
Effects of L-Type Voltage-Gated Calcium Channel (LTCC) Inhibition on Hippocampal Neuronal Death after Pilocarpine-Induced Seizure

 , ,
, ,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement and Care of Experimental Animals
2.2. Seizure Induction
2.3. Amlodipine Administration
2.4. Brain Sample Preparation
2.5. Detection of Zinc Accumulation
2.6. Detection of Oxidative Stress in the Hippocampal Region
2.7. Immunofluorescence Assay
2.8. Immunohistochemistry Assay
2.9. Behavior Test
2.9.1. Barnes Maze Test
2.9.2. Adhesive Removal Test
2.10. Data Analysis
3. Results
3.1. Amlodipine Administration Reduced Cav 1.2 Activation and Zinc Accumulation in Neurons Following Seizure
3.2. Administration of Amlodipine Decreased Reactive Oxidative Stress (ROS) after Seizure
3.3. Amlodipine Administration Increased Neuron Survival after Seizure
3.4. Amlodipine Reduced Blood–Brain Barrier Breakdown after Seizure
3.5. Amlodipine Administration Reduced Astrocyte Over-Activation after Seizure
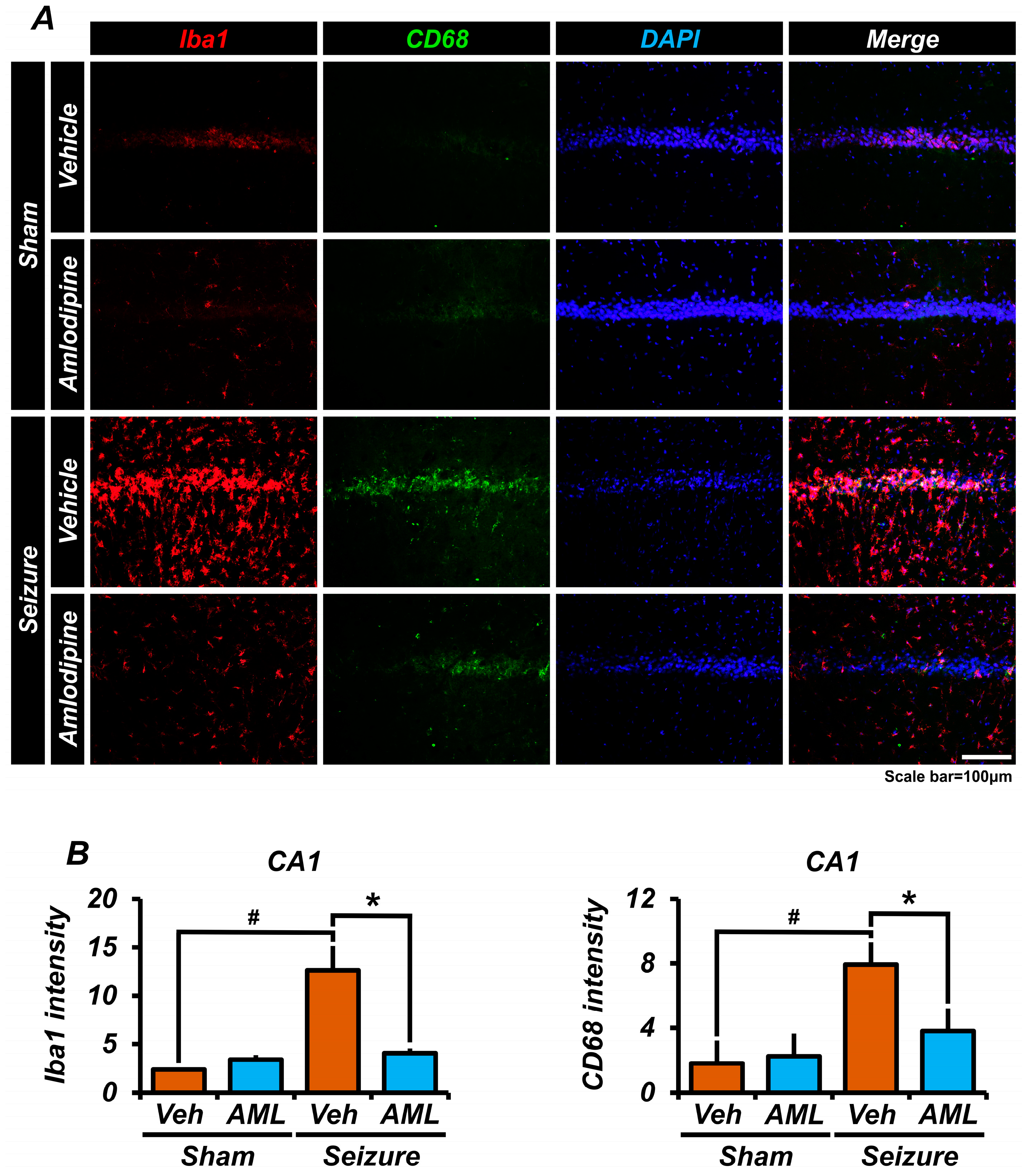
3.6. Amlodipine Administration Reduced Microglia Over-Activation after Seizure
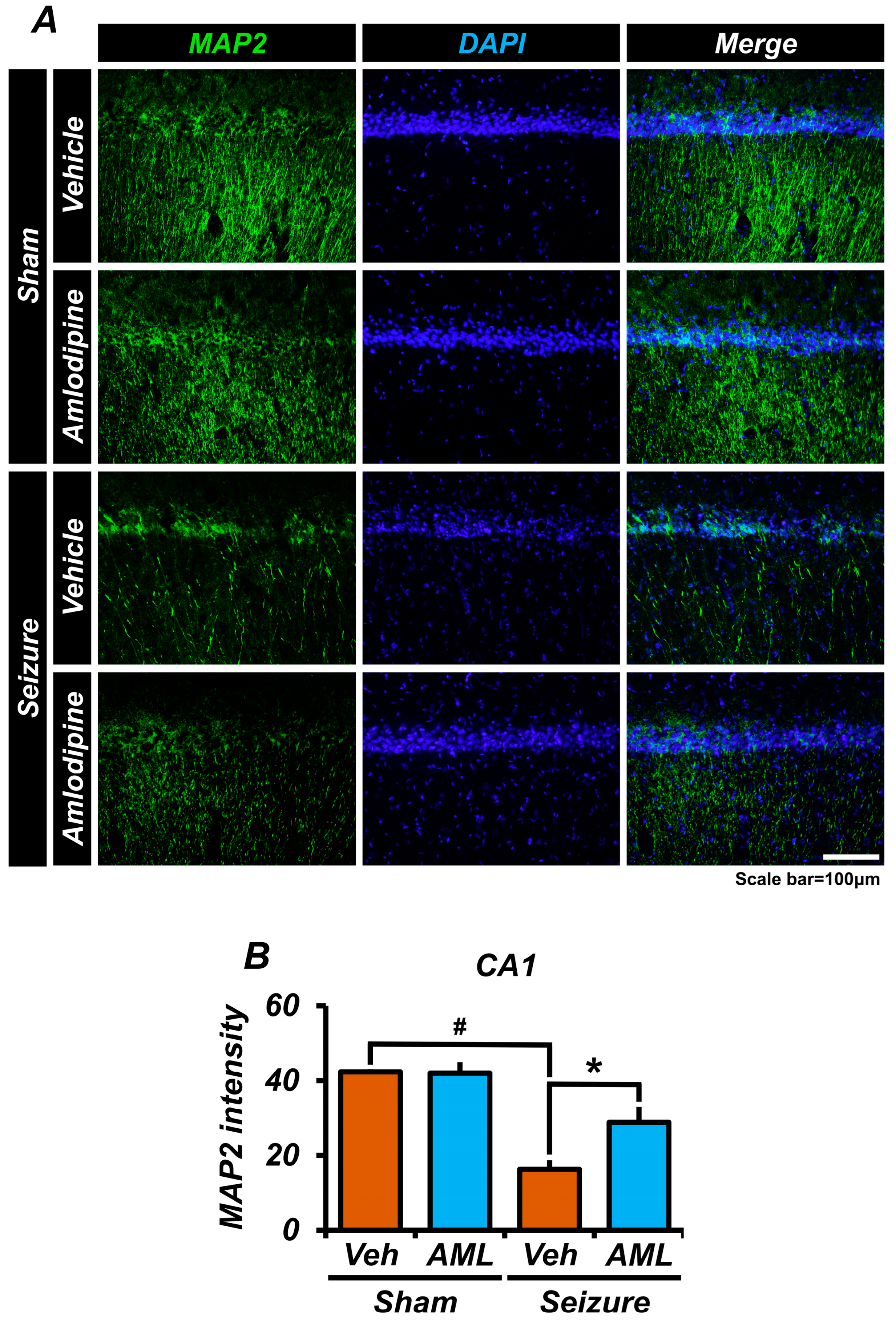
3.7. Amlodipine Administration Reduced Microtubule Disruption after Seizure
3.8. Amlodipine Administration Improved Spatial Cognitive, Memory, and Cognitive Function Recovery after Seizure
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arnold, S.T.; Dodson, W.E. Epilepsy in children. Baillieres Clin. Neurol. 1996, 5, 783–802. [Google Scholar] [PubMed]
- Goldberg, E.M.; Coulter, D.A. Mechanisms of epileptogenesis: A convergence on neural circuit dysfunction. Nat. Rev. Neurosci. 2013, 14, 337–349. [Google Scholar] [CrossRef]
- Shen, Z.; Haragopal, H.; Li, Y.V. Zinc modulates synaptic transmission by differentially regulating synaptic glutamate homeostasis in hippocampus. Eur. J. Neurosci. 2020, 52, 3710–3722. [Google Scholar] [CrossRef]
- Stefan, H. Pathophysiology of human epilepsy: Imaging and physiologic studies. Curr. Opin. Neurol. 2000, 13, 177–181. [Google Scholar] [CrossRef]
- Jeong, J.H.; Lee, S.H.; Kho, A.R.; Hong, D.K.; Kang, D.H.; Kang, B.S.; Park, M.K.; Choi, B.Y.; Choi, H.C.; Lim, M.S.; et al. The Transient Receptor Potential Melastatin 7 (TRPM7) Inhibitors Suppress Seizure-Induced Neuron Death by Inhibiting Zinc Neurotoxicity. Int. J. Mol. Sci. 2020, 21, 7897. [Google Scholar] [CrossRef]
- Lee, S.H.; Choi, B.Y.; Kho, A.R.; Hong, D.K.; Kang, B.S.; Park, M.K.; Lee, S.H.; Choi, H.C.; Song, H.K.; Suh, S.W. Combined Treatment of Dichloroacetic Acid and Pyruvate Increased Neuronal Survival after Seizure. Nutrients 2022, 14, 4804. [Google Scholar] [CrossRef]
- Wahl-Schott, C.; Baumann, L.; Cuny, H.; Eckert, C.; Griessmeier, K.; Biel, M. Switching off calcium-dependent inactivation in L-type calcium channels by an autoinhibitory domain. Proc. Natl. Acad. Sci. USA 2006, 103, 15657–15662. [Google Scholar] [CrossRef]
- Park, S.J.; Min, S.H.; Kang, H.W.; Lee, J.H. Differential zinc permeation and blockade of L-type Ca2+ channel isoforms Cav1.2 and Cav1.3. Biochim. Biophys. Acta 2015, 1848, 2092–2100. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.M.; Bartsch, D. The role of L-type voltage-gated calcium channels Cav1.2 and Cav1.3 in normal and pathological brain function. Cell Tissue Res. 2014, 357, 463–476. [Google Scholar] [CrossRef] [PubMed]
- Striessnig, J.; Koschak, A.; Sinnegger-Brauns, M.J.; Hetzenauer, A.; Nguyen, N.K.; Busquet, P.; Pelster, G.; Singewald, N. Role of voltage-gated L-type Ca2+ channel isoforms for brain function. Biochem. Soc. Trans. 2006, 34, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Sinnegger-Brauns, M.J.; Huber, I.G.; Koschak, A.; Wild, C.; Obermair, G.J.; Einzinger, U.; Hoda, J.C.; Sartori, S.B.; Striessnig, J. Expression and 1,4-dihydropyridine-binding properties of brain L-type calcium channel isoforms. Mol. Pharmacol. 2009, 75, 407–414. [Google Scholar] [CrossRef]
- Clark, N.C.; Nagano, N.; Kuenzi, F.M.; Jarolimek, W.; Huber, I.; Walter, D.; Wietzorrek, G.; Boyce, S.; Kullmann, D.M.; Striessnig, J.; et al. Neurological phenotype and synaptic function in mice lacking the CaV1.3 alpha subunit of neuronal L-type voltage-dependent Ca2+ channels. Neuroscience 2003, 120, 435–442. [Google Scholar] [CrossRef]
- Moosmang, S.; Haider, N.; Klugbauer, N.; Adelsberger, H.; Langwieser, N.; Muller, J.; Stiess, M.; Marais, E.; Schulla, V.; Lacinova, L.; et al. Role of hippocampal Cav1.2 Ca2+ channels in NMDA receptor-independent synaptic plasticity and spatial memory. J. Neurosci. 2005, 25, 9883–9892. [Google Scholar] [CrossRef] [PubMed]
- Frederickson, C.J.; Hernandez, M.D.; McGinty, J.F. Translocation of zinc may contribute to seizure-induced death of neurons. Brain Res. 1989, 480, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Szewczyk, B. Zinc homeostasis and neurodegenerative disorders. Front. Aging Neurosci. 2013, 5, 33. [Google Scholar] [CrossRef]
- Choi, S.; Hong, D.K.; Choi, B.Y.; Suh, S.W. Zinc in the Brain: Friend or Foe? Int. J. Mol. Sci. 2020, 21, 8941. [Google Scholar] [CrossRef] [PubMed]
- Sandstead, H.H. Subclinical zinc deficiency impairs human brain function. J. Trace Elem. Med. Biol. 2012, 26, 70–73. [Google Scholar] [CrossRef]
- Morris, D.R.; Levenson, C.W. Ion channels and zinc: Mechanisms of neurotoxicity and neurodegeneration. J. Toxicol. 2012, 2012, 785647. [Google Scholar] [CrossRef]
- Sensi, S.L.; Yin, H.Z.; Carriedo, S.G.; Rao, S.S.; Weiss, J.H. Preferential Zn2+ influx through Ca2+-permeable AMPA/kainate channels triggers prolonged mitochondrial superoxide production. Proc. Natl. Acad. Sci. USA 1999, 96, 2414–2419. [Google Scholar] [CrossRef]
- Bishop, G.M.; Dringen, R.; Robinson, S.R. Zinc stimulates the production of toxic reactive oxygen species (ROS) and inhibits glutathione reductase in astrocytes. Free Radic. Biol. Med. 2007, 42, 1222–1230. [Google Scholar] [CrossRef]
- Stork, C.J.; Li, Y.V. Elevated Cytoplasmic Free Zinc and Increased Reactive Oxygen Species Generation in the Context of Brain Injury. Acta Neurochir. Suppl. 2016, 121, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Mason, R.P.; Marche, P.; Hintze, T.H. Novel vascular biology of third-generation L-type calcium channel antagonists: Ancillary actions of amlodipine. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 2155–2163. [Google Scholar] [CrossRef] [PubMed]
- Bulsara, K.G.; Cassagnol, M. Amlodipine; StatPearls: Treasure Island, FL, USA, 2024. [Google Scholar]
- Nelson, M.T.; Cheng, H.; Rubart, M.; Santana, L.F.; Bonev, A.D.; Knot, H.J.; Lederer, W.J. Relaxation of arterial smooth muscle by calcium sparks. Science 1995, 270, 633–637. [Google Scholar] [CrossRef]
- Ortner, N.J.; Striessnig, J. L-type calcium channels as drug targets in CNS disorders. Channels 2016, 10, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, D.; Xi, Q.; Pfeffer, L.M.; Jaggar, J.H. Mitochondria control functional CaV1.2 expression in smooth muscle cells of cerebral arteries. Circ. Res. 2010, 107, 631–641. [Google Scholar] [CrossRef]
- Catterall, W.A. Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev. Biol. 2000, 16, 521–555. [Google Scholar] [CrossRef]
- Hockerman, G.H.; Peterson, B.Z.; Johnson, B.D.; Catterall, W.A. Molecular determinants of drug binding and action on L-type calcium channels. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 361–396. [Google Scholar] [CrossRef]
- Hirooka, Y.; Kimura, Y.; Nozoe, M.; Sagara, Y.; Ito, K.; Sunagawa, K. Amlodipine-induced reduction of oxidative stress in the brain is associated with sympatho-inhibitory effects in stroke-prone spontaneously hypertensive rats. Hypertens. Res. 2006, 29, 49–56. [Google Scholar] [CrossRef]
- Lee, Y.J.; Park, H.H.; Koh, S.H.; Choi, N.Y.; Lee, K.Y. Amlodipine besylate and amlodipine camsylate prevent cortical neuronal cell death induced by oxidative stress. J. Neurochem. 2011, 119, 1262–1270. [Google Scholar] [CrossRef]
- Qureshi, I.H.; Riaz, A.; Khan, R.A.; Siddiqui, A.A. Synergistic anticonvulsant effects of pregabalin and amlodipine on acute seizure model of epilepsy in mice. Metab. Brain Dis. 2017, 32, 1051–1060. [Google Scholar] [CrossRef]
- Sathyanarayana Rao, K.N.; Subbalakshmi, N.K. An experimental study of the anticonvulsant effect of amlodipine in mice. Singap. Med. J. 2010, 51, 424–428. [Google Scholar]
- Kaminski, R.; Jasinski, M.; Jagiello-Wojtowicz, E.; Kleinrok, Z.; Czuczwar, S.J. Effect of amlodipine upon the protective activity of antiepileptic drugs against maximal electroshock-induced seizures in mice. Pharmacol. Res. 1999, 40, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Turski, W.A.; Cavalheiro, E.A.; Schwarz, M.; Czuczwar, S.J.; Kleinrok, Z.; Turski, L. Limbic seizures produced by pilocarpine in rats: Behavioural, electroencephalographic and neuropathological study. Behav. Brain Res. 1983, 9, 315–335. [Google Scholar] [CrossRef] [PubMed]
- Cavalheiro, E.A.; Santos, N.F.; Priel, M.R. The pilocarpine model of epilepsy in mice. Epilepsia 1996, 37, 1015–1019. [Google Scholar] [CrossRef] [PubMed]
- Curia, G.; Longo, D.; Biagini, G.; Jones, R.S.; Avoli, M. The pilocarpine model of temporal lobe epilepsy. J. Neurosci. Methods 2008, 172, 143–157. [Google Scholar] [CrossRef]
- Luna-Munguia, H.; Marquez-Bravo, L.; Concha, L. Longitudinal changes in gray and white matter microstructure during epileptogenesis in pilocarpine-induced epileptic rats. Seizure 2021, 90, 130–140. [Google Scholar] [CrossRef]
- Luttjohann, A.; Fabene, P.F.; van Luijtelaar, G. A revised Racine’s scale for PTZ-induced seizures in rats. Physiol. Behav. 2009, 98, 579–586. [Google Scholar] [CrossRef]
- Ihara, Y.; Tomonoh, Y.; Deshimaru, M.; Zhang, B.; Uchida, T.; Ishii, A.; Hirose, S. Retigabine, a Kv7.2/Kv7.3-Channel Opener, Attenuates Drug-Induced Seizures in Knock-In Mice Harboring Kcnq2 Mutations. PLoS ONE 2016, 11, e0150095. [Google Scholar] [CrossRef]
- Frederickson, C.J.; Kasarskis, E.J.; Ringo, D.; Frederickson, R.E. A quinoline fluorescence method for visualizing and assaying the histochemically reactive zinc (bouton zinc) in the brain. J. Neurosci. Methods 1987, 20, 91–103. [Google Scholar] [CrossRef]
- Xu, J.H.; Long, L.; Tang, Y.C.; Hu, H.T.; Tang, F.R. Ca(v)1.2, Ca(v)1.3, and Ca(v)2.1 in the mouse hippocampus during and after pilocarpine-induced status epilepticus. Hippocampus 2007, 17, 235–251. [Google Scholar] [CrossRef]
- Anekonda, T.S.; Quinn, J.F.; Harris, C.; Frahler, K.; Wadsworth, T.L.; Woltjer, R.L. L-type voltage-gated calcium channel blockade with isradipine as a therapeutic strategy for Alzheimer’s disease. Neurobiol. Dis. 2011, 41, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Ito, D.; Imai, Y.; Ohsawa, K.; Nakajima, K.; Fukuuchi, Y.; Kohsaka, S. Microglia-specific localisation of a novel calcium binding protein, Iba1. Mol. Brain Res. 1998, 57, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Holness, C.L.; Simmons, D.L. Molecular cloning of CD68, a human macrophage marker related to lysosomal glycoproteins. Blood 1993, 81, 1607–1613. [Google Scholar] [CrossRef] [PubMed]
- Berg, M.; Bruhn, T.; Frandsen, A.; Schousboe, A.; Diemer, N.H. Kainic acid-induced seizures and brain damage in the rat: Role of calcium homeostasis. J. Neurosci. Res. 1995, 40, 641–646. [Google Scholar] [CrossRef]
- Nunez, J.L.; McCarthy, M.M. Androgens predispose males to GABAA-mediated excitotoxicity in the developing hippocampus. Exp. Neurol. 2008, 210, 699–708. [Google Scholar] [CrossRef]
- Nascimento, V.S.; D’Alva, M.S.; Oliveira, A.A.; Freitas, R.M.; Vasconcelos, S.M.; Sousa, F.C.; Fonteles, M.M. Antioxidant effect of nimodipine in young rats after pilocarpine-induced seizures. Pharmacol. Biochem. Behav. 2005, 82, 11–16. [Google Scholar] [CrossRef]
- Nolte, C.; Gore, A.; Sekler, I.; Kresse, W.; Hershfinkel, M.; Hoffmann, A.; Kettenmann, H.; Moran, A. ZnT-1 expression in astroglial cells protects against zinc toxicity and slows the accumulation of intracellular zinc. Glia 2004, 48, 145–155. [Google Scholar] [CrossRef]
- Shao, Y.Y.; Li, B.; Huang, Y.M.; Luo, Q.; Xie, Y.M.; Chen, Y.H. Thymoquinone Attenuates Brain Injury via an Anti-oxidative Pathway in a Status Epilepticus Rat Model. Transl. Neurosci. 2017, 8, 9–14. [Google Scholar] [CrossRef]
- Quincozes-Santos, A.; Bobermin, L.D.; Tramontina, A.C.; Wartchow, K.M.; Tagliari, B.; Souza, D.O.; Wyse, A.T.; Goncalves, C.A. Oxidative stress mediated by NMDA, AMPA/KA channels in acute hippocampal slices: Neuroprotective effect of resveratrol. Toxicol. Vitr. 2014, 28, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Xian, P.; Wang, T.; Wu, S.; Sun, T.; Wang, W.; Wang, B.; Yang, H.; Yang, Y.; Wang, H.; et al. Antioxidant activity of mesenchymal stem cell-derived extracellular vesicles restores hippocampal neurons following seizure damage. Theranostics 2021, 11, 5986–6005. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.K.; Saadi, A.; Sheeni, Y.; Shekh-Ahmad, T. Specific inhibition of NADPH oxidase 2 modifies chronic epilepsy. Redox Biol. 2022, 58, 102549. [Google Scholar] [CrossRef] [PubMed]
- Pestana, R.R.; Kinjo, E.R.; Hernandes, M.S.; Britto, L.R. Reactive oxygen species generated by NADPH oxidase are involved in neurodegeneration in the pilocarpine model of temporal lobe epilepsy. Neurosci. Lett. 2010, 484, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Kovac, S.; Domijan, A.M.; Walker, M.C.; Abramov, A.Y. Seizure activity results in calcium- and mitochondria-independent ROS production via NADPH and xanthine oxidase activation. Cell Death Dis. 2014, 5, e1442. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Scorziello, A.; Duchen, M.R. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J. Neurosci. 2007, 27, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Eid, T.; Lothman, E.W.; Kohler, C.; Schwarcz, R. Preferential neuronal loss in layer III of the medial entorhinal cortex in rat models of temporal lobe epilepsy. J. Neurosci. 1995, 15, 6301–6313. [Google Scholar] [CrossRef] [PubMed]
- Sankar, R.; Shin, D.H.; Liu, H.; Mazarati, A.; Pereira de Vasconcelos, A.; Wasterlain, C.G. Patterns of status epilepticus-induced neuronal injury during development and long-term consequences. J. Neurosci. 1998, 18, 8382–8393. [Google Scholar] [CrossRef] [PubMed]
- Zucker, D.K.; Wooten, G.F.; Lothman, E.W. Blood-brain barrier changes with kainic acid-induced limbic seizures. Exp. Neurol. 1983, 79, 422–433. [Google Scholar] [CrossRef]
- Krizanac-Bengez, L.; Mayberg, M.R.; Janigro, D. The cerebral vasculature as a therapeutic target for neurological disorders and the role of shear stress in vascular homeostatis and pathophysiology. Neurol. Res. 2004, 26, 846–853. [Google Scholar] [CrossRef]
- Oby, E.; Janigro, D. The blood-brain barrier and epilepsy. Epilepsia 2006, 47, 1761–1774. [Google Scholar] [CrossRef]
- Vezzani, A.; Granata, T. Brain inflammation in epilepsy: Experimental and clinical evidence. Epilepsia 2005, 46, 1724–1743. [Google Scholar] [CrossRef]
- Lee, S.H.; Choi, B.Y.; Kim, J.H.; Kho, A.R.; Sohn, M.; Song, H.K.; Choi, H.C.; Suh, S.W. Late treatment with choline alfoscerate (l-alpha glycerylphosphorylcholine, alpha-GPC) increases hippocampal neurogenesis and provides protection against seizure-induced neuronal death and cognitive impairment. Brain Res. 2017, 1654 Pt A, 66–76. [Google Scholar] [CrossRef]
- Dey, A.; Kang, X.; Qiu, J.; Du, Y.; Jiang, J. Anti-Inflammatory Small Molecules To Treat Seizures and Epilepsy: From Bench to Bedside. Trends Pharmacol. Sci. 2016, 37, 463–484. [Google Scholar] [CrossRef]
- Marchi, N.; Granata, T.; Janigro, D. Inflammatory pathways of seizure disorders. Trends Neurosci. 2014, 37, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Dong, X. Current Strategies for Brain Drug Delivery. Theranostics 2018, 8, 1481–1493. [Google Scholar] [CrossRef]
- Bowyer, J.F.; Robinson, B.; Ali, S.; Schmued, L.C. Neurotoxic-related changes in tyrosine hydroxylase, microglia, myelin, and the blood-brain barrier in the caudate-putamen from acute methamphetamine exposure. Synapse 2008, 62, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Castiglioni, A.J.; Peterson, S.L.; Sanabria, E.L.; Tiffany-Castiglioni, E. Structural changes in astrocytes induced by seizures in a mode of temporal lobe epilepsy. J. Neurosci. Res. 1990, 26, 334–341. [Google Scholar] [CrossRef]
- Jung, K.H.; Chu, K.; Lee, S.T.; Kim, J.H.; Kang, K.M.; Song, E.C.; Kim, S.J.; Park, H.K.; Kim, M.; Lee, S.K.; et al. Region-specific plasticity in the epileptic rat brain: A hippocampal and extrahippocampal analysis. Epilepsia 2009, 50, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Norden, D.M.; Trojanowski, P.J.; Villanueva, E.; Navarro, E.; Godbout, J.P. Sequential activation of microglia and astrocyte cytokine expression precedes increased Iba-1 or GFAP immunoreactivity following systemic immune challenge. Glia 2016, 64, 300–316. [Google Scholar] [CrossRef]
- Benson, M.J.; Manzanero, S.; Borges, K. Complex alterations in microglial M1/M2 markers during the development of epilepsy in two mouse models. Epilepsia 2015, 56, 895–905. [Google Scholar] [CrossRef]
- Boison, D.; Steinhauser, C. Epilepsy and astrocyte energy metabolism. Glia 2018, 66, 1235–1243. [Google Scholar] [CrossRef]
- Schartz, N.D.; Herr, S.A.; Madsen, L.; Butts, S.J.; Torres, C.; Mendez, L.B.; Brewster, A.L. Spatiotemporal profile of Map2 and microglial changes in the hippocampal CA1 region following pilocarpine-induced status epilepticus. Sci. Rep. 2016, 6, 24988. [Google Scholar] [CrossRef] [PubMed]
- Jalava, N.S.; Lopez-Picon, F.R.; Kukko-Lukjanov, T.K.; Holopainen, I.E. Changes in microtubule-associated protein-2 (MAP2) expression during development and after status epilepticus in the immature rat hippocampus. Int. J. Dev. Neurosci. 2007, 25, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C.; Arellano, J.I.; Rodriguez-Sanchez, P.; Avila, J.; DeFelipe, J.; Diez-Guerra, F.J. Microtubule-associated protein 2 phosphorylation is decreased in the human epileptic temporal lobe cortex. Neuroscience 2001, 107, 25–33. [Google Scholar] [CrossRef]
- Ballough, G.P.; Martin, L.J.; Cann, F.J.; Graham, J.S.; Smith, C.D.; Kling, C.E.; Forster, J.S.; Phann, S.; Filbert, M.G. Microtubule-associated protein 2 (MAP-2): A sensitive marker of seizure-related brain damage. J. Neurosci. Methods 1995, 61, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Ingram, D.K.; Joseph, J.A.; Spangler, E.L.; Roberts, D.; Hengemihle, J.; Fanelli, R.J. Chronic nimodipine treatment in aged rats: Analysis of motor and cognitive effects and muscarinic-induced striatal dopamine release. Neurobiol. Aging 1994, 15, 55–61. [Google Scholar] [CrossRef]
- Batuecas, A.; Pereira, R.; Centeno, C.; Pulido, J.A.; Hernandez, M.; Bollati, A.; Bogonez, E.; Satrustegui, J. Effects of chronic nimodipine on working memory of old rats in relation to defects in synaptosomal calcium homeostasis. Eur. J. Pharmacol. 1998, 350, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Quevedo, J.; Vianna, M.; Daroit, D.; Born, A.G.; Kuyven, C.R.; Roesler, R.; Quillfeldt, J.A. L-type voltage-dependent calcium channel blocker nifedipine enhances memory retention when infused into the hippocampus. Neurobiol. Learn. Mem. 1998, 69, 320–325. [Google Scholar] [CrossRef]
- Zhang, J.; Ding, M.P.; Liu, Z.; Xiao, B.; Li, G.L.; Zhou, F.Y. Dynamics of calcium in the hippocampal neuronal culture model of epilepsy. Zhongguo Ying Yong Sheng Li Xue Za Zhi 2007, 23, 200–203. [Google Scholar]
- Yang, F.; Xu, G.L.; Yang, Y.Q.; Shen, D.K.; Feng, P.Z.; Wang, P.; Liu, X.G. Effect of electroacupuncture on epileptic EEG and intracellular Ca2+ content in the hippocampus in epilepsy rats. Zhen Ci Yan Jiu 2009, 34, 163–166. [Google Scholar]
- Raza, M.; Blair, R.E.; Sombati, S.; Carter, D.S.; Deshpande, L.S.; DeLorenzo, R.J. Evidence that injury-induced changes in hippocampal neuronal calcium dynamics during epileptogenesis cause acquired epilepsy. Proc. Natl. Acad. Sci. USA 2004, 101, 17522–17527. [Google Scholar] [CrossRef]
- Su, T.; Cong, W.D.; Long, Y.S.; Luo, A.H.; Sun, W.W.; Deng, W.Y.; Liao, W.P. Altered expression of voltage-gated potassium channel 4.2 and voltage-gated potassium channel 4-interacting protein, and changes in intracellular calcium levels following lithium-pilocarpine-induced status epilepticus. Neuroscience 2008, 157, 566–576. [Google Scholar] [CrossRef]
- Wang, C.; Xie, N.; Wang, Y.; Li, Y.; Ge, X.; Wang, M. Role of the Mitochondrial Calcium Uniporter in Rat Hippocampal Neuronal Death After Pilocarpine-Induced Status Epilepticus. Neurochem. Res. 2015, 40, 1739–1746. [Google Scholar] [CrossRef]
- Friedman, L.K.; Segal, M.; Veliskova, J. GluR2 knockdown reveals a dissociation between [Ca2+]i surge and neurotoxicity. Neurochem. Int. 2003, 43, 179–189. [Google Scholar] [CrossRef]
- Ge, Y.X.; Lin, Y.Y.; Bi, Q.Q.; Chen, Y.J. Brivaracetam Prevents the Over-expression of Synaptic Vesicle Protein 2A and Rescues the Deficits of Hippocampal Long-term Potentiation In Vivo in Chronic Temporal Lobe Epilepsy Rats. Curr. Neurovascular Res. 2020, 17, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Zeng, Y.; Tu, Z.; Li, P.; Chen, H.; Cheng, L.; Tu, S.; Jiang, L. TRPV1 Contributes to the Neuroprotective Effect of Dexmedetomidine in Pilocarpine-Induced Status Epilepticus Juvenile Rats. Biomed. Res. Int. 2020, 2020, 7623635. [Google Scholar] [CrossRef] [PubMed]
- van Luijtelaar, G.; Wiaderna, D.; Elants, C.; Scheenen, W. Opposite effects of T- and L-type Ca2+ channels blockers in generalized absence epilepsy. Eur. J. Pharmacol. 2000, 406, 381–389. [Google Scholar] [CrossRef]
- Zupan, G.; Erakovic, V.; Simonic, A.; Kriz, J.; Varljen, J. The influence of nimodipine, nicardipine and amlodipine on the brain free fatty acid level in rats with penicillin-induced seizures. Prog. Neuropsychopharmacol. Biol. Psychiatry 1999, 23, 951–961. [Google Scholar] [CrossRef]
- Meyer, F.B.; Anderson, R.E.; Sundt, T.M., Jr.; Yaksh, T.L.; Sharbrough, F.W. Suppression of pentylenetetrazole seizures by oral administration of a dihydropyridine Ca2+ antagonist. Epilepsia 1987, 28, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, A.; Saini, H.K.; Garg, S.K. Dose-finding study with nimodipine: A selective central nervous system calcium channel blocker on aminophylline induced seizure models in rats. Brain Res. Bull. 1998, 45, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, R.M.; Mazurek, M.; Turski, W.A.; Kleinrok, Z.; Czuczwar, S.J. Amlodipine enhances the activity of antiepileptic drugs against pentylenetetrazole-induced seizures. Pharmacol. Biochem. Behav. 2001, 68, 661–668. [Google Scholar] [CrossRef]
- Ikegaya, Y.; Nishiyama, N.; Matsuki, N. L-type Ca2+ channel blocker inhibits mossy fiber sprouting and cognitive deficits following pilocarpine seizures in immature mice. Neuroscience 2000, 98, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Woodward, J.J.; Cook, M.E.; Leslie, S.W. Characterization of dihydropyridine-sensitive calcium channels in rat brain synaptosomes. Proc. Natl. Acad. Sci. USA 1988, 85, 7389–7393. [Google Scholar] [CrossRef] [PubMed]
- Erakovic, V.; Zupan, G.; Varljen, J.; Laginja, J.; Simonic, A. Lithium plus pilocarpine induced status epilepticus--biochemical changes. Neurosci. Res. 2000, 36, 157–166. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.-J.; Lee, S.-H.; Kang, B.-S.; Park, M.-K.; Yang, H.-W.; Woo, S.-Y.; Park, S.-W.; Kim, D.-Y.; Jeong, H.-H.; Yang, W.-I.; et al. Effects of L-Type Voltage-Gated Calcium Channel (LTCC) Inhibition on Hippocampal Neuronal Death after Pilocarpine-Induced Seizure. Antioxidants 2024, 13, 389. https://doi.org/10.3390/antiox13040389
Lee C-J, Lee S-H, Kang B-S, Park M-K, Yang H-W, Woo S-Y, Park S-W, Kim D-Y, Jeong H-H, Yang W-I, et al. Effects of L-Type Voltage-Gated Calcium Channel (LTCC) Inhibition on Hippocampal Neuronal Death after Pilocarpine-Induced Seizure. Antioxidants. 2024; 13(4):389. https://doi.org/10.3390/antiox13040389
Chicago/Turabian StyleLee, Chang-Jun, Song-Hee Lee, Beom-Seok Kang, Min-Kyu Park, Hyun-Wook Yang, Seo-Young Woo, Se-Wan Park, Dong-Yeon Kim, Hyun-Ho Jeong, Won-Il Yang, and et al. 2024. "Effects of L-Type Voltage-Gated Calcium Channel (LTCC) Inhibition on Hippocampal Neuronal Death after Pilocarpine-Induced Seizure" Antioxidants 13, no. 4: 389. https://doi.org/10.3390/antiox13040389
APA StyleLee, C. -J., Lee, S. -H., Kang, B. -S., Park, M. -K., Yang, H. -W., Woo, S. -Y., Park, S. -W., Kim, D. -Y., Jeong, H. -H., Yang, W. -I., Kho, A. -R., Choi, B. -Y., Song, H. -K., Choi, H. -C., Kim, Y. -J., & Suh, S. -W. (2024). Effects of L-Type Voltage-Gated Calcium Channel (LTCC) Inhibition on Hippocampal Neuronal Death after Pilocarpine-Induced Seizure. Antioxidants, 13(4), 389. https://doi.org/10.3390/antiox13040389