

Redox Regulation of Phosphatase and Tensin Homolog by Bicarbonate and Hydrogen Peroxide: Implication of Peroxymonocarbonate in Cell Signaling

 , , , and
, , , and 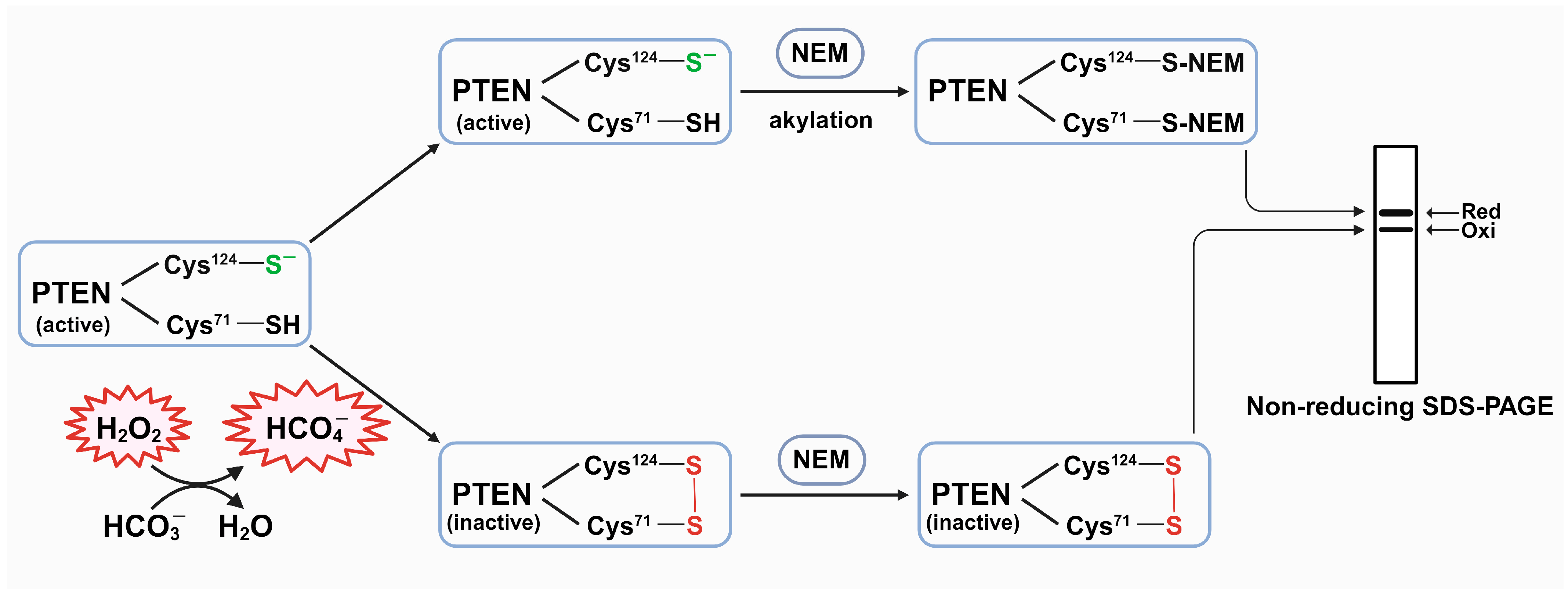{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Immunoblot Analysis of H2O2-Induced Oxidation of PTEN
2.4. Statistical Analysis
3. Results
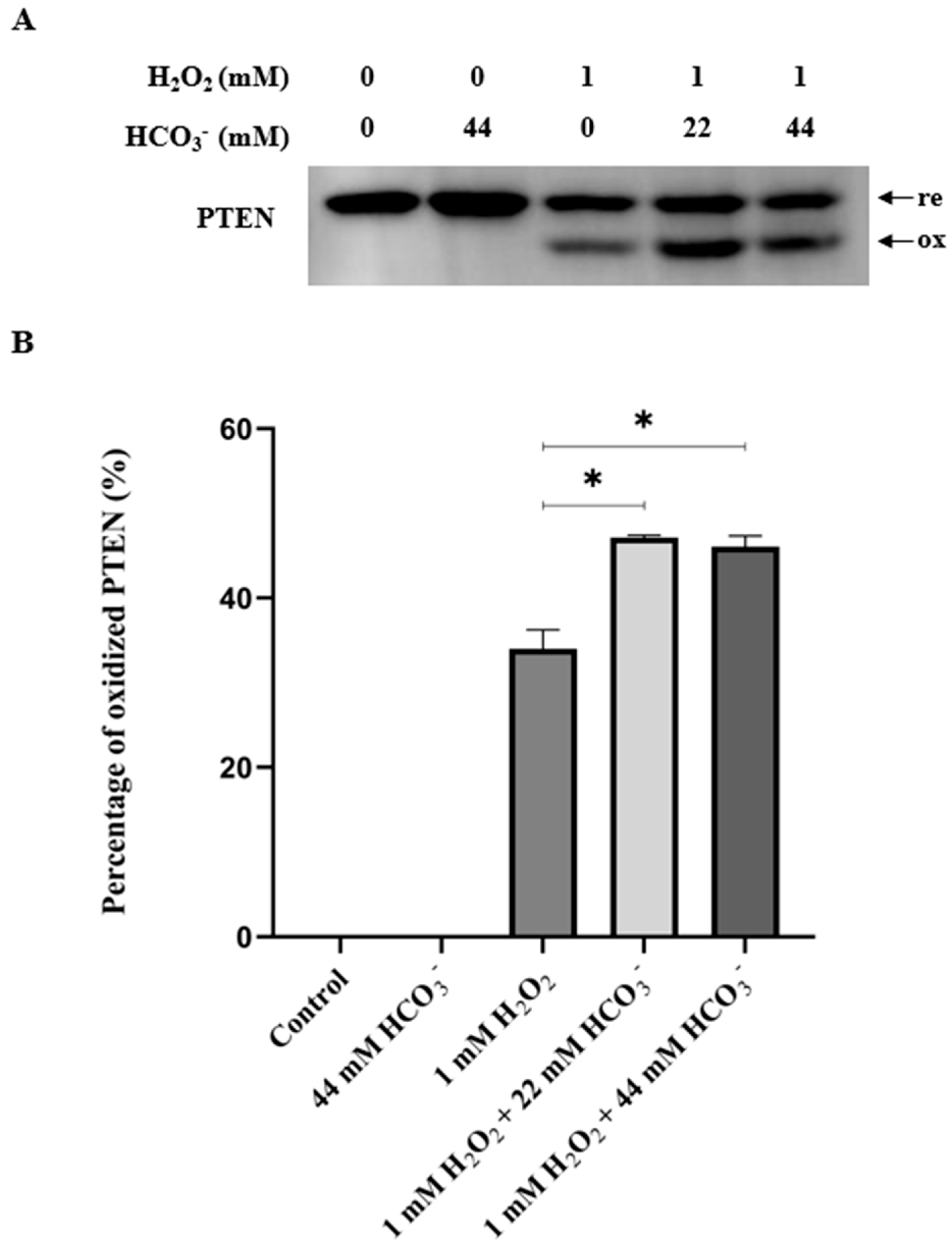
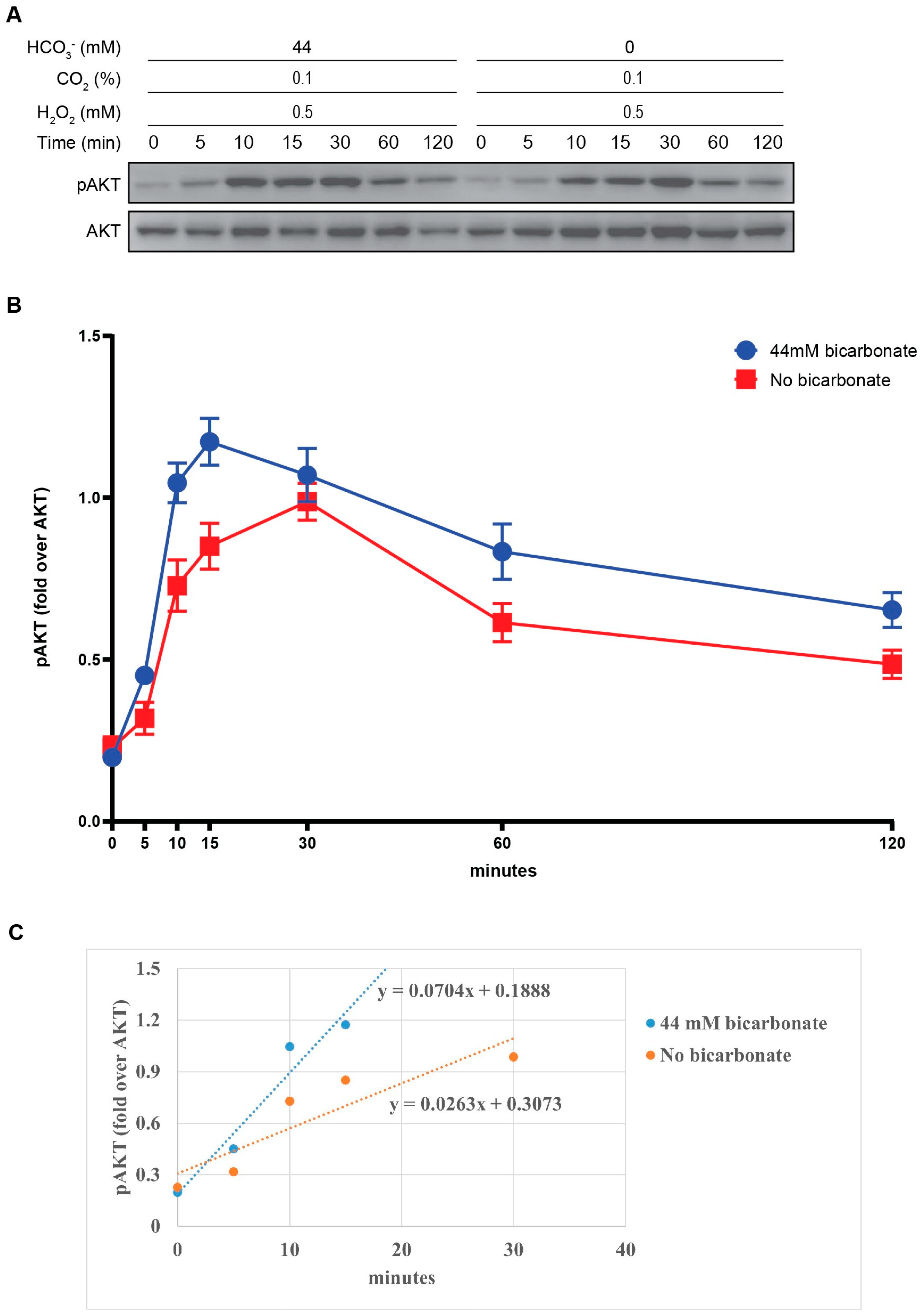
3.1. The Presence of HCO3− Potentiates the PTEN Oxidation by H2O2
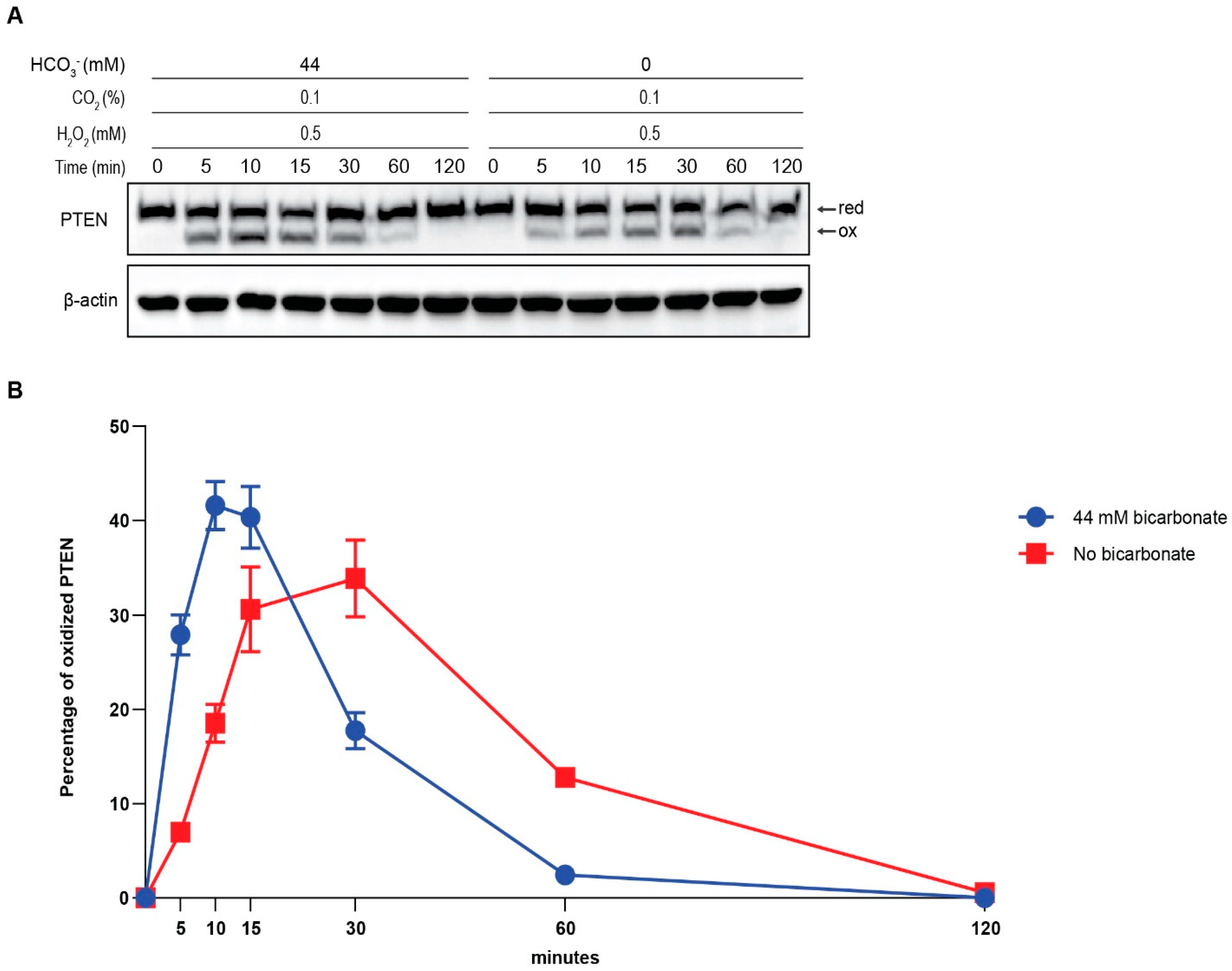
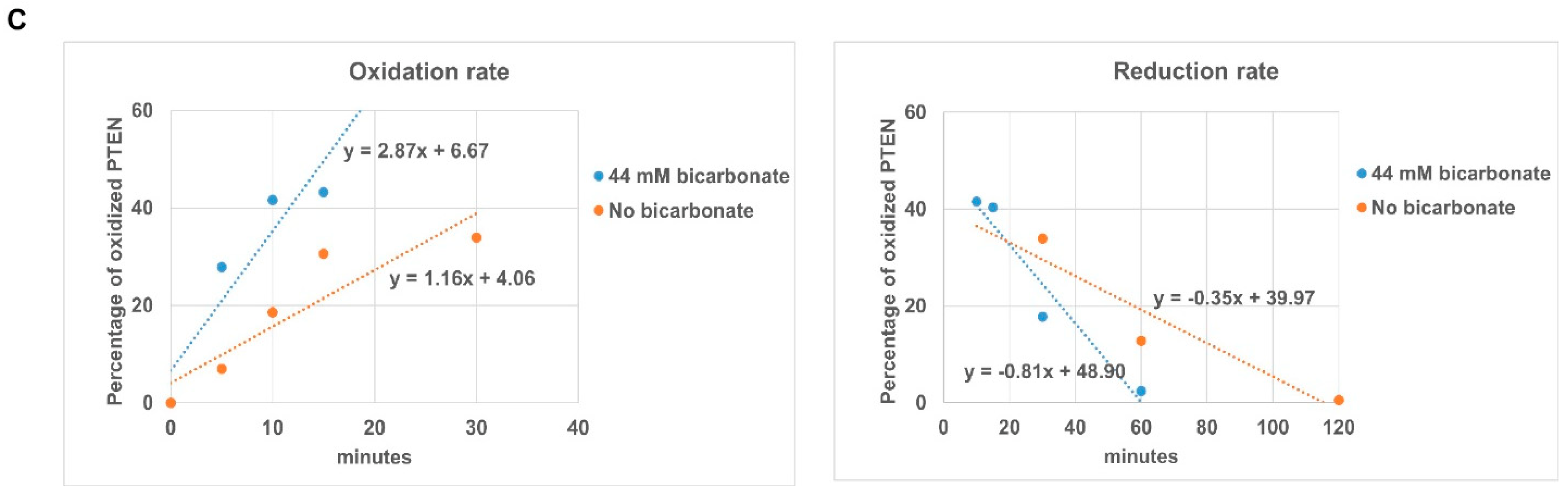
3.2. The Presence of HCO3− Facilitates Redox Regulation of PTEN by H2O2
3.3. The Activation of PI3K/AKT Pathway via PTEN Oxidation by H2O2 and HCO3−
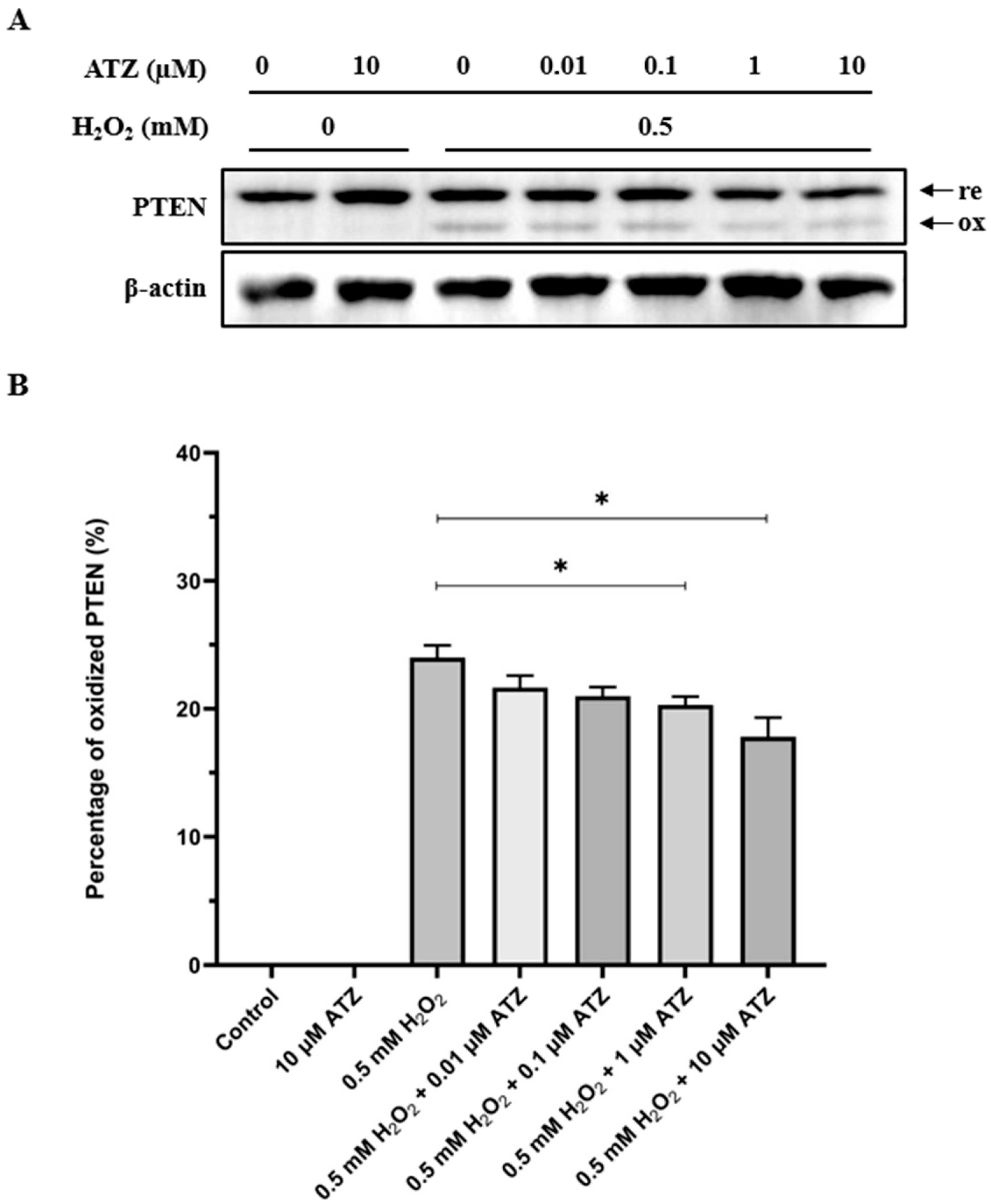
3.4. Oxidation of PTEN Is Decreased in the Presence of CA Inhibitor ATZ
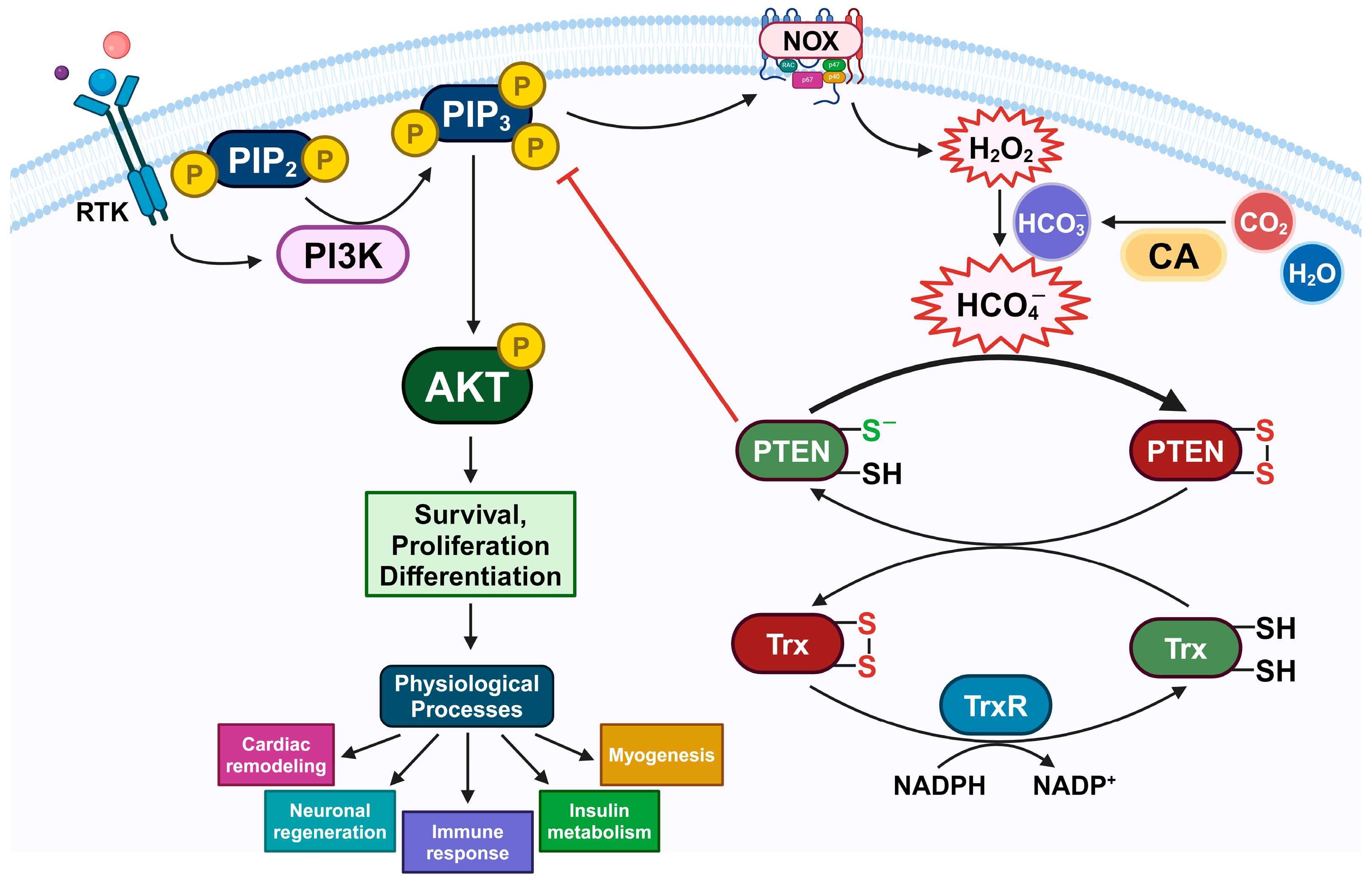
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rhee, S.G.; Bae, Y.S.; Lee, S.-R.; Kwon, J. Hydrogen peroxide: A key messenger that modulates protein phosphorylation through cysteine oxidation. Science’s STKE 2000, 2000, pe1. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Maiorino, M.; Ursini, F. Signaling functions of reactive oxygen species. Biochemistry 2010, 49, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. H2O2, a necessary evil for cell signaling. Science 2006, 312, 1882–1883. [Google Scholar] [CrossRef] [PubMed]
- van der Geer, P.; Hunter, T.; Lindberg, R.A. Receptor protein-tyrosine kinases and their signal transduction pathways. Annu. Rev. Cell Biol. 1994, 10, 251–337. [Google Scholar] [CrossRef] [PubMed]
- Denu, J.M.; Dixon, J.E. Protein tyrosine phosphatases: Mechanisms of catalysis and regulation. Curr. Opin. Chem. Biol. 1998, 2, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Denu, J.M.; Tanner, K.G. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: Evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry 1998, 37, 5633–5642. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-R.; Chen, M.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor: New modes and prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-O.; Yang, H.; Georgescu, M.-M.; Di Cristofano, A.; Maehama, T.; Shi, Y.; Dixon, J.E.; Pandolfi, P.; Pavletich, N.P. Crystal structure of the PTEN tumor suppressor: Implications for its phosphoinositide phosphatase activity and membrane association. Cell 1999, 99, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Stambolic, V.; Suzuki, A.; De La Pompa, J.L.; Brothers, G.M.; Mirtsos, C.; Sasaki, T.; Ruland, J.; Penninger, J.M.; Siderovski, D.P.; Mak, T.W. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 1998, 95, 29–39. [Google Scholar] [CrossRef]
- Leslie, N.R.; Bennett, D.; Lindsay, Y.E.; Stewart, H.; Gray, A.; Downes, C.P. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J. 2003, 22, 5501–5510. [Google Scholar] [CrossRef]
- Rosivatz, E. Inhibiting PTEN. Biochem. Soc. Trans. 2007, 35, 257–259. [Google Scholar] [CrossRef] [PubMed]
- Pulido, R. PTEN inhibition in human disease therapy. Molecules 2018, 23, 285. [Google Scholar] [CrossRef]
- Borges, G.A.; Webber, L.P.; M Marques, A.E.; Guerra, E.N.; Castilho, R.M.; Squarize, C.H. Pharmacological PTEN inhibition: Potential clinical applications and effects in tissue regeneration. Regen. Med. 2020, 15, 1329–1344. [Google Scholar] [CrossRef]
- Lee, S.-R.; Yang, K.-S.; Kwon, J.; Lee, C.; Jeong, W.; Rhee, S.G. Reversible inactivation of the tumor suppressor PTEN by H2O2. J. Biol. Chem. 2002, 277, 20336–20342. [Google Scholar] [CrossRef]
- Rhee, S.G.; Chae, H.Z.; Kim, K. Peroxiredoxins: A historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free. Radic. Biol. Med. 2005, 38, 1543–1552. [Google Scholar] [CrossRef]
- Rhee, S.G.; Woo, H.A.; Kil, I.S.; Bae, S.H. Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J. Biol. Chem. 2012, 287, 4403–4410. [Google Scholar] [CrossRef]
- Perkins, A.; Nelson, K.J.; Parsonage, D.; Poole, L.B.; Karplus, P.A. Peroxiredoxins: Guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem. Sci. 2015, 40, 435–445. [Google Scholar] [CrossRef]
- Jones, D.P.; Griffith, W.P. Alkali-metal peroxocarbonates, M2 [CO3]· n H2O2, M2 [C2O6], M [HCO4]· n H2O, and Li2 [CO4]· H2O. J. Chem. Soc. Dalton Trans. 1980, 2526–2532. [Google Scholar] [CrossRef]
- Flangan, J.; Jones, D.P.; Griffith, W.P.; Skapski, A.C.; West, A.P. On the existence of peroxocarbonates in aqueous solution. J. Chem. Soc. Chem. Commun. 1986, 20–21. [Google Scholar] [CrossRef]
- Richardson, D.E.; Yao, H.; Frank, K.M.; Bennett, D.A. Equilibria, kinetics, and mechanism in the bicarbonate activation of hydrogen peroxide: Oxidation of sulfides by peroxymonocarbonate. J. Am. Chem. Soc. 2000, 122, 1729–1739. [Google Scholar] [CrossRef]
- Bakhmutova-Albert, E.V.; Yao, H.; Denevan, D.E.; Richardson, D.E. Kinetics and mechanism of peroxymonocarbonate formation. Inorg. Chem. 2010, 49, 11287–11296. [Google Scholar] [CrossRef]
- Radi, R. Interplay of carbon dioxide and peroxide metabolism in mammalian cells. J. Biol. Chem. 2022, 298, 102358. [Google Scholar] [CrossRef]
- Trindade, D.F.; Cerchiaro, G.; Augusto, O. A role for peroxymonocarbonate in the stimulation of biothiol peroxidation by the bicarbonate/carbon dioxide pair. Chem. Res. Toxicol. 2006, 19, 1475–1482. [Google Scholar] [CrossRef]
- Zhou, H.; Singh, H.; Parsons, Z.D.; Lewis, S.M.; Bhattacharya, S.; Seiner, D.R.; LaButti, J.N.; Reilly, T.J.; Tanner, J.J.; Gates, K.S. The biological buffer bicarbonate/CO2 potentiates H2O2-mediated inactivation of protein tyrosine phosphatases. J. Am. Chem. Soc. 2011, 133, 15803–15805. [Google Scholar] [CrossRef]
- Dagnell, M.; Cheng, Q.; Rizvi, S.H.M.; Pace, P.E.; Boivin, B.; Winterbourn, C.C.; Arnér, E.S. Bicarbonate is essential for protein-tyrosine phosphatase 1B (PTP1B) oxidation and cellular signaling through EGF-triggered phosphorylation cascades. J. Biol. Chem. 2019, 294, 12330–12338. [Google Scholar] [CrossRef]
- James, J.; Chen, Y.; Hernandez, C.M.; Forster, F.; Dagnell, M.; Cheng, Q.; Saei, A.A.; Gharibi, H.; Lahore, G.F.; Åstrand, A. Redox regulation of PTPN22 affects the severity of T-cell-dependent autoimmune inflammation. Elife 2022, 11, e74549. [Google Scholar] [CrossRef]
- Dorai, T.; Sawczuk, I.S.; Pastorek, J.; Wiernik, P.H.; Dutcher, J.P. The role of carbonic anhydrase IX overexpression in kidney cancer. Eur. J. Cancer 2005, 41, 2935–2947. [Google Scholar] [CrossRef]
- Han, S.-J.; Ahn, Y.; Park, I.; Zhang, Y.; Kim, I.; Kim, H.W.; Ku, C.-S.; Chay, K.-O.; Yang, S.Y.; Ahn, B.W. Assay of the redox state of the tumor suppressor PTEN by mobility shift. Methods 2015, 77, 58–62. [Google Scholar] [CrossRef]
- Nocentini, A.; Supuran, C.T. Carbonic anhydrases: An overview. Carbon. Anhydrases 2019, 3–16. [Google Scholar]
- Tonks, N.K. Protein tyrosine phosphatases: From genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 2006, 7, 833–846. [Google Scholar] [CrossRef]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef]
- Leslie, N.R. The redox regulation of PI 3-kinase-dependent signaling. Antioxid. Redox Signal. 2006, 8, 1765–1774. [Google Scholar] [CrossRef]
- Downes, C.P.; Ross, S.; Maccario, H.; Perera, N.; Davidson, L.; Leslie, N.R. Stimulation of PI 3-kinase signaling via inhibition of the tumor suppressor phosphatase, PTEN. Adv. Enzyme Regul. 2007, 47, 184–194. [Google Scholar] [CrossRef]
- Trinh, V.H.; Nguyen Huu, T.; Sah, D.K.; Choi, J.M.; Yoon, H.J.; Park, S.C.; Jung, Y.S.; Lee, S.-R. Redox Regulation of PTEN by Reactive Oxygen Species: Its Role in Physiological Processes. Antioxidants 2024, 13, 199. [Google Scholar] [CrossRef]
- Cai, Z.; Semenza, G.L. PTEN activity is modulated during ischemia and reperfusion: Involvement in the induction and decay of preconditioning. Circ. Res. 2005, 97, 1351–1359. [Google Scholar] [CrossRef]
- Hervera, A.; De Virgiliis, F.; Palmisano, I.; Zhou, L.; Tantardini, E.; Kong, G.; Hutson, T.; Danzi, M.C.; Perry, R.B.; Santos, C.X.C.; et al. Reactive oxygen species regulate axonal regeneration through the release of exosomal NADPH oxidase 2 complexes into injured axons. Nat. Cell Biol. 2018, 20, 307–319. [Google Scholar] [CrossRef]
- Kwak, H.-J.; Liu, P.; Bajrami, B.; Xu, Y.; Park, S.-Y.; Nombela-Arrieta, C.; Mondal, S.; Sun, Y.; Zhu, H.; Chai, L. Myeloid cell-derived reactive oxygen species externally regulate the proliferation of myeloid progenitors in emergency granulopoiesis. Immunity 2015, 42, 159–171. [Google Scholar] [CrossRef]
- Loh, K.; Deng, H.; Fukushima, A.; Cai, X.; Boivin, B.; Galic, S.; Bruce, C.; Shields, B.J.; Skiba, B.; Ooms, L.M. Reactive oxygen species enhance insulin sensitivity. Cell Metab. 2009, 10, 260–272. [Google Scholar] [CrossRef]
- Kim, J.H.; Choi, T.G.; Park, S.; Yun, H.R.; Nguyen, N.N.Y.; Jo, Y.H.; Jang, M.; Kim, J.; Kim, J.; Kang, I.; et al. Mitochondrial ROS-derived PTEN oxidation activates PI3K pathway for mTOR-induced myogenic autophagy. Cell Death Differ. 2018, 25, 1921–1937. [Google Scholar] [CrossRef]
- Nguyen Huu, T.; Park, J.; Zhang, Y.; Park, I.; Yoon, H.J.; Woo, H.A.; Lee, S.R. Redox Regulation of PTEN by Peroxiredoxins. Antioxidants 2021, 10, 302. [Google Scholar] [CrossRef]
- Cao, J.; Schulte, J.; Knight, A.; Leslie, N.R.; Zagozdzon, A.; Bronson, R.; Manevich, Y.; Beeson, C.; Neumann, C.A. Prdx1 inhibits tumorigenesis via regulating PTEN/AKT activity. Embo J. 2009, 28, 1505–1517. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Park, J.; Han, S.-J.; Yang, S.Y.; Yoon, H.J.; Park, I.; Woo, H.A.; Lee, S.-R. Redox regulation of tumor suppressor PTEN in cell signaling. Redox Biol. 2020, 34, 101553. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H. Will cancer cells be defeated by sodium bicarbonate? Sci. China. Life Sci. 2017, 60, 326. [Google Scholar] [CrossRef] [PubMed]
- Mahadev, K.; Zilbering, A.; Zhu, L.; Goldstein, B.J. Insulin-stimulated hydrogen peroxide reversibly inhibits protein-tyrosine phosphatase 1b in vivo and enhances the early insulin action cascade. J. Biol. Chem. 2001, 276, 21938–21942. [Google Scholar] [CrossRef] [PubMed]
- Meng, T.-C.; Buckley, D.A.; Galic, S.; Tiganis, T.; Tonks, N.K. Regulation of insulin signaling through reversible oxidation of the protein-tyrosine phosphatases TC45 and PTP1B. J. Biol. Chem. 2004, 279, 37716–37725. [Google Scholar] [CrossRef] [PubMed]
- Boosani, C.S.; Gunasekar, P.; Agrawal, D.K. An update on PTEN modulators–a patent review. Expert Opin. Ther. Pat. 2019, 29, 881–889. [Google Scholar] [CrossRef]
- Wykoff, C.C.; Beasley, N.J.; Watson, P.H.; Turner, K.J.; Pastorek, J.; Sibtain, A.; Wilson, G.D.; Turley, H.; Talks, K.L.; Maxwell, P.H. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res. 2000, 60, 7075–7083. [Google Scholar]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trinh, V.H.; Choi, J.-M.; Nguyen Huu, T.; Sah, D.K.; Yoon, H.-J.; Park, S.-C.; Jung, Y.-S.; Ahn, Y.-K.; Lee, K.-H.; Lee, S.-R. Redox Regulation of Phosphatase and Tensin Homolog by Bicarbonate and Hydrogen Peroxide: Implication of Peroxymonocarbonate in Cell Signaling. Antioxidants 2024, 13, 473. https://doi.org/10.3390/antiox13040473
Trinh VH, Choi J-M, Nguyen Huu T, Sah DK, Yoon H-J, Park S-C, Jung Y-S, Ahn Y-K, Lee K-H, Lee S-R. Redox Regulation of Phosphatase and Tensin Homolog by Bicarbonate and Hydrogen Peroxide: Implication of Peroxymonocarbonate in Cell Signaling. Antioxidants. 2024; 13(4):473. https://doi.org/10.3390/antiox13040473
Chicago/Turabian StyleTrinh, Vu Hoang, Jin-Myung Choi, Thang Nguyen Huu, Dhiraj Kumar Sah, Hyun-Joong Yoon, Sang-Chul Park, Yu-Seok Jung, Young-Keun Ahn, Kun-Ho Lee, and Seung-Rock Lee. 2024. "Redox Regulation of Phosphatase and Tensin Homolog by Bicarbonate and Hydrogen Peroxide: Implication of Peroxymonocarbonate in Cell Signaling" Antioxidants 13, no. 4: 473. https://doi.org/10.3390/antiox13040473