
Lovastatin-Induced Mitochondrial Oxidative Stress Leads to the Release of mtDNA to Promote Apoptosis by Activating cGAS-STING Pathway in Human Colorectal Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Cell Transfection and Lentivirus for Gene Knockdown
2.4. Antibodies
2.5. Cell Viability Assay
2.6. Mitochondrial DNA-Depleted ρ0 Cells
2.7. Immunoprecipitation/PCR
2.8. Quantitative Real-Time PCR
2.9. mtDNA Determination in Cytosolic Extracts
2.10. Annexin V/PI Staining
2.11. Seahorse XFp Respirometry Assay
2.12. ROS Measurement
2.13. Mitochondrial ROS Measurement
2.14. Mitochondrial Membrane Potential Measurement
2.15. Immunoblotting
2.16. Mitochondrial DNA Integrity Determination
2.17. Immunocytochemistry Staining
2.18. Total DNA 8-oxoG Content Measurement
2.19. Animal Experiments
2.20. Statistical Analyses
3. Results
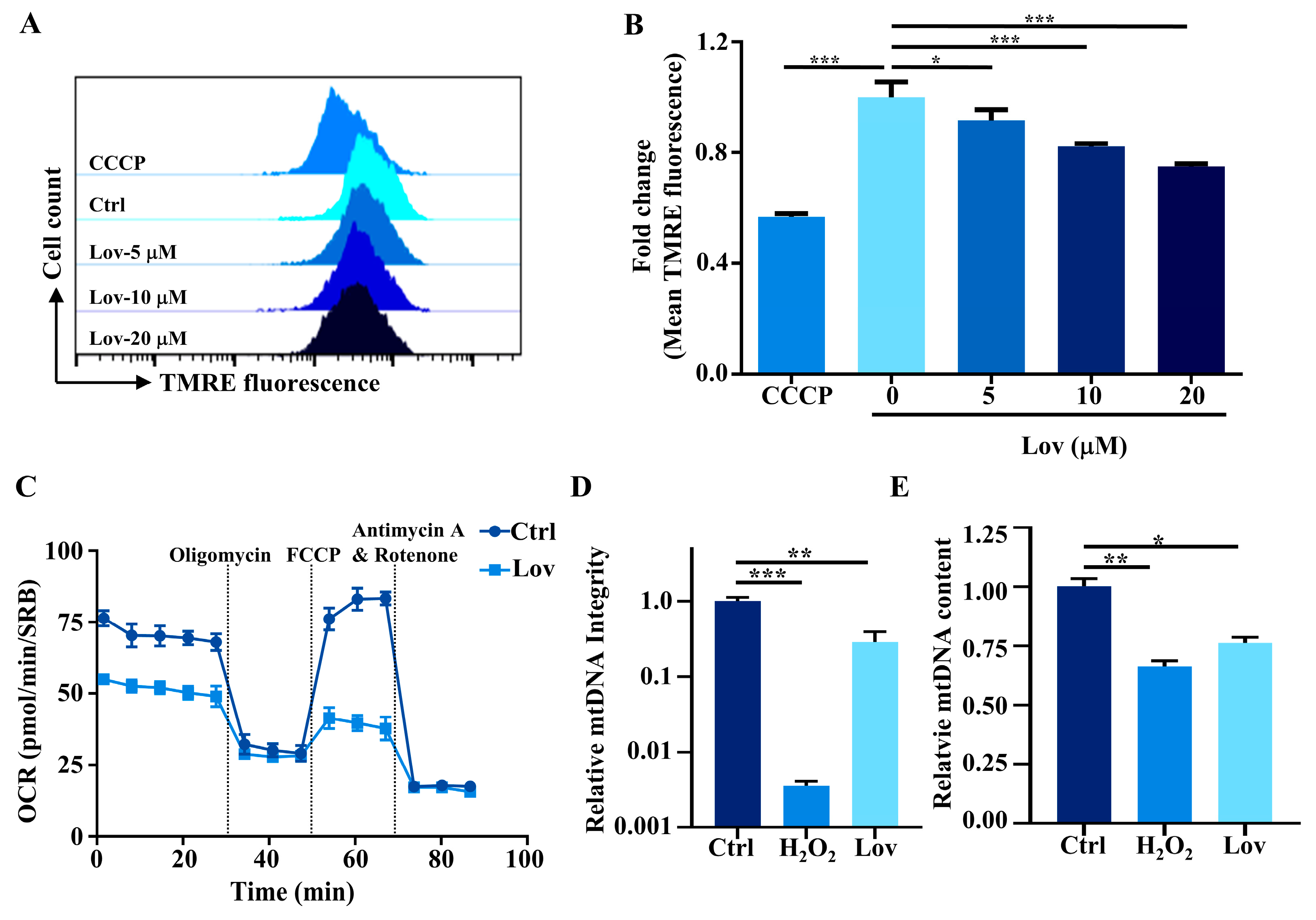
3.1. Lovastatin Exposure Induces Mitochondrial Dysfunction in HCT116 Cells
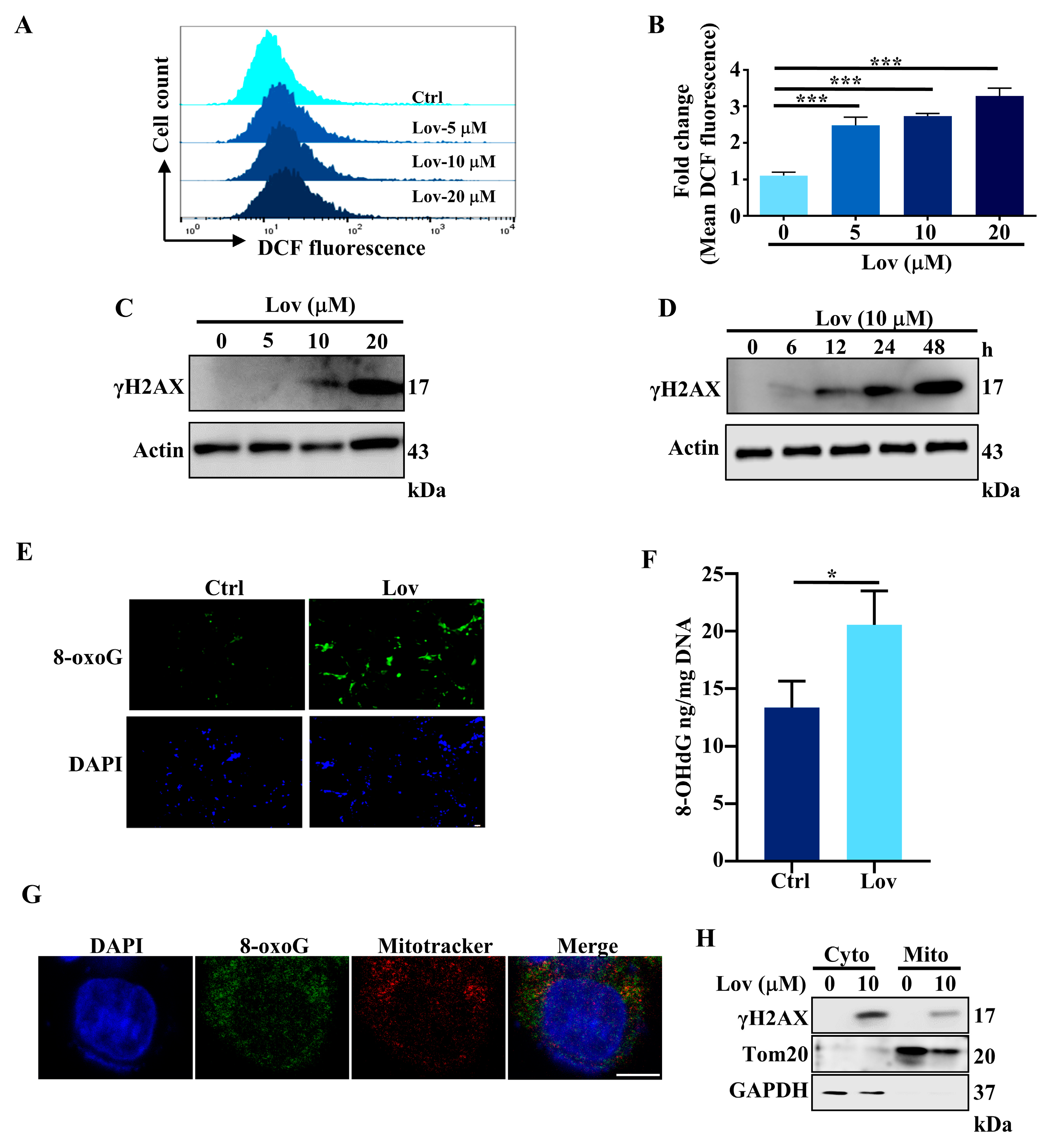
3.2. Lovastatin Exposure Induces DNA Damage in HCT116 Cells
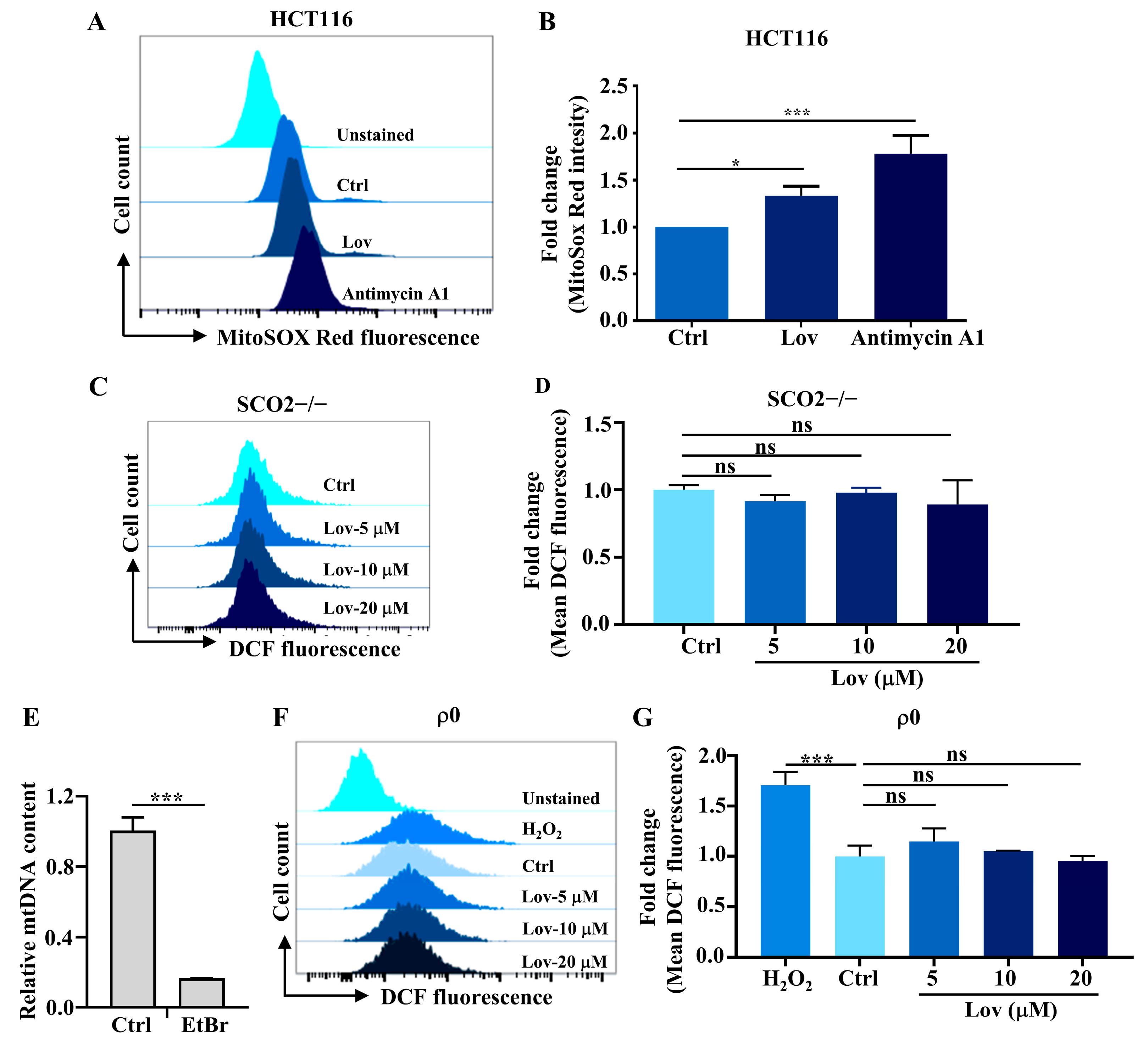
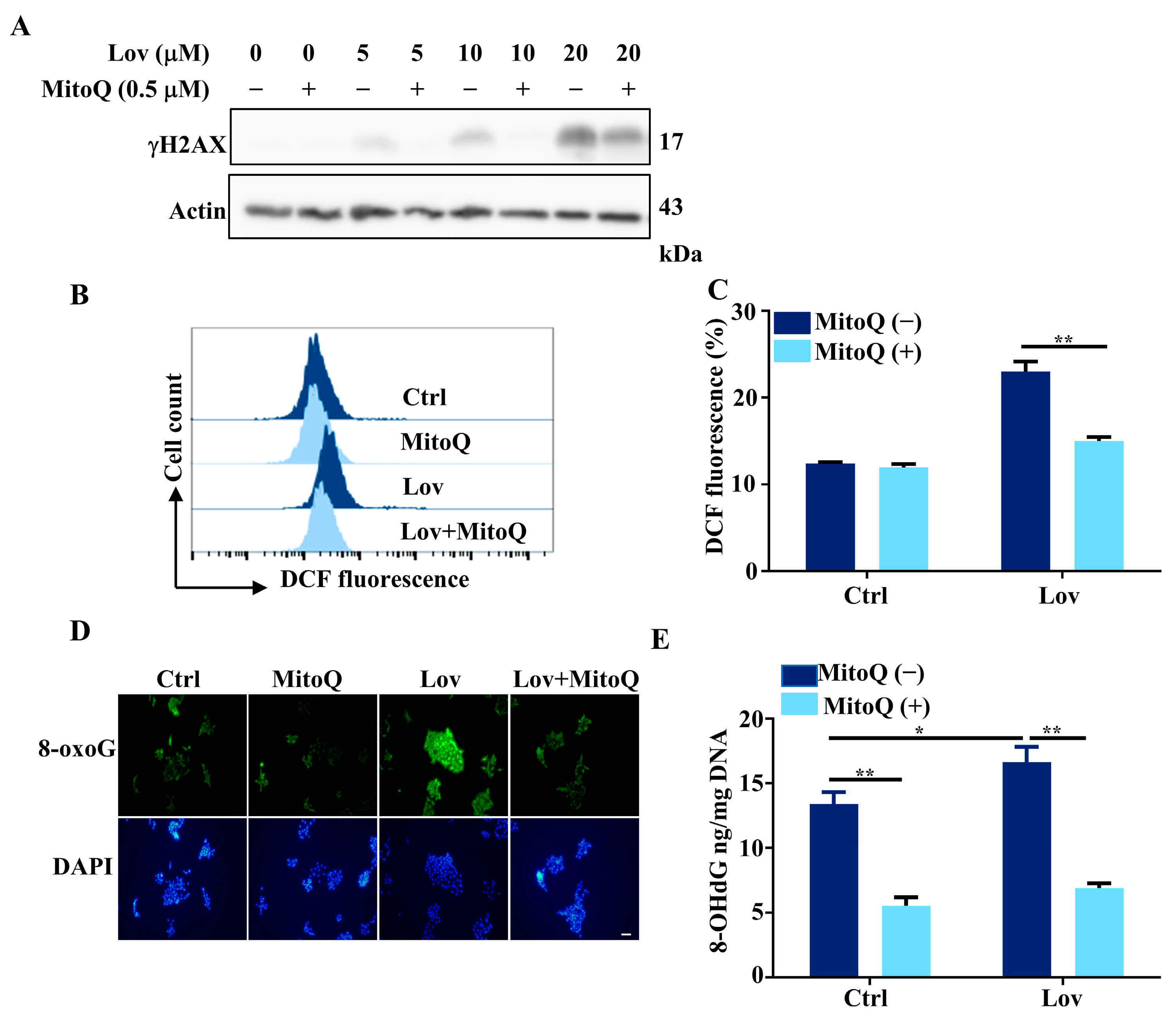
3.3. Mitochondria ROS Generation Contributes to Lovastatin-Induced DNA Damage in HCT116 Cells
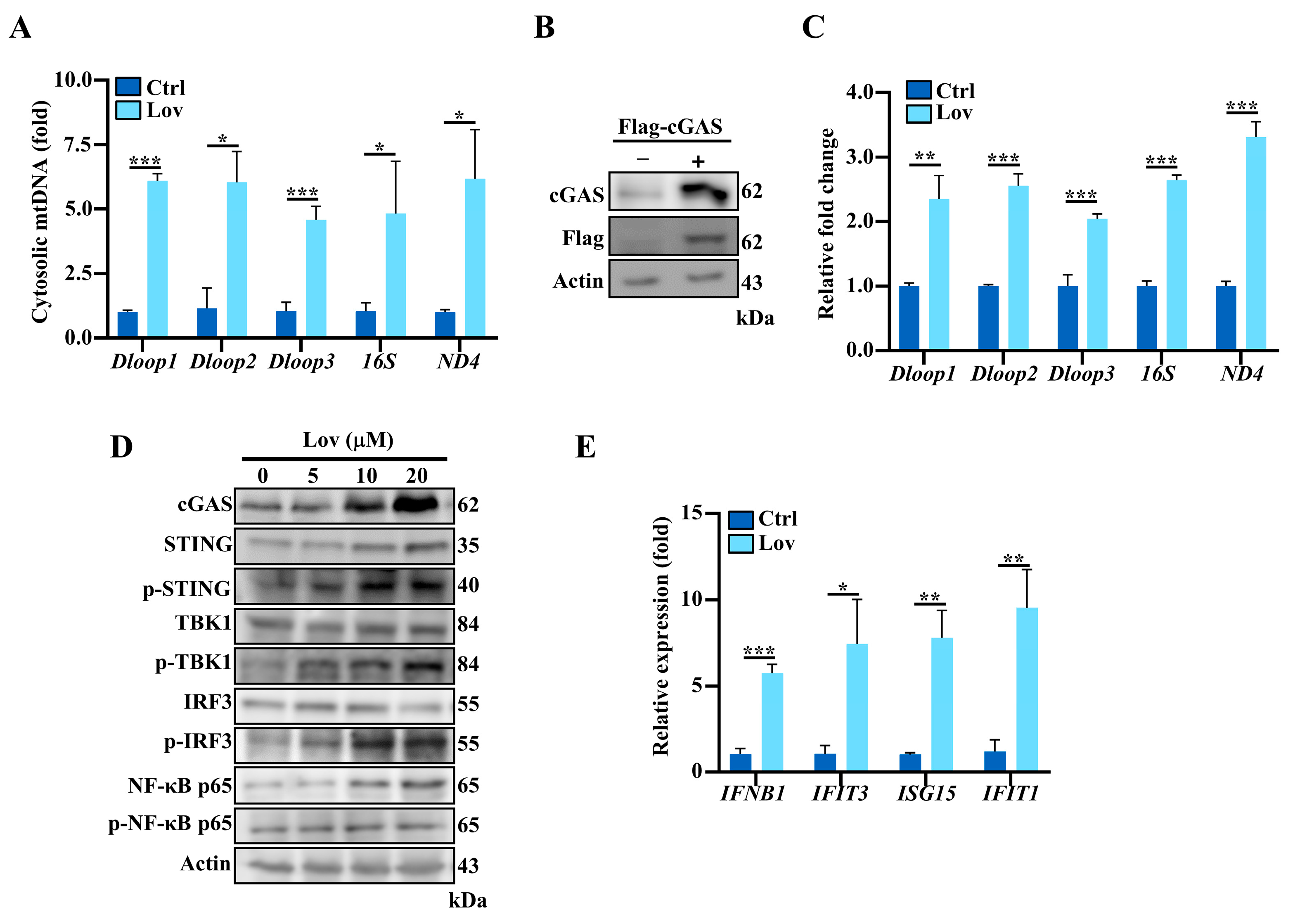
3.4. Lovastatin Promotes Accumulation of Cytosolic mtDNA and Activates cGAS-STING Pathway
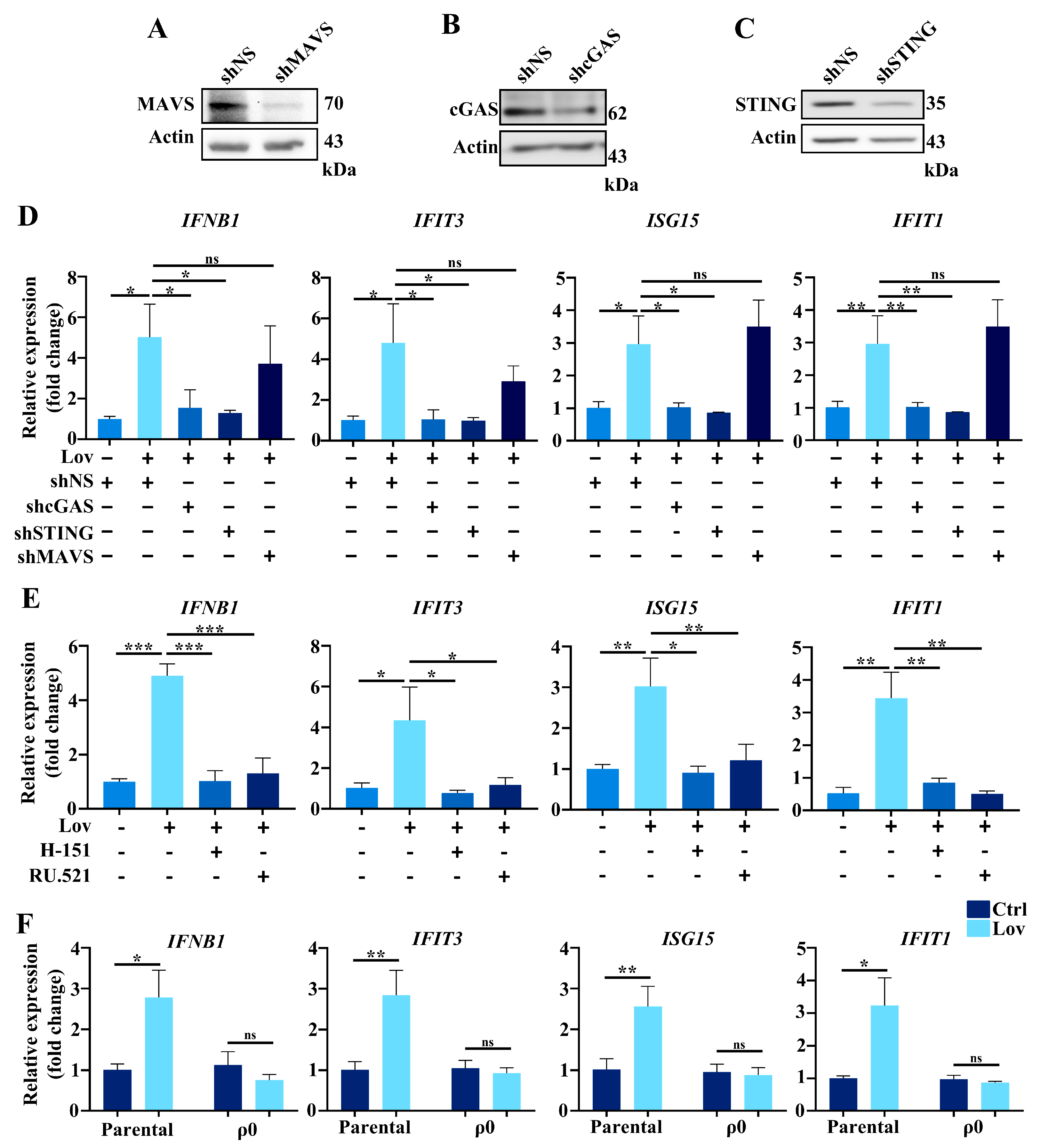
3.5. Type 1 IFNs Signaling Induced by Lovastatin Is Dependent on cGAS/STING
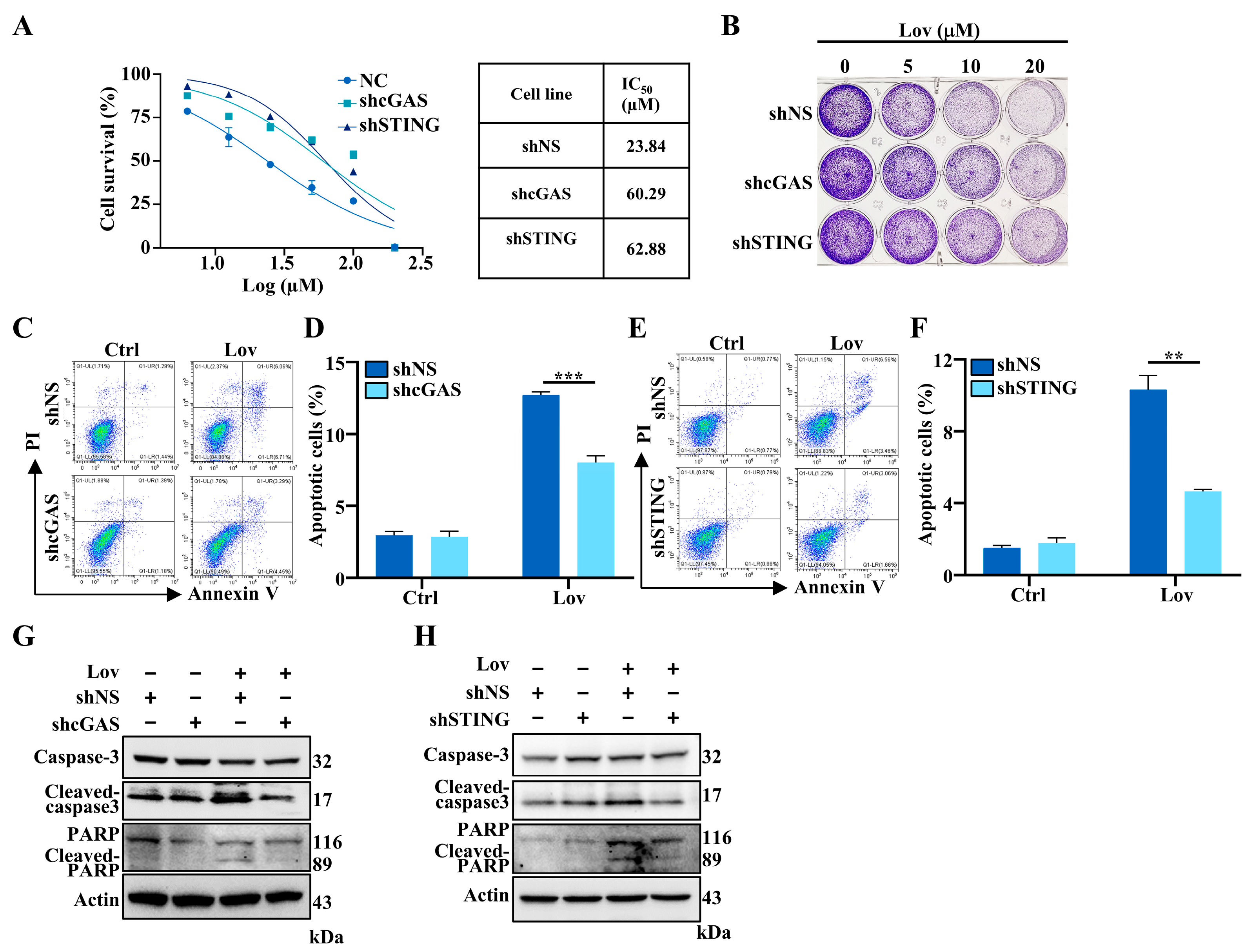
3.6. Lovastatin-Induced Apoptosis Partially Mediated by cGAS-STING Pathway in HCT16 Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jiang, W.; Hu, J.W.; He, X.R.; Jin, W.L.; He, X.Y. Statins: A repurposed drug to fight cancer. J. Exp. Clin. Cancer Res. 2021, 40, 241. [Google Scholar] [CrossRef]
- Yu, D.; Liao, J.K. Emerging views of statin pleiotropy and cholesterol lowering. Cardiovasc. Res. 2022, 118, 413–423. [Google Scholar] [CrossRef]
- Gobel, A.; Rauner, M.; Hofbauer, L.C.; Rachner, T.D. Cholesterol and beyond—The role of the mevalonate pathway in cancer biology. Biochim. Biophys. Acta. Rev. Cancer 2020, 1873, 188351. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.F.; Nordestgaard, B.G.; Bojesen, S.E. Statin use and reduced cancer-related mortality. N. Engl. J. Med. 2012, 367, 1792–1802. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; He, X.; Ding, Y.; Chen, H.; Sun, L. Statin uses and mortality in colorectal cancer patients: An updated systematic review and meta-analysis. Cancer Med. 2019, 8, 3305–3313. [Google Scholar] [CrossRef]
- Dyall, S.D.; Brown, M.T.; Johnson, P.J. Ancient invasions: From endosymbionts to organelles. Science 2004, 304, 253–257. [Google Scholar] [CrossRef]
- Youle, R.J. Mitochondria-Striking a balance between host and endosymbiont. Science 2019, 365, eaaw9855. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Yakes, F.M.; VanHouten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef]
- Hernansanz-Agustin, P.; Enriquez, J.A. Generation of Reactive Oxygen Species by Mitochondria. Antioxidants 2021, 10, 415. [Google Scholar] [CrossRef]
- Coppadoro, A.; Berra, L.; Kumar, A.; Pinciroli, R.; Yamada, M.; Schmidt, U.H.; Bittner, E.A.; Kaneki, M. Critical illness is associated with decreased plasma levels of coenzyme Q10: A cross-sectional study. J. Crit. Care 2013, 28, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Schirris, T.J.J.; Renkema, G.H.; Ritschel, T.; Voermans, N.C.; Bilos, A.; van Engelen, B.G.M.; Brandt, U.; Koopman, W.J.H.; Beyrath, J.D.; Rodenburg, R.J.; et al. Statin-Induced Myopathy Is Associated with Mitochondrial Complex III Inhibition. Cell Metab. 2015, 22, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.J.; Du, J.J.; Lin, W.J.; Long, Z.; Zhang, N.; Huang, X.M.; Xie, Y.; Liu, L.; Ma, W.Z. Regulation of lactate production through p53/beta-enolase axis contributes to statin-associated muscle symptoms. eBioMedicine 2019, 45, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Kwak, H.B.; Thalacker-Mercer, A.; Anderson, E.J.; Lin, C.T.; Kane, D.A.; Lee, N.S.; Cortright, R.N.; Bamman, M.M.; Neufer, P.D. Simvastatin impairs ADP-stimulated respiration and increases mitochondrial oxidative stress in primary human skeletal myotubes. Free Radic. Biol. Med. 2012, 52, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Suematsu, N.; Tsutsui, H.; Wen, J.; Kang, D.; Ikeuchi, M.; Ide, T.; Hayashidani, S.; Shiomi, T.; Kubota, T.; Hamasaki, N.; et al. Oxidative stress mediates tumor necrosis factor-alpha-induced mitochondrial DNA damage and dysfunction in cardiac myocytes. Circulation 2003, 107, 1418–1423. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Wu, Q.; Guo, J.; Ares, I.; Rodriguez, J.L.; Martinez-Larranaga, M.R.; Yuan, Z.; Anadon, A.; Wang, X.; Martinez, M.A. Statins: Adverse reactions, oxidative stress and metabolic interactions. Pharmacol. Ther. 2019, 195, 54–84. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.M.; Huang, J.J.; Du, J.J.; Zhang, N.; Long, Z.; Yang, Y.; Zhong, F.F.; Zheng, B.W.; Shen, Y.F.; Huang, Z.; et al. Autophagy inhibitors increase the susceptibility of KRAS-mutant human colorectal cancer cells to a combined treatment of 2-deoxy-D-glucose and lovastatin. Acta Pharmacol. Sin. 2021, 42, 1875–1887. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef]
- Sun, L.J.; Wu, J.X.; Du, F.H.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Rohl, I.; Hopfner, K.P.; Ludwig, J.; Hornung, V. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature 2013, 498, 380–384. [Google Scholar] [CrossRef]
- Shang, G.J.; Zhang, C.G.; Chen, Z.J.; Bai, X.C.; Zhang, X.W. Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP-AMP. Nature 2019, 567, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Li, X.D.; Wu, J.; Gao, D.; Wang, H.; Sun, L.; Chen, Z.J. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 2013, 341, 1390–1394. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.G.; Shang, G.J.; Gui, X.; Zhang, X.W.; Bai, X.C.; Chen, Z.J. Structural basis of STING binding with and phosphorylation by TBK1. Nature 2019, 567, 394–398. [Google Scholar] [CrossRef]
- Li, T.; Chen, Z.J. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar] [CrossRef]
- Ablasser, A.; Chen, Z.J. cGAS in action: Expanding roles in immunity and inflammation. Science 2019, 363, 6431. [Google Scholar] [CrossRef] [PubMed]
- Hopfner, K.P.; Hornung, V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef] [PubMed]
- Kell, A.M.; Gale, M. RIG-I in RNA virus recognition. Virology 2015, 479, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Dhir, A.; Dhir, S.; Borowski, L.S.; Jimenez, L.; Teitell, M.; Rotig, A.; Crow, Y.J.; Rice, G.I.; Duffy, D.; Tamby, C.; et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 2018, 560, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ye, X.; Dunkert, W.; Song, Y.; Karijolich, J. RIG-I like receptor sensing of host RNAs facilitates the cell-intrinsic immune response to KSHV infection. Nat. Commun. 2018, 9, 4841. [Google Scholar] [CrossRef]
- Marill, J.; Mohamed Anesary, N.; Paris, S. DNA damage enhancement by radiotherapy-activated hafnium oxide nanoparticles improves cGAS-STING pathway activation in human colorectal cancer cells. Radiother. Oncol. 2019, 141, 262–266. [Google Scholar] [CrossRef]
- Li, C.; Liu, W.; Wang, F.; Hayashi, T.; Mizuno, K.; Hattori, S.; Fujisaki, H.; Ikejima, T. DNA damage-triggered activation of cGAS-STING pathway induces apoptosis in human keratinocyte HaCaT cells. Mol. Immunol. 2021, 131, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Gusho, E.; Laimins, L.A. Human papillomaviruses sensitize cells to DNA damage induced apoptosis by targeting the innate immune sensor cGAS. PLoS Pathog. 2022, 18, e1010725. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Huang, J.; Yuan, Z.; Feng, S.; Xie, Y.; Ma, W. Protein kinase C inhibitor chelerythrine selectively inhibits proliferation of triple-negative breast cancer cells. Sci. Rep. 2017, 7, 2022. [Google Scholar] [CrossRef] [PubMed]
- Hashiguchi, K.; Zhang-Akiyama, Q.M. Establishment of human cell lines lacking mitochondrial DNA. Methods Mol. Biol. 2009, 554, 383–391. [Google Scholar] [PubMed]
- Wu, Z.; Oeck, S.; West, A.P.; Mangalhara, K.C.; Sainz, A.G.; Newman, L.E.; Zhang, X.O.; Wu, L.; Yan, Q.; Bosenberg, M.; et al. Mitochondrial DNA Stress Signalling Protects the Nuclear Genome. Nat. Metab. 2019, 1, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Bhattacharjee, S.; Chowdhuri, S.P.; Mallick, A.; Rehman, I.; Basu, S.; Das, B.B. SCAN1-TDP1 trapping on mitochondrial DNA promotes mitochondrial dysfunction and mitophagy. Sci. Adv. 2019, 5, eaax9778. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Wang, P.Y.; Matsumoto, T.; Sung, H.J.; Ma, W.Z.; Choi, J.W.; Anderson, S.A.; Leary, S.C.; Balaban, R.S.; Kang, J.G.; et al. p53 Improves Aerobic Exercise Capacity and Augments Skeletal Muscle Mitochondrial DNA Content. Circ. Res. 2009, 105, 705–712. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef]
- Santos, J.H.; Meyer, J.N.; Mandavilli, B.S.; Van Houten, B. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol. Biol. 2006, 314, 183–199. [Google Scholar]
- Gu, X.; Ma, Y.; Liu, Y.; Wan, Q. Measurement of mitochondrial respiration in adherent cells by Seahorse XF96 Cell Mito Stress Test. STAR Protoc. 2021, 2, 100245. [Google Scholar] [CrossRef]
- Cho, S.I.; Jo, E.R.; Song, H. Urolithin A attenuates auditory cell senescence by activating mitophagy. Sci. Rep. 2022, 12, 7704. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Chen, M.; Jiang, J. Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion 2019, 49, 35–45. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Pilger, A.; Rudiger, H.W. 8-Hydroxy-2′-deoxyguanosine as a marker of oxidative DNA damage related to occupational and environmental exposures. Int. Arch. Occup. Environ. Health 2006, 80, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.J.; Kim, C.W.; Kim, D.R. Cytosolic accumulation of gammaH2AX is associated with tropomyosin-related kinase A-induced cell death in U2OS cells. Exp. Mol. Med. 2008, 40, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.J.; Kim, D.R. Ectopic expression of H2AX protein promotes TrkA-induced cell death via modulation of TrkA tyrosine-490 phosphorylation and JNK activity upon DNA damage. Biochem. Biophys. Res. Commun. 2011, 404, 841–847. [Google Scholar] [CrossRef]
- Jeong, J.H.; Cheol Kang, Y.; Piao, Y.; Kang, S.; Pak, Y.K. miR-24-mediated knockdown of H2AX damages mitochondria and the insulin signaling pathway. Exp. Mol. Med. 2017, 49, e313. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Tolosa, L.; Carmona, A.; Castell, J.V.; Gomez-Lechon, M.J.; Donato, M.T. High-content screening of drug-induced mitochondrial impairment in hepatic cells: Effects of statins. Arch. Toxicol. 2015, 89, 1847–1860. [Google Scholar] [CrossRef]
- Sung, H.J.; Ma, W.; Wang, P.Y.; Hynes, J.; O’Riordan, T.C.; Combs, C.A.; McCoy, J.P., Jr.; Bunz, F.; Kang, J.G.; Hwang, P.M. Mitochondrial respiration protects against oxygen-associated DNA damage. Nat. Commun. 2010, 1, 5. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.; Huynh, N.N.; Hamilton, C.A.; Beattie, E.; Smith, R.A.; Cocheme, H.M.; Murphy, M.P.; Dominiczak, A.F. Mitochondria-targeted antioxidant MitoQ10 improves endothelial function and attenuates cardiac hypertrophy. Hypertension 2009, 54, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.F.; Gruber, J.; Cheah, I.K.; Goo, C.K.; Cheong, W.F.; Shui, G.; Sit, K.P.; Wenk, M.R.; Halliwell, B. The mitochondria-targeted antioxidant MitoQ extends lifespan and improves healthspan of a transgenic Caenorhabditis elegans model of Alzheimer disease. Free Radic. Biol. Med. 2014, 71, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 2020, 183, 636–649. [Google Scholar] [CrossRef] [PubMed]
- Tetikoğlu, S.; Çelik-Uzuner, S. Bee Venom Induces the Interaction between Phosphorylated Histone Variant, H2AX, and the Intracellular Site of beta-Actin in Liver and Breast Cancer Cells. Chem. Biodivers. 2023, 20, e202300401. [Google Scholar] [CrossRef] [PubMed]
- Balka, K.R.; Louis, C.; Saunders, T.L.; Smith, A.M.; Calleja, D.J.; D’Silva, D.B.; Moghaddas, F.; Tailler, M.; Lawlor, K.E.; Zhan, Y.; et al. TBK1 and IKKepsilon Act Redundantly to Mediate STING-Induced NF-kappaB Responses in Myeloid Cells. Cell Rep. 2020, 31, 107492. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Ruiz, P.; Barber, G.N. Intrinsic self-DNA triggers inflammatory disease dependent on STING. J. Immunol. 2014, 193, 4634–4642. [Google Scholar] [CrossRef] [PubMed]
- Rongvaux, A.; Jackson, R.; Harman, C.C.; Li, T.; West, A.P.; de Zoete, M.R.; Wu, Y.; Yordy, B.; Lakhani, S.A.; Kuan, C.Y.; et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 2014, 159, 1563–1577. [Google Scholar] [CrossRef]
- Zhong, W.B.; Tsai, Y.C.; Chin, L.H.; Tseng, J.H.; Tang, L.W.; Horng, S.; Fan, Y.C.; Hsu, S.P. A Synergistic Anti-Cancer Effect of Troglitazone and Lovastatin in a Human Anaplastic Thyroid Cancer Cell Line and in a Mouse Xenograft Model. Int. J. Mol. Sci. 2018, 19, 1834. [Google Scholar] [CrossRef]
- Sen, T.; Rodriguez, B.L.; Chen, L.; Corte, C.M.D.; Morikawa, N.; Fujimoto, J.; Cristea, S.; Nguyen, T.; Diao, L.; Li, L. Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov. 2019, 9, 646–661. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, M.; Amiri, S.; Pecic, S.; Machaj, F.; Rosik, J.; Los, M.J.; Alizadeh, J.; Mahdian, R.; da Silva Rosa, S.C.; Schaafsma, D.; et al. Pleiotropic effects of statins: A focus on cancer. Acta Mol. Basis Dis. 2020, 1866, 165968. [Google Scholar] [CrossRef] [PubMed]
- Antoniades, C.; Channon, K.M. Statins: Pleiotropic regulators of cardiovascular redox state. Antioxid. Redox Signal. 2014, 20, 1195–1197. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Barter, P. Antioxidant effects of statins in the management of cardiometabolic disorders. J. Atheroscler. Thromb. 2014, 21, 997–1010. [Google Scholar] [CrossRef] [PubMed]
- du Souich, P.; Roederer, G.; Dufour, R. Myotoxicity of statins: Mechanism of action. Pharmacol. Ther. 2017, 175, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kwon, O.S.; Cho, S.Y.; Paick, J.S.; Kim, S.W. Chronic administration of atorvastatin could partially ameliorate erectile function in streptozotocin-induced diabetic rats. PLoS ONE 2017, 12, e0172751. [Google Scholar] [CrossRef] [PubMed]
- Hadzi-Petrushev, N.; Dimovska, K.; Jankulovski, N.; Mitrov, D.; Mladenov, M. Supplementation with Alpha-Tocopherol and Ascorbic Acid to Nonalcoholic Fatty Liver Disease’s Statin Therapy in Men. Adv. Pharmacol. Sci. 2018, 2018, 4673061. [Google Scholar] [CrossRef] [PubMed]
- Jiao, X.; Ashtari, N.; Rahimi-Balaei, M.; Chen, Q.M.; Badbezanchi, I.; Shojaei, S.; Marzban, A.; Mirzaei, N.; Chung, S.; Guan, T.; et al. Mevalonate Cascade and Neurodevelopmental and Neurodegenerative Diseases: Future Targets for Therapeutic Application. Curr. Mol. Pharmacol. 2017, 10, 115–140. [Google Scholar]
- Ernster, L.; Dallner, G. Biochemical, physiological and medical aspects of ubiquinone function. Biochim. Biophys. Acta 1995, 1271, 195–204. [Google Scholar] [CrossRef]
- Ross, M.F.; Kelso, G.F.; Blaikie, F.H.; James, A.M.; Cocheme, H.M.; Filipovska, A.; Da Ros, T.; Hurd, T.R.; Smith, R.A.; Murphy, M.P. Lipophilic triphenylphosphonium cations as tools in mitochondrial bioenergetics and free radical biology. Biochemistry 2005, 70, 222–230. [Google Scholar] [CrossRef]
- Jung, E.J.; Kim, D.R. Control of TrkA-Induced Cell Death by JNK Activation and Differential Expression of TrkA upon DNA Damage. Mol. Cells 2010, 30, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Kerur, N.; Fukuda, S.; Banerjee, D.; Kim, Y.; Fu, D.; Apicella, I.; Varshney, A.; Yasuma, R.; Fowler, B.J.; Baghdasaryan, E.; et al. cGAS drives noncanonical-inflammasome activation in age-related macular degeneration. Nat. Med. 2018, 24, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, A.; Baruah, P.S.; Smith, J.C.; Wang, Z.; Sayles, N.M.; Andrews, P.; Kendall, J.; Leu, J.; Chunduri, N.K.; Levy, D.; et al. Single-Chromosomal Gains Can Function as Metastasis Suppressors and Promoters in Colon Cancer. Dev. Cell 2020, 52, 413–428.e6. [Google Scholar] [CrossRef] [PubMed]
- Basit, A.; Cho, M.G.; Kim, E.Y.; Kwon, D.; Kang, S.J.; Lee, J.H. The cGAS/STING/TBK1/IRF3 innate immunity pathway maintains chromosomal stability through regulation of p21 levels. Exp. Mol. Med. 2020, 52, 643–657. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Harding, S.M.; Natesan, R.; Tian, L.; Benci, J.L.; Li, W.; Minn, A.J.; Asangani, I.A.; Greenberg, R.A. Cell Cycle Checkpoints Cooperate to Suppress DNA- and RNA-Associated Molecular Pattern Recognition and Anti-Tumor Immune Responses. Cell Rep. 2020, 32, 108080. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Guan, J.; Lu, S.; Jin, Q.; Rousseau, B.; Lu, T.; Stephens, D.; Zhang, H.; Zhu, J.; Yang, M.; et al. DNA Sensing in Mismatch Repair-Deficient Tumor Cells Is Essential for Anti-tumor Immunity. Cancer Cell 2021, 39, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Krivega, M.; Stiefel, C.M.; Karbassi, S.; Andersen, L.L.; Chunduri, N.K.; Donnelly, N.; Pichlmair, A.; Storchova, Z. Genotoxic stress in constitutive trisomies induces autophagy and the innate immune response via the cGAS-STING pathway. Commun. Biol. 2021, 4, 831. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Gao, Y.; Su, Y.; Zhou, Y.; Yang, T.; Li, Y.; Wang, Y.; Sun, Y.; Chen, L.; Zhang, F.; et al. Oroxylin A regulates cGAS DNA hypermethylation induced by methionine metabolism to promote HSC senescence. Pharmacol. Res. 2023, 187, 106590. [Google Scholar] [CrossRef] [PubMed]
- Hao, F. Entanglement of Methylation Changes and cGAS-STING Signaling in Non-Small-Cell Lung Cancer. Comb. Chem. High Throughput Screen. 2023, 26, 224–235. [Google Scholar] [CrossRef]
- Kodach, L.L.; Jacobs, R.J.; Voorneveld, P.W.; Wildenberg, M.E.; Verspaget, H.W.; van Wezel, T.; Morreau, H.; Hommes, D.W.; Peppelenbosch, M.P.; van den Brink, G.R.; et al. Statins augment the chemosensitivity of colorectal cancer cells inducing epigenetic reprogramming and reducing colorectal cancer cell ‘stemness’ via the bone morphogenetic protein pathway. Gut 2011, 60, 1544–1553. [Google Scholar] [CrossRef]
- Kwon, J.; Bakhoum, S.F. The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov. 2020, 10, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Furda, A.M.; Marrangoni, A.M.; Lokshin, A.; Van Houten, B. Oxidants and not alkylating agents induce rapid mtDNA loss and mitochondrial dysfunction. DNA Repair 2012, 11, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, K.J.; Carroll, P.; Martin, C.A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Harding, S.M.; Benci, J.L.; Irianto, J.; Discher, D.E.; Minn, A.J.; Greenberg, R.A. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017, 548, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, S.; Spitkovsky, D.; Radicella, J.P.; Epe, B.; Wiesner, R.J. Reactive oxygen species derived from the mitochondrial respiratory chain are not responsible for the basal levels of oxidative base modifications observed in nuclear DNA of Mammalian cells. Free Radic. Biol. Med. 2004, 36, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Cleaver, J.E.; Brennan-Minnella, A.M.; Swanson, R.A.; Fong, K.W.; Chen, J.J.; Chou, K.M.; Chen, Y.W.; Revet, I.; Bezrookove, V. Mitochondrial reactive oxygen species are scavenged by Cockayne syndrome B protein in human fibroblasts without nuclear DNA damage. Proc. Natl. Acad. Sci. USA 2014, 111, 13487–13492. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Chen, Y.J.; Dobbs, N.; Sakai, T.; Liou, J.; Miner, J.J.; Yan, N. STING-mediated disruption of calcium homeostasis chronically activates ER stress and primes T cell death. J. Exp. Med. 2019, 216, 867–883. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Shi, Q.; Zhang, S.; Lin, H.; Han, C.; Qian, X.; Huang, Y.; Ren, X.; Sun, J.; Feng, N.; et al. Inhibition of the cGAS-STING Pathway Attenuates Lung Ischemia/Reperfusion Injury via Regulating Endoplasmic Reticulum Stress in Alveolar Epithelial Type II Cells of Rats. J. Inflamm. Res. 2022, 15, 5103–5119. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zhou, H.; Wu, H.; Wu, Q.; Duan, M.; Deng, W.; Tang, Q. STING-IRF3 contributes to lipopolysaccharide-induced cardiac dysfunction, inflammation, apoptosis and pyroptosis by activating NLRP3. Redox Biol. 2019, 24, 101215. [Google Scholar] [CrossRef]
- Cui, Y.; Zhao, D.; Sreevatsan, S.; Liu, C.; Yang, W.; Song, Z.; Yang, L.; Barrow, P.; Zhou, X. Mycobacterium bovis Induces Endoplasmic Reticulum Stress Mediated-Apoptosis by Activating IRF3 in a Murine Macrophage Cell Line. Front. Cell. Infect. Microbiol. 2016, 6, 182. [Google Scholar] [CrossRef]
- Dedoni, S.; Olianas, M.C.; Onali, P. Interferon-beta induces apoptosis in human SH-SY5Y neuroblastoma cells through activation of JAK-STAT signaling and down-regulation of PI3K/Akt pathway. J. Neurochem. 2010, 115, 1421–1433. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Park, J.; Takamatsu, H.; Konaka, H.; Aoki, W.; Aburaya, S.; Ueda, M.; Nishide, M.; Koyama, S.; Hayama, Y.; et al. Apoptosis-derived membrane vesicles drive the cGAS-STING pathway and enhance type I IFN production in systemic lupus erythematosus. Ann. Rheum. Dis. 2018, 77, 1507–1515. [Google Scholar] [CrossRef] [PubMed]
- Raish, M.; Ahmad, A.; Ansari, M.A.; Alkharfy, K.M.; Aljenoobi, F.I.; Jan, B.L.; Al-Mohizea, A.M.; Khan, A.; Ali, N. Momordica charantia polysaccharides ameliorate oxidative stress, inflammation, and apoptosis in ethanol-induced gastritis in mucosa through NF-kB signaling pathway inhibition. Int. J. Biol. Macromol. 2018, 111, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.; Xu, L.; Guo, W.; Wang, Y.; Wu, J.; Li, X.; Cai, X.; Hu, J.; Wang, M.; Xu, Q. SHP2-mediated inhibition of DNA repair contributes to cGAS–STING activation and chemotherapeutic sensitivity in colon cancer. Cancer Res. 2021, 81, 3215–3228. [Google Scholar] [CrossRef] [PubMed]
- Stamm, J.A.; Ornstein, D.L. The role of statins in cancer prevention and treatment. Oncology (Williston Park) 2005, 19, 739–750. [Google Scholar] [PubMed]
- Yen, C.S.; Chen, J.C.; Chang, Y.F.; Hsu, Y.F.; Chiu, P.T.; Shiue, C.; Chuang, Y.F.; Ou, G.; Hsu, M.J. Lovastatin causes FaDu hypopharyngeal carcinoma cell death via AMPK-p63-survivin signaling cascade. Sci. Rep. 2016, 6, 25082. [Google Scholar] [CrossRef] [PubMed]
- Han, J.X.; Tao, Z.H.; Wang, J.L.; Zhang, L.; Yu, C.Y.; Kang, Z.R.; Xie, Y.; Li, J.; Lu, S.; Cui, Y.; et al. Microbiota-derived tryptophan catabolites mediate the chemopreventive effects of statins on colorectal cancer. Nat. Microbiol. 2023, 8, 919–933. [Google Scholar] [CrossRef] [PubMed]
- Pecoraro, V.; Moja, L.; Dall’Olmo, L.; Cappellini, G.; Garattini, S. Most appropriate animal models to study the efficacy of statins: A systematic review. Eur. J. Clin. Investig. 2014, 44, 848–871. [Google Scholar] [CrossRef]
- Björkhem-Bergman, L.; Lindh, J.D.; Bergman, P. What is a relevant statin concentration in cell experiments claiming pleiotropic effects? Br. J. Clin. Pharmacol. 2011, 72, 164. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, X.; Liang, N.; Zhang, F.; Lin, W.; Ma, W. Lovastatin-Induced Mitochondrial Oxidative Stress Leads to the Release of mtDNA to Promote Apoptosis by Activating cGAS-STING Pathway in Human Colorectal Cancer Cells. Antioxidants 2024, 13, 679. https://doi.org/10.3390/antiox13060679
Huang X, Liang N, Zhang F, Lin W, Ma W. Lovastatin-Induced Mitochondrial Oxidative Stress Leads to the Release of mtDNA to Promote Apoptosis by Activating cGAS-STING Pathway in Human Colorectal Cancer Cells. Antioxidants. 2024; 13(6):679. https://doi.org/10.3390/antiox13060679
Chicago/Turabian StyleHuang, Xiaoming, Ning Liang, Fuming Zhang, Wanjun Lin, and Wenzhe Ma. 2024. "Lovastatin-Induced Mitochondrial Oxidative Stress Leads to the Release of mtDNA to Promote Apoptosis by Activating cGAS-STING Pathway in Human Colorectal Cancer Cells" Antioxidants 13, no. 6: 679. https://doi.org/10.3390/antiox13060679