, J.M. The Structural Basis of Necroptotic Cell Death Signaling. Trends Biochem. Sci. 2019, 44, 53–63. [Google Scholar] [CrossRef]
- Lee, E.-W.; Seo, J.; Jeong, M.; Lee, S.; Song, J. The Roles of FADD in Extrinsic Apoptosis and Necroptosis. BMB Rep. 2012, 45, 496–508. [Google Scholar] [CrossRef]
- Kaiser, W.J.; Sridharan, H.; Huang, C.; Mandal, P.; Upton, J.W.; Gough, P.J.; Sehon, C.A.; Marquis, R.W.; Bertin, J.; Mocarski, E.S. Toll-like Receptor 3-Mediated Necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 2013, 288, 31268–31279. [Google Scholar] [CrossRef]
- Chen, X.-Y.; Dai, Y.-H.; Wan, X.-X.; Hu, X.-M.; Zhao, W.-J.; Ban, X.-X.; Wan, H.; Huang, K.; Zhang, Q.; Xiong, K. ZBP1-Mediated Necroptosis: Mechanisms and Therapeutic Implications. Molecules 2022, 28, 52. [Google Scholar] [CrossRef]
- Kang, T.-B.; Yang, S.-H.; Toth, B.; Kovalenko, A.; Wallach, D. Caspase-8 Blocks Kinase RIPK3-Mediated Activation of the NLRP3 Inflammasome. Immunity 2013, 38, 27–40. [Google Scholar] [CrossRef]
- Duong, B.H.; Onizawa, M.; Oses-Prieto, J.A.; Advincula, R.; Burlingame, A.; Malynn, B.A.; Ma, A. A20 Restricts Ubiquitination of Pro-Interleukin-1β Protein Complexes and Suppresses NLRP3 Inflammasome Activity. Immunity 2015, 42, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Conos, S.A.; Chen, K.W.; De Nardo, D.; Hara, H.; Whitehead, L.; Núñez, G.; Masters, S.L.; Murphy, J.M.; Schroder, K.; Vaux, D.L.; et al. Active MLKL Triggers the NLRP3 Inflammasome in a Cell-Intrinsic Manner. Proc. Natl. Acad. Sci. USA 2017, 114, E961–E969. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhou, T.; Sun, X.; Zheng, Y.; Cheng, B.; Li, M.; Liu, X.; He, C. Necroptosis in Microglia Contributes to Neuroinflammation and Retinal Degeneration Through TLR4 Activation. Cell Death Differ. 2018, 25, 180–189. [Google Scholar] [CrossRef]
- Orning, P.; Weng, D.; Starheim, K.; Ratner, D.; Best, Z.; Lee, B.; Brooks, A.; Xia, S.; Wu, H.; Kelliher, M.A.; et al. Pathogen Blockade of TAK1 Triggers Caspase-8-Dependent Cleavage of Gasdermin D and Cell Death. Science 2018, 362, 1064–1069. [Google Scholar] [CrossRef]
- Meng, Y.; Sandow, J.J.; Czabotar, P.E.; Murphy, J.M. The Regulation of Necroptosis by Post-Translational Modifications. Cell Death Differ. 2021, 28, 861–883. [Google Scholar] [CrossRef]
- Goodall, M.L.; Fitzwalter, B.E.; Zahedi, S.; Wu, M.; Rodriguez, D.; Mulcahy-Levy, J.M.; Green, D.R.; Morgan, M.; Cramer, S.D.; Thorburn, A. The Autophagy Machinery Controls Cell Death Switching Between Apoptosis and Necroptosis. Dev. Cell 2016, 37, 337–349. [Google Scholar] [CrossRef]
- Bray, K.; Mathew, R.; Lau, A.; Kamphorst, J.J.; Fan, J.; Chen, J.; Chen, H.-Y.; Ghavami, A.; Stein, M.; DiPaola, R.S.; et al. Autophagy Suppresses RIP Kinase-Dependent Necrosis Enabling Survival to mTOR Inhibition. PLoS ONE 2012, 7, e41831. [Google Scholar] [CrossRef]
- Seo, J.; Lee, E.-W.; Sung, H.; Seong, D.; Dondelinger, Y.; Shin, J.; Jeong, M.; Lee, H.-K.; Kim, J.-H.; Han, S.Y.; et al. CHIP Controls Necroptosis through Ubiquitylation- and Lysosome-Dependent Degradation of RIPK3. Nat. Cell Biol. 2016, 18, 291–302. [Google Scholar] [CrossRef]
- Liu, S.; Li, Y.; Choi, H.M.C.; Sarkar, C.; Koh, E.Y.; Wu, J.; Lipinski, M.M. Lysosomal Damage after Spinal Cord Injury Causes Accumulation of RIPK1 and RIPK3 Proteins and Potentiation of Necroptosis. Cell Death Dis. 2018, 9, 476. [Google Scholar] [CrossRef]
- Oshima, R.; Hasegawa, T.; Tamai, K.; Sugeno, N.; Yoshida, S.; Kobayashi, J.; Kikuchi, A.; Baba, T.; Futatsugi, A.; Sato, I.; et al. ESCRT-0 Dysfunction Compromises Autophagic Degradation of Protein Aggregates and Facilitates ER Stress-Mediated Neurodegeneration via Apoptotic and Necroptotic Pathways. Sci. Rep. 2016, 6, 24997. [Google Scholar] [CrossRef]
- Lee, S.B.; Kim, J.J.; Han, S.-A.; Fan, Y.; Guo, L.-S.; Aziz, K.; Nowsheen, S.; Kim, S.S.; Park, S.-Y.; Luo, Q.; et al. The AMPK-Parkin Axis Negatively Regulates Necroptosis and Tumorigenesis by Inhibiting the Necrosome. Nat. Cell Biol. 2019, 21, 940–951. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.-N.; Guy, C.; Olauson, H.; Becker, J.U.; Yang, M.; Fitzgerald, P.; Linkermann, A.; Green, D.R. ESCRT-III Acts Downstream of MLKL to Regulate Necroptotic Cell Death and Its Consequences. Cell 2017, 169, 286–300.e16. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.G.; Oberst, A.; Quarato, G.; Milasta, S.; Haller, M.; Wang, R.; Karvela, M.; Ichim, G.; Yatim, N.; Albert, M.L.; et al. Widespread Mitochondrial Depletion via Mitophagy Does Not Compromise Necroptosis. Cell Rep. 2013, 5, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Basit, F.; van Oppen, L.M.; Schöckel, L.; Bossenbroek, H.M.; van Emst-de Vries, S.E.; Hermeling, J.C.; Grefte, S.; Kopitz, C.; Heroult, M.; Hgm Willems, P.; et al. Mitochondrial Complex I Inhibition Triggers a Mitophagy-Dependent ROS Increase Leading to Necroptosis and Ferroptosis in Melanoma Cells. Cell Death Dis. 2017, 8, e2716. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lee, S.; Min, S.; Kang, S.W. RIP3-Dependent Accumulation of Mitochondrial Superoxide Anions in TNF-α-Induced Necroptosis. Mol. Cells 2022, 45, 193–201. [Google Scholar] [CrossRef]
- Zhang, Y.; Su, S.S.; Zhao, S.; Yang, Z.; Zhong, C.-Q.; Chen, X.; Cai, Q.; Yang, Z.-H.; Huang, D.; Wu, R.; et al. RIP1 Autophosphorylation Is Promoted by Mitochondrial ROS and Is Essential for RIP3 Recruitment into Necrosome. Nat. Commun. 2017, 8, 14329. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, Y.; Zhang, Y.; He, X.; Zhong, C.-Q.; Ni, H.; Chen, X.; Liang, Y.; Wu, J.; Zhao, S.; et al. RIP3 Targets Pyruvate Dehydrogenase Complex to Increase Aerobic Respiration in TNF-Induced Necroptosis. Nat. Cell Biol. 2018, 20, 186–197. [Google Scholar] [CrossRef]
- Reynoso, E.; Liu, H.; Li, L.; Yuan, A.L.; Chen, S.; Wang, Z. Thioredoxin-1 Actively Maintains the Pseudokinase MLKL in a Reduced State to Suppress Disulfide Bond-Dependent MLKL Polymer Formation and Necroptosis. J. Biol. Chem. 2017, 292, 17514–17524. [Google Scholar] [CrossRef]
- Rius-Pérez, S.; Pérez, S.; Toledano, M.B.; Sastre, J. P53 Drives Necroptosis via Downregulation of Sulfiredoxin and Peroxiredoxin 3. Redox Biol. 2022, 56, 102423. [Google Scholar] [CrossRef]
- Lu, W.; Karuppagounder, S.S.; Springer, D.A.; Allen, M.D.; Zheng, L.; Chao, B.; Zhang, Y.; Dawson, V.L.; Dawson, T.M.; Lenardo, M. Genetic Deficiency of the Mitochondrial Protein PGAM5 Causes a Parkinson’s-like Movement Disorder. Nat. Commun. 2014, 5, 4930. [Google Scholar] [CrossRef]
- Lu, W.; Sun, J.; Yoon, J.S.; Zhang, Y.; Zheng, L.; Murphy, E.; Mattson, M.P.; Lenardo, M.J. Mitochondrial Protein PGAM5 Regulates Mitophagic Protection Against Cell Necroptosis. PLoS ONE 2016, 11, e0147792. [Google Scholar] [CrossRef] [PubMed]
- Weindel, C.G.; Martinez, E.L.; Zhao, X.; Mabry, C.J.; Bell, S.L.; Vail, K.J.; Coleman, A.K.; VanPortfliet, J.J.; Zhao, B.; Wagner, A.R.; et al. Mitochondrial ROS Promotes Susceptibility to Infection via Gasdermin D-Mediated Necroptosis. Cell 2022, 185, 3214–3231.e23. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Tang, M.-B.; Luo, H.-Y.; Shi, C.-H.; Xu, Y.-M. Necroptosis in Neurodegenerative Diseases: A Potential Therapeutic Target. Cell Death Dis. 2017, 8, e2905. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.-R.; Wang, J.; Zhou, S.-K.; Yang, L.; Yin, J.; Cao, J.-P.; Cheng, Y.-B. Necrostatin-1 Protection of Dopaminergic Neurons. Neural Regen. Res. 2015, 10, 1120–1124. [Google Scholar] [CrossRef] [PubMed]
- Iannielli, A.; Bido, S.; Folladori, L.; Segnali, A.; Cancellieri, C.; Maresca, A.; Massimino, L.; Rubio, A.; Morabito, G.; Caporali, L.; et al. Pharmacological Inhibition of Necroptosis Protects from Dopaminergic Neuronal Cell Death in Parkinson’s Disease Models. Cell Rep. 2018, 22, 2066–2079. [Google Scholar] [CrossRef]
- Yang, S.-H.; Lee, D.K.; Shin, J.; Lee, S.; Baek, S.; Kim, J.; Jung, H.; Hah, J.-M.; Kim, Y. Nec-1 Alleviates Cognitive Impairment with Reduction of Aβ and Tau Abnormalities in APP/PS1 Mice. EMBO Mol. Med. 2017, 9, 61–77. [Google Scholar] [CrossRef]
- Caccamo, A.; Branca, C.; Piras, I.S.; Ferreira, E.; Huentelman, M.J.; Liang, W.S.; Readhead, B.; Dudley, J.T.; Spangenberg, E.E.; Green, K.N.; et al. Necroptosis Activation in Alzheimer’s Disease. Nat. Neurosci. 2017, 20, 1236–1246. [Google Scholar] [CrossRef]
- Geng, L.; Gao, W.; Saiyin, H.; Li, Y.; Zeng, Y.; Zhang, Z.; Li, X.; Liu, Z.; Gao, Q.; An, P.; et al. MLKL Deficiency Alleviates Neuroinflammation and Motor Deficits in the α-Synuclein Transgenic Mouse Model of Parkinson’s Disease. Mol. Neurodegener. 2023, 18, 94. [Google Scholar] [CrossRef]
- Leem, Y.-H.; Kim, D.-Y.; Park, J.-E.; Kim, H.-S. Necrosulfonamide Exerts Neuroprotective Effect by Inhibiting Necroptosis, Neuroinflammation, and α-Synuclein Oligomerization in a Subacute MPTP Mouse Model of Parkinson’s Disease. Sci. Rep. 2023, 13, 8783. [Google Scholar] [CrossRef]
- Oñate, M.; Catenaccio, A.; Salvadores, N.; Saquel, C.; Martinez, A.; Moreno-Gonzalez, I.; Gamez, N.; Soto, P.; Soto, C.; Hetz, C.; et al. The Necroptosis Machinery Mediates Axonal Degeneration in a Model of Parkinson Disease. Cell Death Differ. 2020, 27, 1169–1185. [Google Scholar] [CrossRef]
- Jayaraman, A.; Htike, T.T.; James, R.; Picon, C.; Reynolds, R. TNF-Mediated Neuroinflammation Is Linked to Neuronal Necroptosis in Alzheimer’s Disease Hippocampus. Acta Neuropathol. Commun. 2021, 9, 159. [Google Scholar] [CrossRef] [PubMed]
- Koper, M.J.; Van Schoor, E.; Ospitalieri, S.; Vandenberghe, R.; Vandenbulcke, M.; von Arnim, C.A.F.; Tousseyn, T.; Balusu, S.; De Strooper, B.; Thal, D.R. Necrosome Complex Detected in Granulovacuolar Degeneration Is Associated with Neuronal Loss in Alzheimer’s Disease. Acta Neuropathol. 2020, 139, 463–484. [Google Scholar] [CrossRef] [PubMed]
- Wiersma, V.I.; van Ziel, A.M.; Vazquez-Sanchez, S.; Nölle, A.; Berenjeno-Correa, E.; Bonaterra-Pastra, A.; Clavaguera, F.; Tolnay, M.; Musters, R.J.P.; van Weering, J.R.T.; et al. Granulovacuolar Degeneration Bodies Are Neuron-Selective Lysosomal Structures Induced by Intracellular Tau Pathology. Acta Neuropathol. 2019, 138, 943–970. [Google Scholar] [CrossRef] [PubMed]
- Salvadores, N.; Moreno-Gonzalez, I.; Gamez, N.; Quiroz, G.; Vegas-Gomez, L.; Escandón, M.; Jimenez, S.; Vitorica, J.; Gutierrez, A.; Soto, C.; et al. Aβ Oligomers Trigger Necroptosis-Mediated Neurodegeneration via Microglia Activation in Alzheimer’s Disease. Acta Neuropathol. Commun. 2022, 10, 31. [Google Scholar] [CrossRef]
- Balusu, S.; Horré, K.; Thrupp, N.; Craessaerts, K.; Snellinx, A.; Serneels, L.; T’Syen, D.; Chrysidou, I.; Arranz, A.M.; Sierksma, A.; et al. MEG3 Activates Necroptosis in Human Neuron Xenografts Modeling Alzheimer’s Disease. Science 2023, 381, 1176–1182. [Google Scholar] [CrossRef]
- Chan, H.-H.; Leong, C.-O.; Lim, C.-L.; Koh, R.-Y. Roles of Receptor-Interacting Protein Kinase 1 in SH-SY5Y Cells with Beta Amyloid-Induced Neurotoxicity. J. Cell Mol. Med. 2022, 26, 1434–1444. [Google Scholar] [CrossRef]
- Dong, Y.; Yu, H.; Li, X.; Bian, K.; Zheng, Y.; Dai, M.; Feng, X.; Sun, Y.; He, Y.; Yu, B.; et al. Hyperphosphorylated Tau Mediates Neuronal Death by Inducing Necroptosis and Inflammation in Alzheimer’s Disease. J. Neuroinflammation 2022, 19, 205. [Google Scholar] [CrossRef]
- Yu, H.; Morihara, R.; Ota-Elliott, R.; Bian, Z.; Bian, Y.; Hu, X.; Sun, H.; Fukui, Y.; Abe, K.; Ishiura, H.; et al. Injection of Exogenous Amyloid-β Oligomers Aggravated Cognitive Deficits, and Activated Necroptosis, in APP23 Transgenic Mice. Brain Res. 2023, 1821, 148565. [Google Scholar] [CrossRef]
- Tu, J.-L.; Chen, W.-P.; Cheng, Z.-J.; Zhang, G.; Luo, Q.-H.; Li, M.; Liu, X. EGb761 Ameliorates Cell Necroptosis by Attenuating RIP1-Mediated Mitochondrial Dysfunction and ROS Production in Both In Vivo and In Vitro Models of Alzheimer’s Disease. Brain Res. 2020, 1736, 146730. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Hirschhorn, T.; Stockwell, B.R. The Development of the Concept of Ferroptosis. Free Radic. Biol. Med. 2019, 133, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.-J.; Ikeda, M.; Ide, T.; Hur, K.Y.; Lee, M.-S. Mitochondrial Event as an Ultimate Step in Ferroptosis. Cell Death Discov. 2022, 8, 414. [Google Scholar] [CrossRef] [PubMed]
- Lyamzaev, K.G.; Panteleeva, A.A.; Simonyan, R.A.; Avetisyan, A.V.; Chernyak, B.V. Mitochondrial Lipid Peroxidation Is Responsible for Ferroptosis. Cells 2023, 12, 611. [Google Scholar] [CrossRef] [PubMed]
- Merkel, M.; Goebel, B.; Boll, M.; Adhikari, A.; Maurer, V.; Steinhilber, D.; Culmsee, C. Mitochondrial Reactive Oxygen Species Formation Determines ACSL4/LPCAT2-Mediated Ferroptosis. Antioxidants 2023, 12, 1590. [Google Scholar] [CrossRef] [PubMed]
- Tsurusaki, S.; Tsuchiya, Y.; Koumura, T.; Nakasone, M.; Sakamoto, T.; Matsuoka, M.; Imai, H.; Yuet-Yin Kok, C.; Okochi, H.; Nakano, H.; et al. Hepatic Ferroptosis Plays an Important Role as the Trigger for Initiating Inflammation in Nonalcoholic Steatohepatitis. Cell Death Dis. 2019, 10, 449. [Google Scholar] [CrossRef]
- Li, W.; Feng, G.; Gauthier, J.M.; Lokshina, I.; Higashikubo, R.; Evans, S.; Liu, X.; Hassan, A.; Tanaka, S.; Cicka, M.; et al. Ferroptotic Cell Death and TLR4/Trif Signaling Initiate Neutrophil Recruitment after Heart Transplantation. J. Clin. Investig. 2019, 129, 2293–2304. [Google Scholar] [CrossRef]
- Gupta, U.; Ghosh, S.; Wallace, C.T.; Shang, P.; Xin, Y.; Nair, A.P.; Yazdankhah, M.; Strizhakova, A.; Ross, M.A.; Liu, H.; et al. Increased LCN2 (Lipocalin 2) in the RPE Decreases Autophagy and Activates Inflammasome-Ferroptosis Processes in a Mouse Model of Dry AMD. Autophagy 2023, 19, 92–111. [Google Scholar] [CrossRef]
- Xie, S.-S.; Deng, Y.; Guo, S.-L.; Li, J.-Q.; Zhou, Y.-C.; Liao, J.; Wu, D.-D.; Lan, W.-F. Endothelial Cell Ferroptosis Mediates Monocrotaline-Induced Pulmonary Hypertension in Rats by Modulating NLRP3 Inflammasome Activation. Sci. Rep. 2022, 12, 3056. [Google Scholar] [CrossRef]
- Li, C.; Deng, X.; Xie, X.; Liu, Y.; Friedmann Angeli, J.P.; Lai, L. Activation of Glutathione Peroxidase 4 as a Novel Anti-Inflammatory Strategy. Front. Pharmacol. 2018, 9, 1120. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Xiao, Z.; Kong, B.; Fang, J.; Qin, T.; Dai, C.; Shuai, W.; Huang, H. Ferrostatin-1 Alleviates Lipopolysaccharide-Induced Cardiac Dysfunction. Bioengineered 2021, 12, 9367–9376. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zeng, L.; Zhu, S.; Xie, Y.; Liu, J.; Wen, Q.; Cao, L.; Xie, M.; Ran, Q.; Kroemer, G.; et al. Lipid Peroxidation Drives Gasdermin D-Mediated Pyroptosis in Lethal Polymicrobial Sepsis. Cell Host Microbe 2018, 24, 97–108.e4. [Google Scholar] [CrossRef] [PubMed]
- Fan, R.; Sui, J.; Dong, X.; Jing, B.; Gao, Z. Wedelolactone Alleviates Acute Pancreatitis and Associated Lung Injury via GPX4 Mediated Suppression of Pyroptosis and Ferroptosis. Free Radic. Biol. Med. 2021, 173, 29–40. [Google Scholar] [CrossRef]
- Fu, Y.; He, Y.; Phan, K.; Bhatia, S.; Pickford, R.; Wu, P.; Dzamko, N.; Halliday, G.M.; Kim, W.S. Increased Unsaturated Lipids Underlie Lipid Peroxidation in Synucleinopathy Brain. Acta Neuropathol. Commun. 2022, 10, 165. [Google Scholar] [CrossRef]
- Emre, C.; Do, K.V.; Jun, B.; Hjorth, E.; Alcalde, S.G.; Kautzmann, M.-A.I.; Gordon, W.C.; Nilsson, P.; Bazan, N.G.; Schultzberg, M. Age-Related Changes in Brain Phospholipids and Bioactive Lipids in the APP Knock-in Mouse Model of Alzheimer’s Disease. Acta Neuropathol. Commun. 2021, 9, 116. [Google Scholar] [CrossRef]
- Ayton, S.; Wang, Y.; Diouf, I.; Schneider, J.A.; Brockman, J.; Morris, M.C.; Bush, A.I. Brain Iron Is Associated with Accelerated Cognitive Decline in People with Alzheimer Pathology. Mol. Psychiatry 2020, 25, 2932–2941. [Google Scholar] [CrossRef]
- Mahoney-Sanchez, L.; Bouchaoui, H.; Boussaad, I.; Jonneaux, A.; Timmerman, K.; Berdeaux, O.; Ayton, S.; Krüger, R.; Duce, J.A.; Devos, D.; et al. Alpha Synuclein Determines Ferroptosis Sensitivity in Dopaminergic Neurons via Modulation of Ether-Phospholipid Membrane Composition. Cell Rep. 2022, 40, 111231. [Google Scholar] [CrossRef]
- Greenough, M.A.; Lane, D.J.R.; Balez, R.; Anastacio, H.T.D.; Zeng, Z.; Ganio, K.; McDevitt, C.A.; Acevedo, K.; Belaidi, A.A.; Koistinaho, J.; et al. Selective Ferroptosis Vulnerability Due to Familial Alzheimer’s Disease Presenilin Mutations. Cell Death Differ. 2022, 29, 2123–2136. [Google Scholar] [CrossRef]
- Park, M.W.; Cha, H.W.; Kim, J.; Kim, J.H.; Yang, H.; Yoon, S.; Boonpraman, N.; Yi, S.S.; Yoo, I.D.; Moon, J.-S. NOX4 Promotes Ferroptosis of Astrocytes by Oxidative Stress-Induced Lipid Peroxidation via the Impairment of Mitochondrial Metabolism in Alzheimer’s Diseases. Redox Biol. 2021, 41, 101947. [Google Scholar] [CrossRef]
- Marmolejo-Garza, A.; Krabbendam, I.E.; Luu, M.D.A.; Brouwer, F.; Trombetta-Lima, M.; Unal, O.; O’Connor, S.J.; Majerníková, N.; Elzinga, C.R.S.; Mammucari, C.; et al. Negative Modulation of Mitochondrial Calcium Uniporter Complex Protects Neurons against Ferroptosis. Cell Death Dis. 2023, 14, 772. [Google Scholar] [CrossRef]
- Chen, T.; Majerníková, N.; Marmolejo-Garza, A.; Trombetta-Lima, M.; Sabogal-Guáqueta, A.M.; Zhang, Y.; Ten Kate, R.; Zuidema, M.; Mulder, P.P.M.F.A.; den Dunnen, W.; et al. Mitochondrial Transplantation Rescues Neuronal Cells from Ferroptosis. Free Radic. Biol. Med. 2023, 208, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.K.; Zelic, M.; Han, Y.; Teeple, E.; Chen, L.; Sadeghi, M.; Shankara, S.; Guo, L.; Li, C.; Pontarelli, F.; et al. Microglia Ferroptosis Is Regulated by SEC24B and Contributes to Neurodegeneration. Nat. Neurosci. 2023, 26, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.K.; Ugalde, C.L.; Rolland, A.-S.; Skidmore, J.; Devos, D.; Hammond, T.R. Therapeutic Inhibition of Ferroptosis in Neurodegenerative Disease. Trends Pharmacol. Sci. 2023, 44, 674–688. [Google Scholar] [CrossRef]
- Ruiz, L.M.; Libedinsky, A.; Elorza, A.A. Role of Copper on Mitochondrial Function and Metabolism. Front. Mol. Biosci. 2021, 8, 711227. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper Induces Cell Death by Targeting Lipoylated TCA Cycle Proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, Q.; Lu, L.; Su, Y.; Shi, W.; Zhang, H.; Liu, R.; Pu, Y.; Yin, L. Copper Induces Cognitive Impairment in Mice via Modulation of Cuproptosis and CREB Signaling. Nutrients 2023, 15, 972. [Google Scholar] [CrossRef]
- Zhao, S.; Zhang, L.; Ji, W.; Shi, Y.; Lai, G.; Chi, H.; Huang, W.; Cheng, C. Machine Learning-Based Characterization of Cuprotosis-Related Biomarkers and Immune Infiltration in Parkinson’s Disease. Front. Genet. 2022, 13, 1010361. [Google Scholar] [CrossRef]
- Wu, J.; Qin, C.; Cai, Y.; Zhou, J.; Xu, D.; Lei, Y.; Fang, G.; Chai, S.; Xiong, N. Machine Learning Screening for Parkinson’s Disease-Related Cuproptosis-Related Typing Development and Validation and Exploration of Personalized Drugs for Cuproptosis Genes. Ann. Transl. Med. 2023, 11, 11. [Google Scholar] [CrossRef]
- Zhang, M.; Meng, W.; Liu, C.; Wang, H.; Li, R.; Wang, Q.; Gao, Y.; Zhou, S.; Du, T.; Yuan, T.; et al. Identification of Cuproptosis Clusters and Integrative Analyses in Parkinson’s Disease. Brain Sci. 2023, 13, 1015. [Google Scholar] [CrossRef]
- Lai, Y.; Lin, C.; Lin, X.; Wu, L.; Zhao, Y.; Lin, F. Identification and Immunological Characterization of Cuproptosis-Related Molecular Clusters in Alzheimer’s Disease. Front. Aging Neurosci. 2022, 14, 932676. [Google Scholar] [CrossRef]
- Deng, H.; Zhu, S.; Yang, H.; Cui, H.; Guo, H.; Deng, J.; Ren, Z.; Geng, Y.; Ouyang, P.; Xu, Z.; et al. The Dysregulation of Inflammatory Pathways Triggered by Copper Exposure. Biol. Trace Elem Res. 2023, 201, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Yu, F.; Gong, P.; Qiu, Y.; Zhou, W.; Cui, Y.; Li, J.; Chen, H. Subneurotoxic Copper(II)-Induced NF-κB-Dependent Microglial Activation Is Associated with Mitochondrial ROS. Toxicol. Appl. Pharmacol. 2014, 276, 95–103. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhang, Y.; Lu, L.; Zhang, H.; Zhao, C.; Pu, Y.; Yin, L. Copper Induces Microglia-Mediated Neuroinflammation through ROS/NF-κB Pathway and Mitophagy Disorder. Food Chem. Toxicol. 2022, 168, 113369. [Google Scholar] [CrossRef] [PubMed]
- Deigendesch, N.; Zychlinsky, A.; Meissner, F. Copper Regulates the Canonical NLRP3 Inflammasome. J. Immunol. 2018, 200, 1607–1617. [Google Scholar] [CrossRef] [PubMed]
- Kitazawa, M.; Hsu, H.-W.; Medeiros, R. Copper Exposure Perturbs Brain Inflammatory Responses and Impairs Clearance of Amyloid-Beta. Toxicol. Sci. 2016, 152, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Gong, P.; Hu, Z.; Qiu, Y.; Cui, Y.; Gao, X.; Chen, H.; Li, J. Cu(II) Enhances the Effect of Alzheimer’s Amyloid-β Peptide on Microglial Activation. J. Neuroinflammation 2015, 12, 122. [Google Scholar] [CrossRef]
- Lim, S.L.; Rodriguez-Ortiz, C.J.; Hsu, H.-W.; Wu, J.; Zumkehr, J.; Kilian, J.; Vidal, J.; Ayata, P.; Kitazawa, M. Chronic Copper Exposure Directs Microglia towards Degenerative Expression Signatures in Wild-Type and J20 Mouse Model of Alzheimer’s Disease. J. Trace Elem. Med. Biol. 2020, 62, 126578. [Google Scholar] [CrossRef]
- Sales, T.A.; Prandi, I.G.; de Castro, A.A.; Leal, D.H.S.; da Cunha, E.F.F.; Kuca, K.; Ramalho, T.C. Recent Developments in Metal-Based Drugs and Chelating Agents for Neurodegenerative Diseases Treatments. Int. J. Mol. Sci. 2019, 20, 1829. [Google Scholar] [CrossRef]
- Zahid, A.; Li, B.; Kombe, A.J.K.; Jin, T.; Tao, J. Pharmacological Inhibitors of the NLRP3 Inflammasome. Front. Immunol. 2019, 10, 2538. [Google Scholar] [CrossRef]
- Gao, C.; Jiang, J.; Tan, Y.; Chen, S. Microglia in Neurodegenerative Diseases: Mechanism and Potential Therapeutic Targets. Signal Transduct. Target. Ther. 2023, 8, 359. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, B.; Sinha, S.C.; Amin, S.; Gan, L. Mechanism and Therapeutic Potential of Targeting cGAS-STING Signaling in Neurological Disorders. Mol. Neurodegener. 2023, 18, 79. [Google Scholar] [CrossRef] [PubMed]
- Long, H.; Simmons, A.; Mayorga, A.; Burgess, B.; Nguyen, T.; Budda, B.; Rychkova, A.; Rhinn, H.; Tassi, I.; Ward, M.; et al. Preclinical and First-in-Human Evaluation of AL002, a Novel TREM2 Agonistic Antibody for Alzheimer’s Disease. Alzheimers Res. Ther. 2024, 16, 235. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Cai, L.; Yao, J.; Li, C.; Wang, X. Agonists and Inhibitors of the cGAS-STING Pathway. Molecules 2024, 29, 3121. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. Biotechs Step on cGAS for Autoimmune Diseases. Nat. Rev. Drug Discov. 2023, 22, 939–941. [Google Scholar] [CrossRef]
- Ashok, A.; Andrabi, S.S.; Mansoor, S.; Kuang, Y.; Kwon, B.K.; Labhasetwar, V. Antioxidant Therapy in Oxidative Stress-Induced Neurodegenerative Diseases: Role of Nanoparticle-Based Drug Delivery Systems in Clinical Translation. Antioxidants 2022, 11, 408. [Google Scholar] [CrossRef]
- Kwon, H.J.; Cha, M.-Y.; Kim, D.; Kim, D.K.; Soh, M.; Shin, K.; Hyeon, T.; Mook-Jung, I. Mitochondria-Targeting Ceria Nanoparticles as Antioxidants for Alzheimer’s Disease. ACS Nano 2016, 10, 2860–2870. [Google Scholar] [CrossRef]
- Swarnakar, N.K.; Jain, A.K.; Singh, R.P.; Godugu, C.; Das, M.; Jain, S. Oral Bioavailability, Therapeutic Efficacy and Reactive Oxygen Species Scavenging Properties of Coenzyme Q10-Loaded Polymeric Nanoparticles. Biomaterials 2011, 32, 6860–6874. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, P.; Yue, C.; Jin, Z.; Liu, Q.; Du, X.; He, Q. Sustained Release of Bioactive Hydrogen by Pd Hydride Nanoparticles Overcomes Alzheimer’s Disease. Biomaterials 2019, 197, 393–404. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abadin, X.; de Dios, C.; Zubillaga, M.; Ivars, E.; Puigròs, M.; Marí, M.; Morales, A.; Vizuete, M.; Vitorica, J.; Trullas, R.; et al. Neuroinflammation in Age-Related Neurodegenerative Diseases: Role of Mitochondrial Oxidative Stress. Antioxidants 2024, 13, 1440. https://doi.org/10.3390/antiox13121440
Abadin X, de Dios C, Zubillaga M, Ivars E, Puigròs M, Marí M, Morales A, Vizuete M, Vitorica J, Trullas R, et al. Neuroinflammation in Age-Related Neurodegenerative Diseases: Role of Mitochondrial Oxidative Stress. Antioxidants. 2024; 13(12):1440. https://doi.org/10.3390/antiox13121440
Chicago/Turabian StyleAbadin, Xenia, Cristina de Dios, Marlene Zubillaga, Elia Ivars, Margalida Puigròs, Montserrat Marí, Albert Morales, Marisa Vizuete, Javier Vitorica, Ramon Trullas, and et al. 2024. "Neuroinflammation in Age-Related Neurodegenerative Diseases: Role of Mitochondrial Oxidative Stress" Antioxidants 13, no. 12: 1440. https://doi.org/10.3390/antiox13121440
APA StyleAbadin, X., de Dios, C., Zubillaga, M., Ivars, E., Puigròs, M., Marí, M., Morales, A., Vizuete, M., Vitorica, J., Trullas, R., Colell, A., & Roca-Agujetas, V. (2024). Neuroinflammation in Age-Related Neurodegenerative Diseases: Role of Mitochondrial Oxidative Stress. Antioxidants, 13(12), 1440. https://doi.org/10.3390/antiox13121440