
Deletion of Glyoxalase 1 Exacerbates Acetaminophen-Induced Hepatotoxicity in Mice

 , ,
, ,  ,
,  and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animals
2.3. Behavioral Assessment of GKO Mice
2.4. Animal Handling
2.5. ALT Level Assessment in Serum
2.6. Histology
2.7. Measurement of Lipid Content
2.8. APAP and Metabolites in Serum and Liver Samples
2.9. Measurement of Liver GSH Content
2.10. Lipid Peroxidation Assay
2.11. Evaluation of Protein Carbonyl Content
2.12. Determination of CYP2E1 Activity
2.13. Determination of Liver Superoxide Dismutase (SOD) Activity
2.14. Determination of Liver Catalase Activity
2.15. Determination of AGE Levels by AGE ELISA
2.16. Western Blot Analysis
2.17. Statistical Analysis
3. Results and Discussion
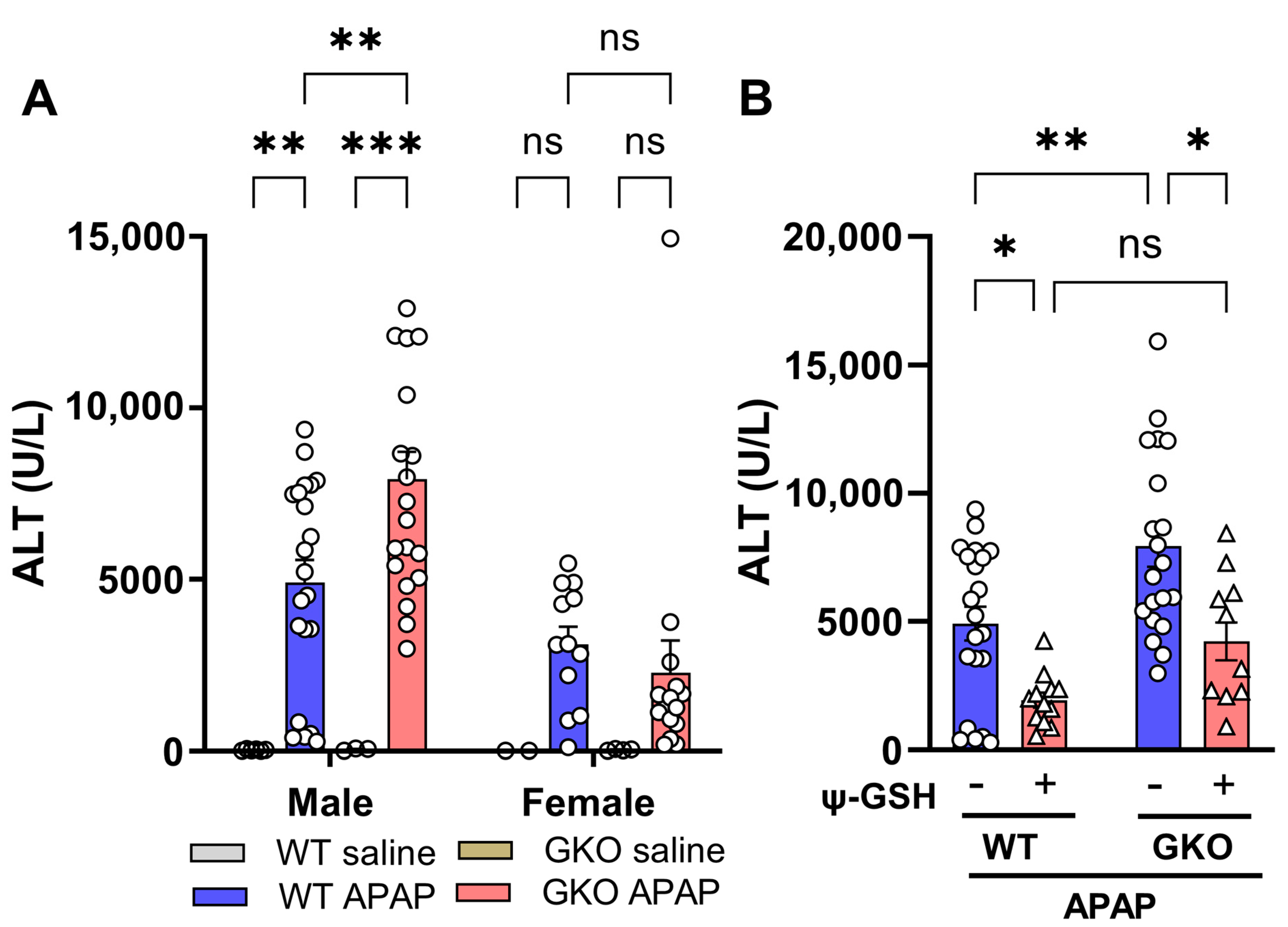
3.1. Liver Toxicity Induced by APAP Overdose Is Potentiated in Glo-1-Knockout Mice
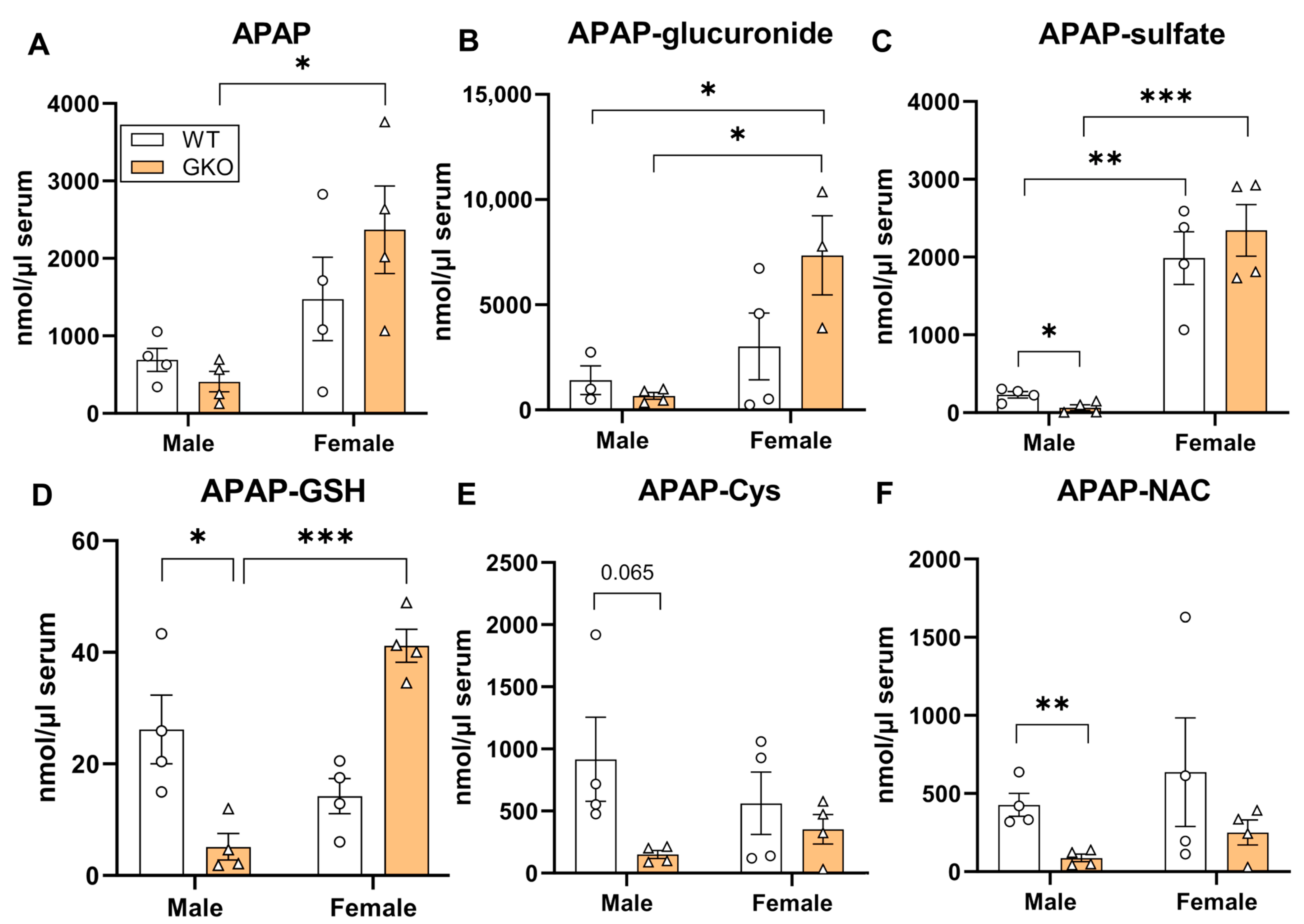
3.2. Deletion of Glo-1 Reduced Overall Metabolic Detoxification of APAP
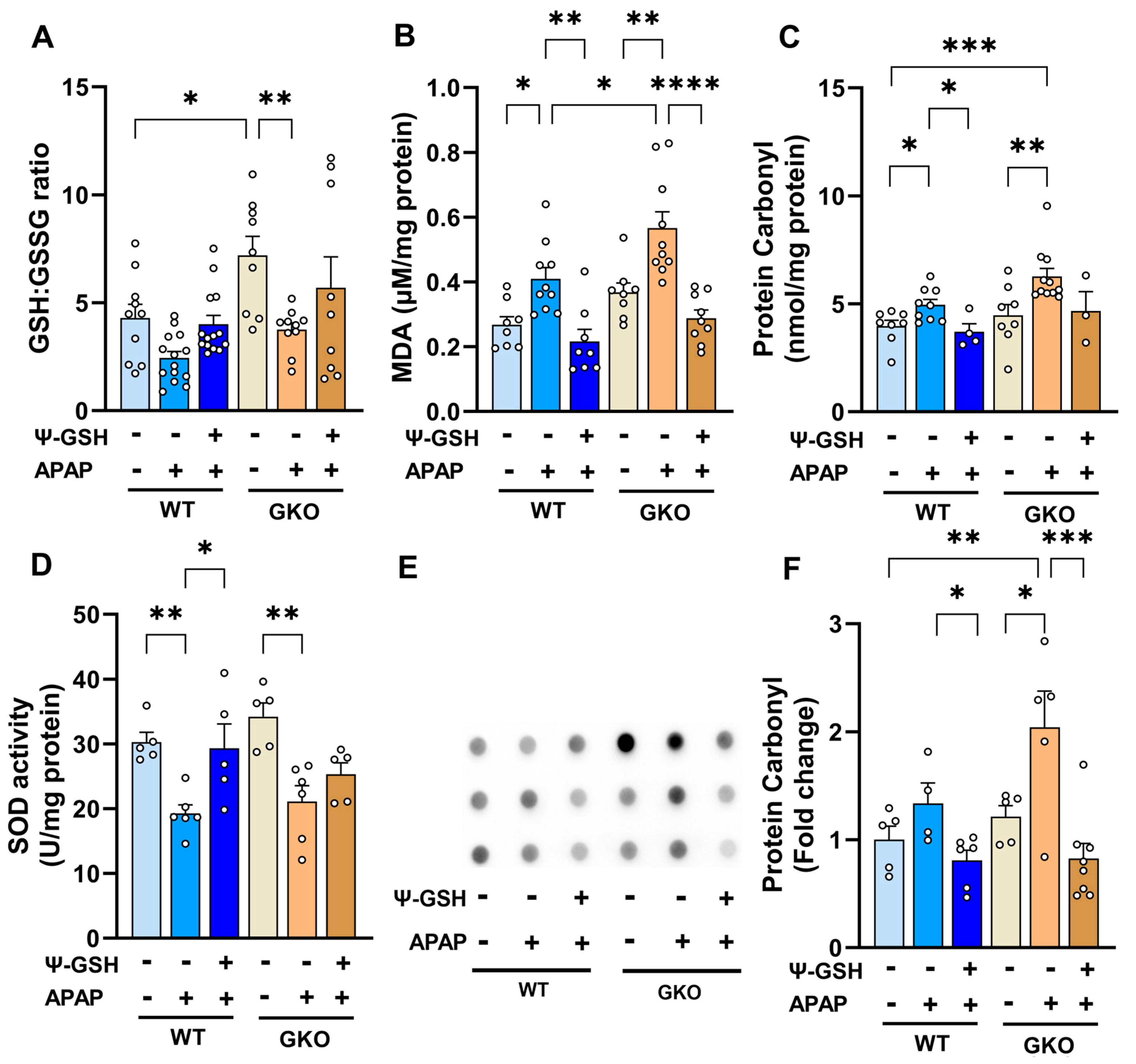
3.3. Increased Oxidative Stress Observed in APAP-Treated Glo-1 KO Mice
3.4. Increased Methylglyoxal-Derived AGE Levels in the Livers of Glo-1-Deleted Mice
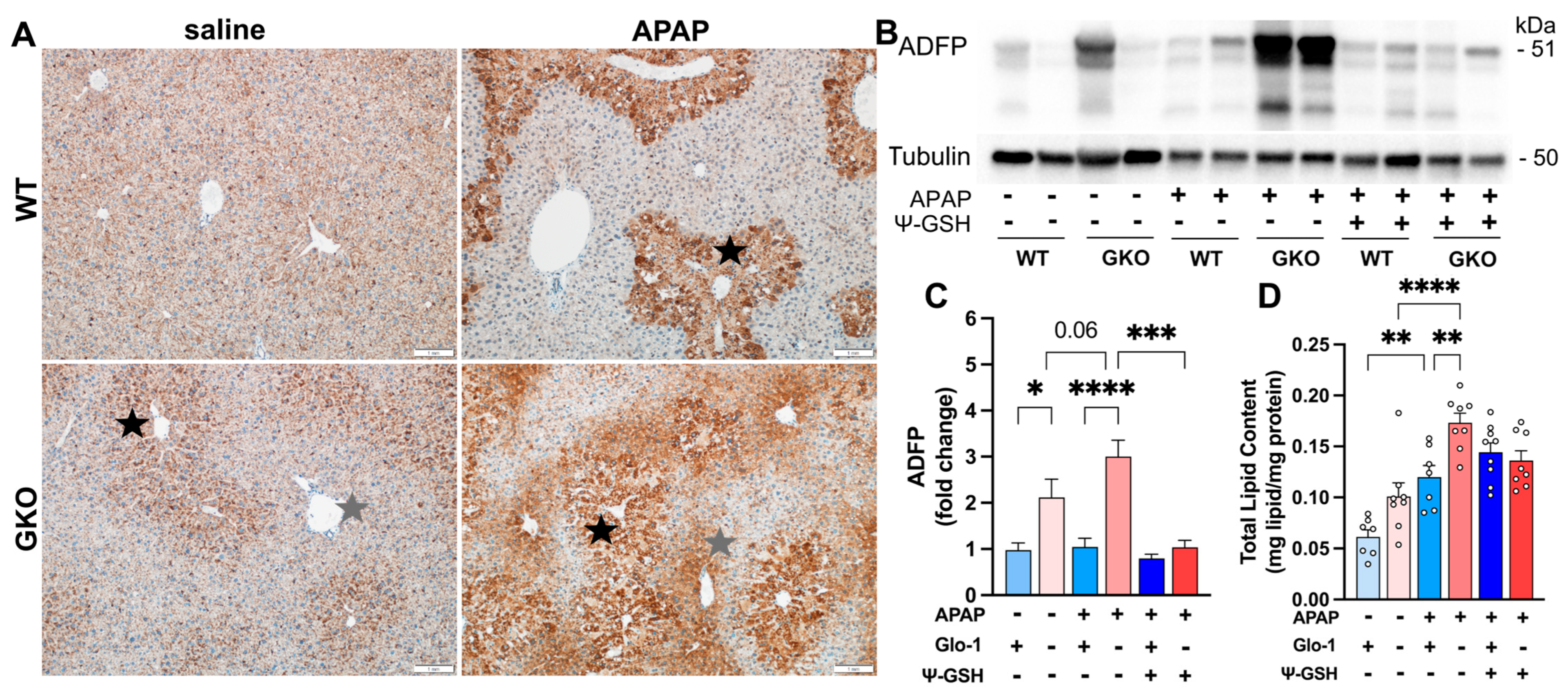
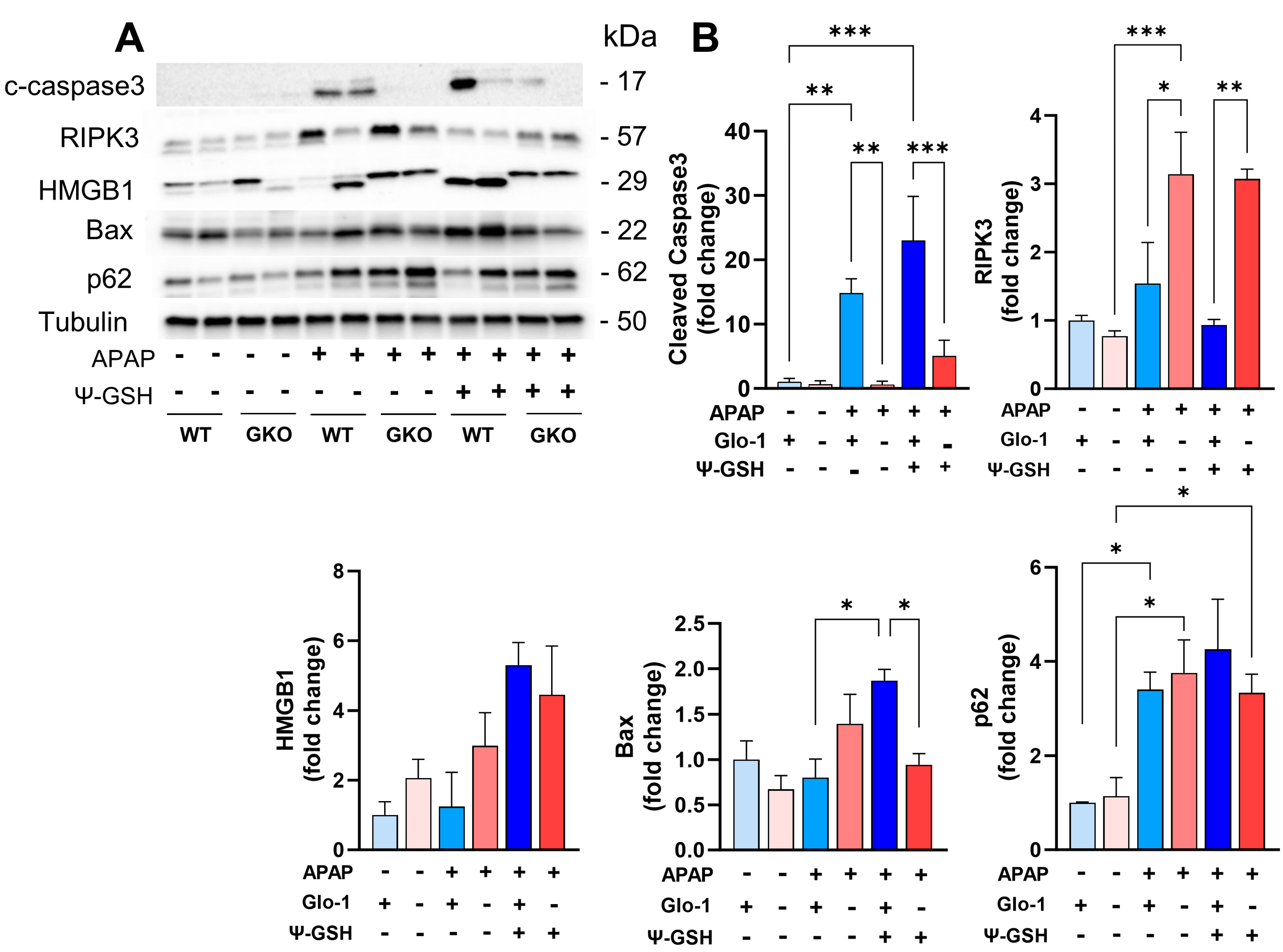
3.5. Distinct Cell Death Mechanisms Activated by High-Dose APAP in the Absence of Glo-1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cichoż-Lach, H.; Michalak, A. Oxidative stress as a crucial factor in liver diseases. World J. Gastroenterol. 2014, 20, 8082–8091. [Google Scholar] [CrossRef] [PubMed]
- Maessen, D.E.; Stehouwer, C.D.; Schalkwijk, C.G. The role of methylglyoxal and the glyoxalase system in diabetes and other age-related diseases. Clin. Sci. 2015, 128, 839–861. [Google Scholar] [CrossRef] [PubMed]
- Hollenbach, M. The Role of Glyoxalase-I (Glo-I), Advanced Glycation Endproducts (AGEs), and Their Receptor (RAGE) in Chronic Liver Disease and Hepatocellular Carcinoma (HCC). Int. J. Mol. Sci. 2017, 18, 2466. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zhou, C.; Huang, M.; Tang, C.; Liu, X.; Yue, Y.; Diao, Q.; Zheng, Z.; Liu, D. Glyoxalase system: A systematic review of its biological activity, related-diseases, screening methods and small molecule regulators. Biomed. Pharmacother. 2020, 131, 110663. [Google Scholar] [CrossRef] [PubMed]
- Basta, G.; Navarra, T.; De Simone, P.; Del Turco, S.; Gastaldelli, A.; Filipponi, F. What is the role of the receptor for advanced glycation end products-ligand axis in liver injury? Liver Transpl. 2011, 17, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Ekong, U.; Zeng, S.; Dun, H.; Feirt, N.; Guo, J.; Ippagunta, N.; Guarrera, J.V.; Lu, Y.; Weinberg, A.; Qu, W.; et al. Blockade of the receptor for advanced glycation end products attenuates acetaminophen-induced hepatotoxicity in mice. J. Gastroenterol. Hepatol. 2006, 21, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Serban, A.I.; Stanca, L.; Geicu, O.I.; Munteanu, M.C.; Dinischiotu, A. RAGE and TGF-β1 Cross-Talk Regulate Extracellular Matrix Turnover and Cytokine Synthesis in AGEs Exposed Fibroblast Cells. PLoS ONE 2016, 11, e0152376. [Google Scholar] [CrossRef] [PubMed]
- Scirè, A.; Cianfruglia, L.; Minnelli, C.; Romaldi, B.; Laudadio, E.; Galeazzi, R.; Antognelli, C.; Armeni, T. Glyoxalase 2: Towards a Broader View of the Second Player of the Glyoxalase System. Antioxidants 2022, 11, 2131. [Google Scholar] [CrossRef] [PubMed]
- Hollenbach, M.; Thonig, A.; Pohl, S.; Ripoll, C.; Michel, M.; Zipprich, A. Expression of glyoxalase-I is reduced in cirrhotic livers: A possible mechanism in the development of cirrhosis. PLoS ONE 2017, 12, e0171260. [Google Scholar] [CrossRef]
- Shi, L.; Hao, Z.; Zhang, S.; Wei, M.; Lu, B.; Wang, Z.; Ji, L. Baicalein and baicalin alleviate acetaminophen-induced liver injury by activating Nrf2 antioxidative pathway: The involvement of ERK1/2 and PKC. Biochem. Pharmacol. 2018, 150, 9–23. [Google Scholar] [CrossRef]
- Saito, C.; Zwingmann, C.; Jaeschke, H. Novel mechanisms of protection against acetaminophen hepatotoxicity in mice by glutathione and N-acetylcysteine. Hepatology 2010, 51, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Trenti, T.; Bertolotti, M.; Castellana, C.N.; Ferrari, A.; Pini, L.A.; Sternieri, E. Plasma glutathione level in paracetamol daily abuser patients. Changes in plasma cysteine and thiol groups after reduced glutathione administration. Toxicol. Lett. 1992, 64–65, 757–761. [Google Scholar] [CrossRef] [PubMed]
- More, S.S.; Nugent, J.; Vartak, A.P.; Nye, S.M.; Vince, R. Hepatoprotective Effect of ψ-Glutathione in a Murine Model of Acetaminophen-Induced Liver Toxicity. Chem. Res. Toxicol. 2017, 30, 777–784. [Google Scholar] [CrossRef]
- More, S.S.; Vince, R. Potential of a γ-glutamyl-transpeptidase-stable glutathione analogue against amyloid-β toxicity. ACS Chem. Neurosci. 2012, 3, 204–210. [Google Scholar] [CrossRef]
- Christopher Kwon, Y.I.; Xie, W.; Zhu, H.; Xie, J.; Shinn, K.; Juckel, N.; Vince, R.; More, S.S.; Lee, M.K. γ-Glutamyl-Transpeptidase-Resistant Glutathione Analog Attenuates Progression of Alzheimer’s Disease-like Pathology and Neurodegeneration in a Mouse Model. Antioxidants 2021, 10, 1796. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.; Kwon, D.M.; Kwon, K.; Park, C. Generation and characterization of mouse knockout for glyoxalase 1. Biochem. Biophys. Res. Commun. 2017, 490, 460–465. [Google Scholar] [CrossRef] [PubMed]
- McMahon, A.; Lu, H.; Butovich, I.A. The spectrophotometric sulfo-phospho-vanillin assessment of total lipids in human meibomian gland secretions. Lipids 2013, 48, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Xie, J.; Vince, R.; More, S.S. Guanabenz Attenuates Acetaminophen-Induced Liver Toxicity and Synergizes Analgesia in Mice. Chem. Res. Toxicol. 2020, 33, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Cederbaum, A.I. Adenovirus-Mediated Expression of CYP2E1 Produces Liver Toxicity in Mice. Toxicol. Sci. 2006, 91, 365–371. [Google Scholar] [CrossRef]
- Duan, L.; Davis, J.S.; Woolbright, B.L.; Du, K.; Cahkraborty, M.; Weemhoff, J.; Jaeschke, H.; Bourdi, M. Differential susceptibility to acetaminophen-induced liver injury in sub-strains of C57BL/6 mice: 6N versus 6J. Food Chem. Toxicol. 2016, 98, 107–118. [Google Scholar] [CrossRef]
- de Almeida, G.R.L.; Szczepanik, J.C.; Selhorst, I.; Cunha, M.P.; Dafre, A.L. The expanding impact of methylglyoxal on behavior-related disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 2023, 120, 110635. [Google Scholar] [CrossRef] [PubMed]
- Distler, M.G.; Plant, L.D.; Sokoloff, G.; Hawk, A.J.; Aneas, I.; Wuenschell, G.E.; Termini, J.; Meredith, S.C.; Nobrega, M.A.; Palmer, A.A. Glyoxalase 1 increases anxiety by reducing GABAA receptor agonist methylglyoxal. J. Clin. Investig. 2012, 122, 2306–2315. [Google Scholar] [CrossRef] [PubMed]
- Hinson, J.A.; Reid, A.B.; McCullough, S.S.; James, L.P. Acetaminophen-induced hepatotoxicity: Role of metabolic activation, reactive oxygen/nitrogen species, and mitochondrial permeability transition. Drug Metab. Rev. 2004, 36, 805–822. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.W.; Bucci, T.J.; Benson, R.W.; Warbritton, A.R.; McRae, T.A.; Pumford, N.R.; Hinson, J.A. Immunohistochemical localization and quantification of the 3-(cystein-S-yl)-acetaminophen protein adduct in acetaminophen hepatotoxicity. Am. J. Pathol. 1991, 138, 359–371. [Google Scholar] [PubMed]
- Spanos, C.; Maldonado, E.M.; Fisher, C.P.; Leenutaphong, P.; Oviedo-Orta, E.; Windridge, D.; Salguero, F.J.; Bermúdez-Fajardo, A.; Weeks, M.E.; Evans, C.; et al. Proteomic identification and characterization of hepatic glyoxalase 1 dysregulation in non-alcoholic fatty liver disease. Proteome Sci. 2018, 16, 4. [Google Scholar] [CrossRef]
- Videla, L.A.; Rodrigo, R.; Orellana, M.; Fernandez, V.; Tapia, G.; Quiñones, L.; Varela, N.; Contreras, J.; Lazarte, R.; Csendes, A.; et al. Oxidative stress-related parameters in the liver of non-alcoholic fatty liver disease patients. Clin. Sci. 2004, 106, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Okumura, T. Role of lipid droplet proteins in liver steatosis. J. Physiol. Biochem. 2011, 67, 629–636. [Google Scholar] [CrossRef]
- Ma, J.; Wei, Z.; Wang, Q.; Lu, X.; Zhou, Z.; Li, R.; Shu, Q.; Liu, Y.; Wang, J.; Liu, N.; et al. Association of serum creatinine with hepatic steatosis and fibrosis: A cross-sectional study. BMC Gastroenterol. 2022, 22, 358. [Google Scholar] [CrossRef]
- Monteillet, L.; Gjorgjieva, M.; Silva, M.; Verzieux, V.; Imikirene, L.; Duchampt, A.; Guillou, H.; Mithieux, G.; Rajas, F. Intracellular lipids are an independent cause of liver injury and chronic kidney disease in non alcoholic fatty liver disease-like context. Mol. Metab. 2018, 16, 100–115. [Google Scholar] [CrossRef]
- Tan, H.; Stathakis, P.; Varghese, B.; Buckley, N.A.; Chiew, A.L. Delayed Acetaminophen Absorption Resulting in Acute Liver Failure. Case Rep. Crit. Care 2022, 2022, 3672248. [Google Scholar] [CrossRef]
- Ramachandran, A.; Jaeschke, H. Mechanisms of acetaminophen hepatotoxicity and their translation to the human pathophysiology. J. Clin. Transl. Res. 2017, 3, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Bajt, M.L.; Ramachandran, A.; Yan, H.M.; Lebofsky, M.; Farhood, A.; Lemasters, J.J.; Jaeschke, H. Apoptosis-inducing factor modulates mitochondrial oxidant stress in acetaminophen hepatotoxicity. Toxicol. Sci. 2011, 122, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H. Glutathione disulfide formation and oxidant stress during acetaminophen-induced hepatotoxicity in mice in vivo: The protective effect of allopurinol. J. Pharmacol. Exp. Ther. 1990, 255, 935–941. [Google Scholar] [PubMed]
- Ramachandran, A.; Lebofsky, M.; Weinman, S.A.; Jaeschke, H. The impact of partial manganese superoxide dismutase (SOD2)-deficiency on mitochondrial oxidant stress, DNA fragmentation and liver injury during acetaminophen hepatotoxicity. Toxicol. Appl. Pharmacol. 2011, 251, 226–233. [Google Scholar] [CrossRef] [PubMed]
- McGill, M.R.; Lebofsky, M.; Norris, H.R.; Slawson, M.H.; Bajt, M.L.; Xie, Y.; Williams, C.D.; Wilkins, D.G.; Rollins, D.E.; Jaeschke, H. Plasma and liver acetaminophen-protein adduct levels in mice after acetaminophen treatment: Dose-response, mechanisms, and clinical implications. Toxicol. Appl. Pharmacol. 2013, 269, 240–249. [Google Scholar] [CrossRef]
- Morgenstern, J.; Fleming, T.; Schumacher, D.; Eckstein, V.; Freichel, M.; Herzig, S.; Nawroth, P. Loss of Glyoxalase 1 Induces Compensatory Mechanism to Achieve Dicarbonyl Detoxification in Mammalian Schwann Cells. J. Biol. Chem. 2017, 292, 3224–3238. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, D.; Morgenstern, J.; Oguchi, Y.; Volk, N.; Kopf, S.; Groener, J.B.; Nawroth, P.P.; Fleming, T.; Freichel, M. Compensatory mechanisms for methylglyoxal detoxification in experimental & clinical diabetes. Mol. Metab. 2018, 18, 143–152. [Google Scholar] [CrossRef] [PubMed]
- More, S.S.; Vince, R. Inhibition of glyoxalase I: The first low-nanomolar tight-binding inhibitors. J. Med. Chem. 2009, 52, 4650–4656. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Toyoshima, M.; Hisano, Y.; Balan, S.; Iwayama, Y.; Aono, H.; Futamura, Y.; Osada, H.; Owada, Y.; Yoshikawa, T. Glyoxalase I disruption and external carbonyl stress impair mitochondrial function in human induced pluripotent stem cells and derived neurons. Transl. Psychiatry 2021, 11, 275. [Google Scholar] [CrossRef]
- Yuan, J.; Najafov, A.; Py, B.F. Roles of Caspases in Necrotic Cell Death. Cell 2016, 167, 1693–1704. [Google Scholar] [CrossRef]
- Jaeschke, H.; Ramachandran, A. Acetaminophen-induced apoptosis: Facts versus fiction. J. Clin. Transl. Res. 2020, 6, 36–47. [Google Scholar] [PubMed]
- Chao, X.; Wang, H.; Jaeschke, H.; Ding, W.X. Role and mechanisms of autophagy in acetaminophen-induced liver injury. Liver Int. 2018, 38, 1363–1374. [Google Scholar] [CrossRef] [PubMed]
- Magna, M.; Pisetsky, D.S. The Role of HMGB1 in the Pathogenesis of Inflammatory and Autoimmune Diseases. Mol. Med. 2014, 20, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; McGill, M.R.; Xie, Y.; Ni, H.M.; Ding, W.X.; Jaeschke, H. Receptor interacting protein kinase 3 is a critical early mediator of acetaminophen-induced hepatocyte necrosis in mice. Hepatology 2013, 58, 2099–2108. [Google Scholar] [CrossRef]
- Minsart, C.; Liefferinckx, C.; Lemmers, A.; Dressen, C.; Quertinmont, E.; Leclercq, I.; Devière, J.; Moreau, R.; Gustot, T. New insights in acetaminophen toxicity: HMGB1 contributes by itself to amplify hepatocyte necrosis in vitro through the TLR4-TRIF-RIPK3 axis. Sci. Rep. 2020, 10, 5557. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Mu, N.; Gu, C.; Liu, M.; Yang, Z.; Yin, Y.; Chen, M.; Wang, Y.; Han, Y.; Yu, L.; et al. Metformin mediates cardioprotection against aging-induced ischemic necroptosis. Aging Cell 2020, 19, e13096. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Williams, C.D.; McGill, M.R.; Jaeschke, H. Lower susceptibility of female mice to acetaminophen hepatotoxicity: Role of mitochondrial glutathione, oxidant stress and c-jun N-terminal kinase. Toxicol. Appl. Pharmacol. 2014, 281, 58–66. [Google Scholar] [CrossRef]
- Bao, Y.; Wang, P.; Shao, X.; Zhu, J.; Xiao, J.; Shi, J.; Zhang, L.; Zhu, H.J.; Ma, X.; Manautou, J.E.; et al. Acetaminophen-Induced Liver Injury Alters Expression and Activities of Cytochrome P450 Enzymes in an Age-Dependent Manner in Mouse Liver. Drug Metab. Dispos. 2020, 48, 326–336. [Google Scholar] [CrossRef]
- Rabbani, N.; Xue, M.; Thornalley, P.J. Dicarbonyls and glyoxalase in disease mechanisms and clinical therapeutics. Glycoconj. J. 2016, 33, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Matsui, T. Role of receptor for advanced glycation end products (RAGE) in liver disease. Eur. J. Med. Res. 2015, 20, 15. [Google Scholar] [CrossRef]
- Chang, B.H.; Li, L.; Paul, A.; Taniguchi, S.; Nannegari, V.; Heird, W.C.; Chan, L. Protection against fatty liver but normal adipogenesis in mice lacking adipose differentiation-related protein. Mol. Cell Biol. 2006, 26, 1063–1076. [Google Scholar] [CrossRef] [PubMed]
- Moraru, A.; Wiederstein, J.; Pfaff, D.; Fleming, T.; Miller, A.K.; Nawroth, P.; Teleman, A.A. Elevated Levels of the Reactive Metabolite Methylglyoxal Recapitulate Progression of Type 2 Diabetes. Cell Metab. 2018, 27, 926–934.e928. [Google Scholar] [CrossRef] [PubMed]
- Lodd, E.; Wiggenhauser, L.M.; Morgenstern, J.; Fleming, T.H.; Poschet, G.; Büttner, M.; Tabler, C.T.; Wohlfart, D.P.; Nawroth, P.P.; Kroll, J. The combination of loss of glyoxalase1 and obesity results in hyperglycemia. JCI Insight 2019, 4, 126154. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, K.; Kanno, K.; Hyogo, H.; Yamagishi, S.; Takeuchi, M.; Tazuma, S.; Chayama, K. Advanced glycation end products enhance the proliferation and activation of hepatic stellate cells. J. Gastroenterol. 2008, 43, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Akazawa, Y.; Nakao, K. To die or not to die: Death signaling in nonalcoholic fatty liver disease. J. Gastroenterol. 2018, 53, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Ferret, P.J.; Hammoud, R.; Tulliez, M.; Tran, A.; Trébéden, H.; Jaffray, P.; Malassagne, B.; Calmus, Y.; Weill, B.; Batteux, F. Detoxification of reactive oxygen species by a nonpeptidyl mimic of superoxide dismutase cures acetaminophen-induced acute liver failure in the mouse. Hepatology 2001, 33, 1173–1180. [Google Scholar] [CrossRef] [PubMed]
- Mirochnitchenko, O.; Weisbrot-Lefkowitz, M.; Reuhl, K.; Chen, L.; Yang, C.; Inouye, M. Acetaminophen toxicity. Opposite effects of two forms of glutathione peroxidase. J. Biol. Chem. 1999, 274, 10349–10355. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wei, S.; Zhou, H.; Shen, G.; Gan, X.; Zhou, S.; Qiu, J.; Shi, C.; Lu, L. Hyperglycemia exacerbates acetaminophen-induced acute liver injury by promoting liver-resident macrophage proinflammatory response via AMPK/PI3K/AKT-mediated oxidative stress. Cell Death Discov. 2019, 5, 119. [Google Scholar] [CrossRef]
- Khayyat, A.; Tobwala, S.; Hart, M.; Ercal, N. N-acetylcysteine amide, a promising antidote for acetaminophen toxicity. Toxicol. Lett. 2016, 241, 133–142. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dobariya, P.; Xie, W.; Rao, S.P.; Xie, J.; Seelig, D.M.; Vince, R.; Lee, M.K.; More, S.S. Deletion of Glyoxalase 1 Exacerbates Acetaminophen-Induced Hepatotoxicity in Mice. Antioxidants 2024, 13, 648. https://doi.org/10.3390/antiox13060648
Dobariya P, Xie W, Rao SP, Xie J, Seelig DM, Vince R, Lee MK, More SS. Deletion of Glyoxalase 1 Exacerbates Acetaminophen-Induced Hepatotoxicity in Mice. Antioxidants. 2024; 13(6):648. https://doi.org/10.3390/antiox13060648
Chicago/Turabian StyleDobariya, Prakashkumar, Wei Xie, Swetha Pavani Rao, Jiashu Xie, Davis M. Seelig, Robert Vince, Michael K. Lee, and Swati S. More. 2024. "Deletion of Glyoxalase 1 Exacerbates Acetaminophen-Induced Hepatotoxicity in Mice" Antioxidants 13, no. 6: 648. https://doi.org/10.3390/antiox13060648
APA StyleDobariya, P., Xie, W., Rao, S. P., Xie, J., Seelig, D. M., Vince, R., Lee, M. K., & More, S. S. (2024). Deletion of Glyoxalase 1 Exacerbates Acetaminophen-Induced Hepatotoxicity in Mice. Antioxidants, 13(6), 648. https://doi.org/10.3390/antiox13060648