
Anticancer Therapies Based on Oxidative Damage: Lycium barbarum Inhibits the Proliferation of MCF-7 Cells by Activating Pyroptosis through Endoplasmic Reticulum Stress


 ,
,  , , ,
, , ,  , , ,
, , ,  ,
, 
Abstract

1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. Determination of Total Phenolic Content (TPC), Total Flavonoid Content (TFC), and Total Chlorophyll and Carotenoid Content
2.3. Determination of Antioxidant Activity through DPPH and ABTS Assays
2.4. Metal Binding Studies
2.5. UHPLC-HRMS/MS Analysis
2.6. Quantitative Analysis
2.7. Cell Cultures and Drug Treatment
2.8. Cell Viability Assay and Phase-Contrast Analysis
2.9. Colony Formation Assay
2.10. Wound Healing Assay
2.11. Determination of Hypodiploid Nuclei
2.12. PI/Hoechst 33342 Double Staining Assay
2.13. Western Blotting Analysis
2.14. Measurement of LDH
2.15. ROS Detection
2.16. RNA Extraction, Reverse Transcription, and Real-Time PCR
2.17. RT-PCR and XBP1 Splicing Assay
2.18. Determination of Protein Misfolding
2.19. Measurement of Intracellular Ca2+ Signaling
2.20. Statistical Analysis
3. Results
3.1. Total Phenolic, Flavonoid, Chlorophyll, and Carotenoid Content, and Antioxidant Activity
3.2. Phytochemical Profile of LBE

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peak | Compound | Rt (min) | [M–H]− | [M+H]+ | MS/MS | Molecular Formula | Error (ppm) | Classification | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Gallic acid | 2.37 | 169.0136 | - | 125.0233 | C7H5O5 | 1.55 | Phenolic acids | [72] |
| 2 | p-coumaric acid-hexoside | 3.41 | 325.0934 | - | 119.0491 163.0392 | C15H18O8 | 0.76 | Hydroxycinnamic acid derivates | [72] |
| 3 | Hydroxybenzoic acid | 5.15 | 137.0235 | - | 93.0333 | C7H6O3 | 0.74 | Phenolic acids | [72] |
| 4 | Benzoic acid | 6.68 | 121.0285 | - | / | C7H6O2 | −0.33 | Organic acids | [72] |
| 5 | Blechnic acid | 7.37 | 371.0988 | - | 119.0491 163.0392 | C16H20O10 | 2.69 | Flavonoids | [78] |
| 6 | di-O-caffeoylquinic acid | 8.60 | 515.1413 | - | 353.0885 323.0759 | C22H28O14 | 1.77 | Hydroxycinnamic acid derivates | [72] |
| 7 | Caffeic acid | 9.06 | 179.0344 | - | 135.0441 | C9H8O4 | 1.13 | Hydroxycinnamic acids | [72] |
| 8 | p-coumaric acid-hexoside (isomer I) | 9.35 | 325.0934 | - | 145.0285 163.0391 | C15H18O8 | 4.42 | Hydroxycinnamic acid derivates | [72] |
| 9 | di-O-caffeoylquinic acid | 10.10 | 515.1415 | - | 191.0554 179.0333 | C22H28O14 | 3.78 | Hydroxycinnamic acid derivates | [72] |
| 10 | N-Caffeoyl, N′-dihydrocaffeoyl spermidine dihexose | 10.46 | - | 796.3496 | 163.0388 220.0966 310.2125 472.2498 | C37H53N3O16 | −1.31 | Hydroxycinnamic acid amides | [65,70] |
| 11 | N1-dihydrocaffeoyl, N10-caffeoyl spermidine hexose | 11.25 | - | 634.2977 | 163.0389 220.0967 165.0546 472.2523 310.2133 | C31H43N3O11 | −0.54 | Hydroxycinnamic acid amides | [65,70] |
| 12 | Ferulic acid hexoside | 11.26 | 355.1041 | - | 175.0392 160.0157 | C16H20O9 | 6.70 | Hydroxycinnamic acid derivates | [74] |
| 13 | p-coumaric acid-hexoside (isomer II) | 11.40 | 325.0933 | - | 145.0285 163.0390 119.0491 | C15H18O8 | 2.26 | Hydroxycinnamic acid derivates | [72] |
| 14 | N1-dihydrocaffeoyl, N10-caffeoyl spermidine | 11.76 | 470.2301 | - | 135.0442 308.1985 163.0402 291.1843 | C25H33N3O6 | −4.32 | Hydroxycinnamic acid amides | [65] |
| 15 | N1-dihydrocaffeoyl, N10-caffeoyl spermidine (isomer I) | 11.77 | - | 472.2445 | 163.0389 220.0967 310.2130 293.1865 236.1276 | C25H33N3O6 | 1.08 | Hydroxycinnamic acid amides | [65,70] |
| 16 | p-coumaric acid | 11.88 | 163.0392 | - | 119.0490 | C9H8O3 | 0.94 | Hydroxycinnamic acids | [72] |
| 17 | p-coumaric acid (isomer I) | 11.97 | - | 165.0547 | 147.0439 119.0492 | C9H8O3 | −0.96 | Hydroxycinnamic acids | [79] |
| 18 | N1,N10-bis-(caffeoyl) spermidine dihexose | 12.07 | - | 794.3350 | 163.0389 220.0969 308.1956 632.3118 470.2307 | C40H49N4O13 | −0.28 | Hydroxycinnamic acid amides | [65,70] |
| 19 | p-coumaroyl-quinic acid | 12.76 | 337.0935 | - | 191.0553 93.0333 | C16H18O8 | 3.09 | Hydroxycinnamic acid derivates | [72] |
| 20 | O-trans-feruloyl-O-β-d-glucopyranosyl-α-d-glucopyranoside | 12.80 | 517.1569 | - | 193.0500 175.0392 | C22H30O14 | 2.32 | Hydroxycinnamic acid derivates | [72] |
| 21 | Rutin hexose | 13.35 | 771.2005 | - | 609.1435 462.0828 301.0348 | C33H40O21 | 2.72 | Flavonoids | [74] |
| 22 | Scopoletin | 13.70 | 191.0343 | - | 176.0107 148.0156 | C10H8O4 | 2.18 | Coumarins | [56] |
| 23 | Ferulic acid | 14.11 | 193.0501 | - | 134.0363 178.0264 149.0598 | C10H10O4 | 2.42 | Hydroxycinnamic acids | [72] |
| 24 | N-acetyl-DL-tryptophan | 14.23 | 245.0931 | - | 203.0820 74.0235 116.0493 | C13H14N2O3 | 3.93 | Tryptophan derivates | [74] |
| 25 | Azelaic acid | 17.44 | 187.0968 | - | 125.0961 97.0646 | C9H16O4 | 2.02 | Fatty acids | [74] |
| 26 | Rutin | 18.05 | 609.1463 | - | 300.0275 301.0356 | C27H30O16 | 0.41 | Flavonoids | [72] |
| 27 | Rutin (isomer I) | 18.07 | - | 611.1603 | 303.0497 465.1002 | C27H30O16 | −0.63 | Flavonoids | [79] |
| 28 | Quercetin-O-hexoside | 18.24 | 463.0886 | - | 300.0275 301.0358 | C21H20O12 | 2.69 | Flavonoids | [72] |
| 29 | N-p-cis/trans-coumaroyl-tyramine | 18.81 | 282.1139 | - | 119.0491 243.0793 | C17H17NO3 | 4.28 | Hydroxycinnamic acid amides | [72] |
| 30 | Naringenin-O-hexoside (Prunin) | 18.99 | 433.1145 | - | 271.0610 | C21H22O10 | 2.38 | Flavonoids | [72] |
| 31 | Kaempferol O-hexoside-rhamnoside (Nictoflorin) | 19.90 | 593.1516 | - | 285.0405 255.0298 | C27H30O15 | 2.04 | Flavonoids | [72] |
| 32 | Kaempferol O-hexoside-rhamnoside (Nictoflorin) (isomer I) | 19.96 | - | 595.1658 | 287.0550 258.2194 | C27H30O15 | −0.37 | Flavonoids | [79] |
| 33 | N-cis/trans-feruloyltyramine | 20.05 | 312.1245 | - | 148.0520 297.1012 | C18H19NO4 | 3.75 | Hydroxycinnamic acid amides | [72] |
| 34 | Kaempferol-O-hexoside | 20.06 | 447.0939 | - | 284.0340 285.0403 255.0299 | C21H20O11 | −0.87 | Flavonoids | [72] |
| 35 | Isorhamnetin-O-rutinoside | 20.45 | 623.1626 | - | 315.0511 300.0198 | C28H32O16 | 2.13 | Flavonoids | [72] |
| 36 | Isorhamnetin-O-hexoside | 20.68 | 477.1046 | - | 314.0432 243.0302 271.0262 | C22H22O12 | 1.09 | Flavonoids | [72] |
| 37 | N-p-cis/trans-coumaroyl-tyramine (isomer I) | 21.60 | 282.1139 | - | 119.0491 | C17H17NO3 | 4.28 | Hydroxycinnamic acid amides | [72] |
| 38 | N-p-cis/trans-coumaroyl-tyramine (isomer II) | 21.63 | - | 284.1280 | 147.0439 121.0649 164.0692 | C17H17NO3 | −0.88 | Hydroxycinnamic acid amides | [72] |
| 39 | N-cis/trans-feruloyltyramine | 22.62 | 312.1245 | - | 148.0520 297.1007 | C18H19NO4 | 4.15 | Hydroxycinnamic acid amides | [72] |
| 40 | Quercetin | 23.36 | 301.2023 | - | 151.0028 178.9983 | C15H10O7 | 4.13 | Flavonoids | [72] |
| 41 | Naringenin | 23.82 | 271.0615 | - | 119.0491 151.0028 | C15H12O5 | 5.03 | Flavonoids | [72] |
| 42 | Trihydroxy-octadecadienoic acid (TriHODE) | 24.46 | 327.2179 | - | 171.1019 211.1336 291.2002 | C18H32O5 | 4.33 | Fatty acids | CD |
| 43 | Trihydroxy-octadecenoic acid | 24.84 | 329.2334 | - | 171.1018 139.1117 211.1336 | C18H34O5 | 3.76 | Fatty acids | [77] CD |
| 44 | Trihydroxy-octadecadienoic acid (TriHODE) (isomer I) | 25.48 | 327.2180 | - | 201.1126 213.1127 291.1967 | C18H32O5 | 3.68 | Fatty acids | CD |
| 45 | Trihydroxy-octadecadienoic acid (TriHODE) (isomer II) | 26.11 | 327.2180 | - | 171.1019 201.1127 291.1961 | C18H32O5 | 4.05 | Fatty acids | CD |
| 46 | 13-Oxo-octadecadienoic acid (13-Oxo-ODE) | 26.61 | - | 295.2268 | 277.2162 179.1434 | C18H30O3 | 0.95 | Fatty acids | [77] CD |
| 47 | Dihydroxy octadecadienoic acid (DiHODE) | 26.63 | 311.2230 | - | 293.2125 275.2019 | C18H32O4 | 4.29 | Fatty acids | [77] CD |
| 48 | Dihydroxy octadecadienoic acid (DiHODE) (isomer I) | 26.76 | 311.2230 | - | 293.2125 275.2019 | C18H32O4 | 4.69 | Fatty acids | [77] CD |
| 49 | Dihydroxy octadecadienoic acid (DiHODE) (isomer II) | 26.94 | 311.2231 | - | 293.2123 275.2020 | C18H32O4 | 4.49 | Fatty acids | [77] CD |
| 50 | 13-Oxo-octadecadienoic acid (13-Oxo-ODE) (isomer I) | 27.40 | 293.2126 | - | 275.2016 171.1017 | C18H30O3 | 4.79 | Fatty acids | [77] CD |
| 51 | tris-(Dihydrocaffeoyl) spermine | 27.41 | - | 693.3461 | 293.0565 | C37H48N4O9 | −5.43 | Hydroxycinnamic acid amides | [65,70] |
| 52 | Hydroxy-octadecadienoic acid (HODE) | 27.86 | 295.2279 | - | 171.1018 277.2176 | C18H32O3 | 4.05 | Fatty acids | [77] CD |
| 53 | 13-Oxo-octadecadienoic acid (13-Oxo-ODE) (isomer II) | 28.24 | 293.2124 | - | 275.2015 | C18H30O3 | 4.37 | Fatty acids | [77] CD |
| 54 | Palmitic acid | 28.91 | 255.2330 | - | - | C16H31O2 | 4.07 | Fatty acids | [72,74] |
| 55 | Linoleamide | 29.36 | - | 280.2636 | 263.2368 | C18H33NO | −0.34 | Fatty acids | [80] |
| 56 | Palmitic amide | 29.84 | - | 256.2636 | 88.0761 102.0913 172.1689 | C16H33NO | 0.47 | Fatty acids | [80] |
| 57 | Oleamide | 30.06 | - | 282.2789 | 265.2529 247.2418 | C18H36NO | 0.66 | Fatty acids | [80] CD |
| 58 | Oleic acid | 30.86 | 281.2487 | - | - | C18H34O2 | 4.09 | Fatty acids | [68] CD |
| 59 | Protocatechuate | 31.01 | 153.0184 | - | 109.0283 | C7H5O4− | 0.89 | Phenolic acids | [72] |
| 60 | Violaxanthin | 34.24 | - | 601.4229 | - | C40H56O4 | −4.10 | Xanthophylls | [81] |
| Peak | Compounds | µg CAE g−1 dw |
|---|---|---|
| 2 | p-Coumaric acid-hexoside (isomer I) | 27.98 ± 1.38 |
| 6 | 3,4-di-O-caffeoylquinic acid | 6.20 ± 0.18 |
| 7 | Caffeic acid | 1.07 ± 0.12 |
| 8 | p-coumaric acid-hexoside (isomer II) | 35.55 ± 2.84 |
| 9 | 3,5-di-O-caffeoylquinic acid | 7.68 ± 1.98 |
| 12 | Ferulic acid hexoside | <LOQ |
| 13 | p-coumaric acid-hexoside (isomer III) | <LOQ |
| 14 | N1-dihydrocaffeoyl, N10-caffeoyl spermidine | <LOQ |
| 16 | p-coumaric acid | 214.43 ± 1.42 |
| 19 | p-coumaroyl-quinic acid | 2.38 ± 0.076 |
| 23 | Ferulic acid | 0.99 ± 0.18 |
| 29 | N-p-cis/trans-coumaroyl-tyramine | <LOQ |
| 33 | N-cis/trans-feruloyltyramine | 407.24 ± 20.86 |
| 37 | N-p-cis/trans-coumaroyl-tyramine (isomer I) | 53.05 ± 3.07 |
| 39 | N-cis/trans-feruloyltyramine | 522.52 ± 24.10 |
| Total Hydroxycinnamic Acids | 1279.10 ± 0.05 | |
| µg RE g−1 dw | ||
| 21 | Rutin hexose | 18.49 ± 7.96 |
| 26 | Rutin | 1607.36 ± 89.85 |
| 28 | Quercetin-3-O-hexoside | <LOQ |
| 30 | Naringenin-7-O-hexoside (Prunin) | 1.15 ± 1.58 |
| 31 | Kaempferol 3-O-hexoside-7-O-rhamnoside (Nictoflorin) (isomer I) | 298.88 ± 17.93 |
| 34 | Kaempferol-3-O-hexoside | <LOQ |
| 35 | Isorhamnetin-3-O-rutinoside | 182.55 ± 36.32 |
| 36 | Isorhamnetin-3-O-hexoside | <LOQ |
| 40 | Quercetin | <LOQ |
| 41 | Naringenin | 14.91 ± 8.61 |
| Total Flavonoids | 2123.34 ± 0.06 | |
| mg OAE g−1 dw | ||
| 42 | Trihydroxy-octadecadienoic acid (TriHODE) | 3.22 ± 0.30 |
| 43 | Trihydroxy-octadecenoic acid | 1201.76 ± 30.44 |
| 44 | Trihydroxy-octadecadienoic acid (TriHODE) (isomer I) | 276.61 ± 6.20 |
| 45 | Trihydroxy-octadecadienoic acid (TriHODE) (isomer II) | 440.45 ± 10.03 |
| 47 | Dihydroxy octadecadienoic acid (DiHODE) | 538.99 ± 6.96 |
| 48 | Dihydroxy octadecadienoic acid (DiHODE) (isomer I) | 435.75 ± 2.64 |
| 49 | Dihydroxy octadecadienoic acid (DiHODE) (isomer II) | 433.99 ± 7.60 |
| 50 | 13-Oxo-octadecadienoic acid (13-Oxo-ODE) (isomer I) | 472.07 ± 18.54 |
| 52 | Hydroxy-octadecadienoic acid (HODE) | 1737.23 ± 23.17 |
| 53 | 13-Oxo-octadecadienoic acid (13-Oxo-ODE) (isomer II) | 220.76 ± 9.00 |
| 54 | Palmitic acid | <LOQ |
| 58 | Oleic acid | 26.25 ± 0.33 |
| Total Fatty Acids | 5787.08 ± 24.83 |
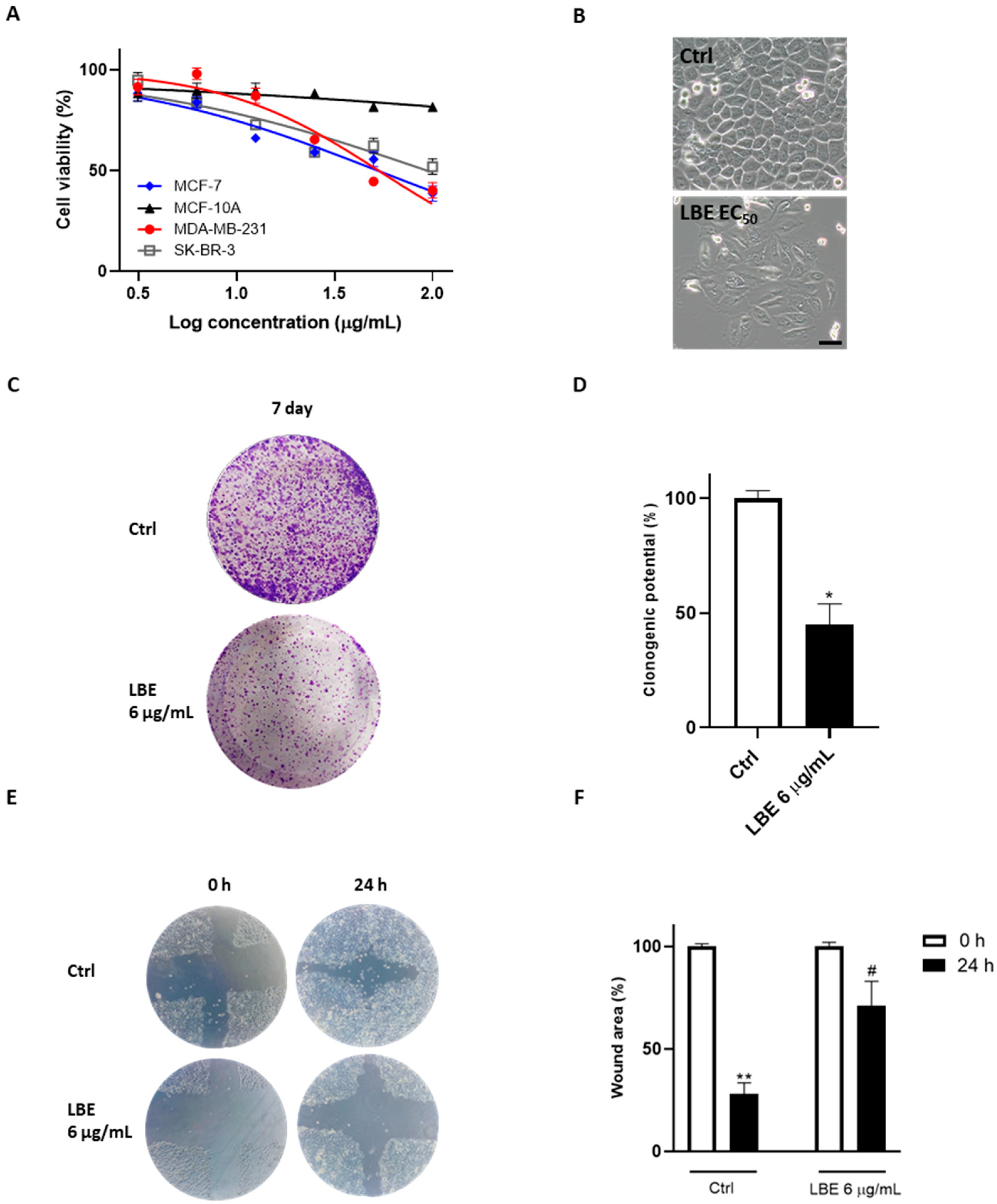
3.3. LBE Induces Pyroptosis in MCF-7 Cells, Saving Healthy Cells
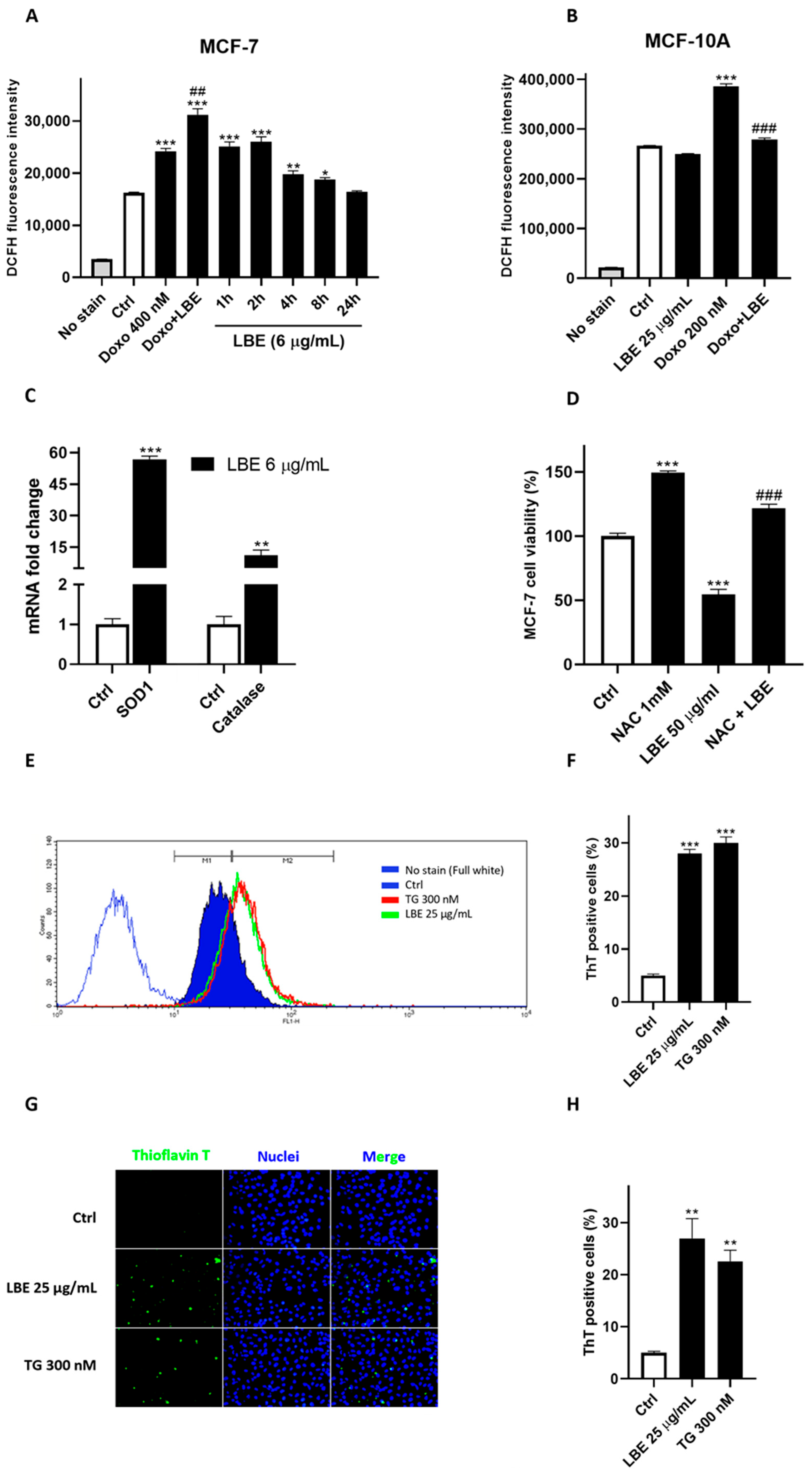
3.4. LBE Elicits Oxidative Stress in MCF-7 Cancer Cells and Exhibits Antioxidant Effects in Healthy Cells
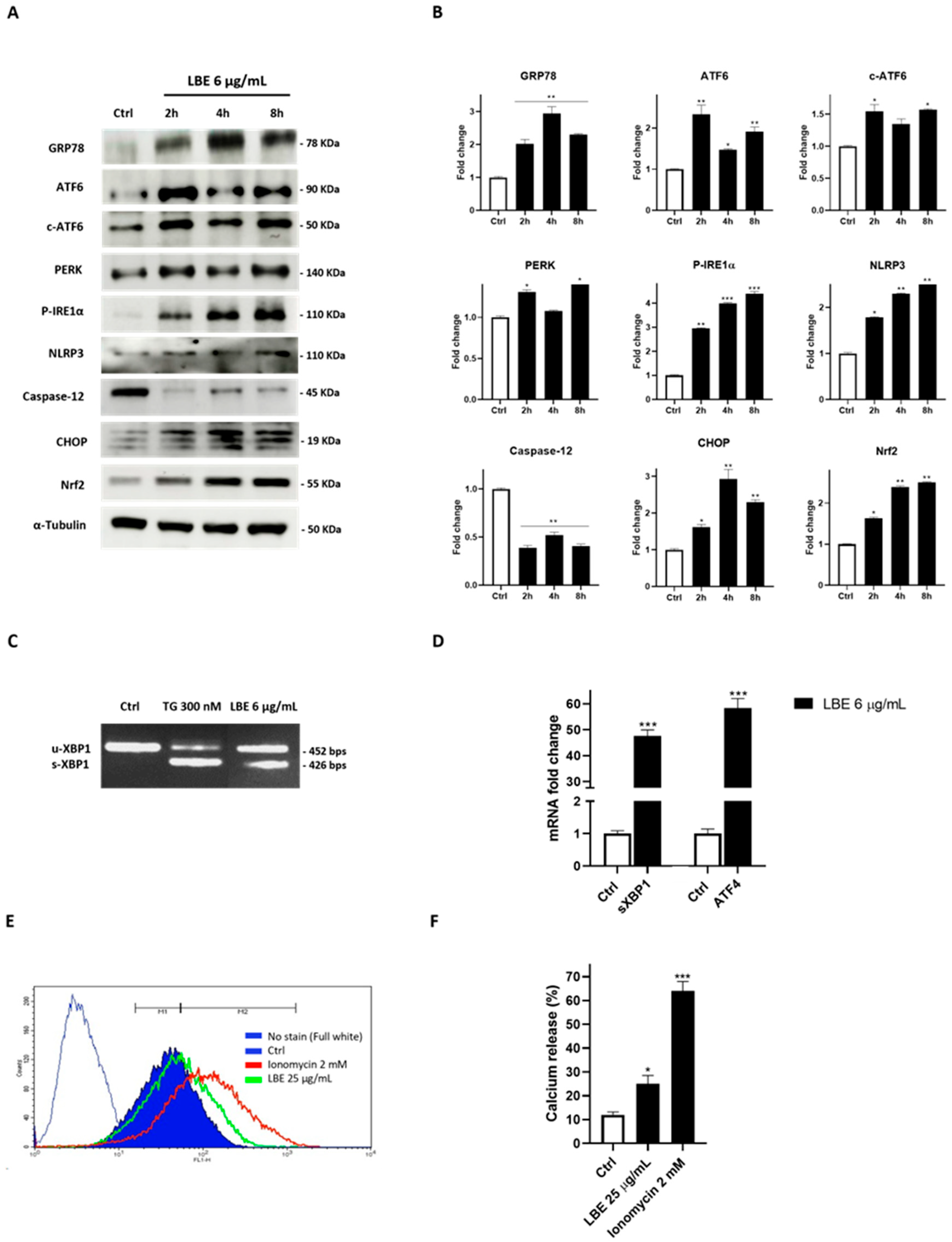
3.5. LBE Induces ER Stress in MCF-7 Cancer Cells, Promoting Inflammasome Activation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guida, F.; Kidman, R.; Ferlay, J.; Schüz, J.; Soerjomataram, I.; Kithaka, B.; Ginsburg, O.; Mailhot Vega, R.B.; Galukande, M.; Parham, G.; et al. Global and regional estimates of orphans attributed to maternal cancer mortality in 2020. Nat. Med. 2022, 28, 2563–2572. [Google Scholar] [CrossRef]
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Miller, K.D.; Siegel, R.L.; Lin, C.C.; Mariotto, A.B.; Kramer, J.L.; Rowland, J.H.; Stein, K.D.; Alteri, R.; Jemal, A. Cancer treatment and survivorship statistics, 2016. CA Cancer J. Clin. 2019, 66, 271–289. [Google Scholar] [CrossRef]
- Maiuolo, J.; Gliozzi, M.; Carresi, C.; Musolino, V.; Oppedisano, F.; Scarano, F.; Nucera, S.; Scicchitano, M.; Bosco, F.; Macri, R.; et al. Nutraceuticals and Cancer: Potential for Natural Polyphenols. Nutrients 2021, 13, 3834. [Google Scholar] [CrossRef]
- Saitta, M.; Lo Curto, S.; Salvo, F.; Di Bella, G.; Dugo, G. Gas chromatographic–tandem mass spectrometric identification of phenolic compounds in Sicilian olive oils. Anal. Chim. Acta 2002, 466, 335–344. [Google Scholar] [CrossRef]
- Pepe, G.; Pagano, F.; Adesso, S.; Sommella, E.; Ostacolo, C.; Manfra, M.; Chieppa, M.; Sala, M.; Russo, M.; Marzocco, S.; et al. Bioavailable Citrus sinensis Extract: Polyphenolic Composition and Biological Activity. Molecules 2017, 22, 623. [Google Scholar] [CrossRef]
- Novi, S.; Vestuto, V.; Campiglia, P.; Tecce, N.; Bertamino, A.; Tecce, M.F. Anti-Angiogenic Effects of Natural Compounds in Diet-Associated Hepatic Inflammation. Nutrients 2023, 15, 2748. [Google Scholar] [CrossRef]
- Vestuto, V.; Amodio, G.; Pepe, G.; Basilicata, M.G.; Belvedere, R.; Napolitano, E.; Guarnieri, D.; Pagliara, V.; Paladino, S.; Rodriquez, M.; et al. Cocoa Extract Provides Protection against 6-OHDA Toxicity in SH-SY5Y Dopaminergic Neurons by Targeting PERK. Biomedicines 2022, 10, 2009. [Google Scholar] [CrossRef]
- Yordi, E.G.; Perez, E.M.; Matos, M.J.; Villares, E.U. Antioxidant and Prooxidant Effects of Polyphenolic Compounds and Structure-Activity Relationship Evidence. In Nutrition, Well-Being and Health; IntechOpen: London, UK, 2012; pp. 23–48. [Google Scholar]
- Leon-Gonzales, A.J.; Auger, C.; Schini-Kerth, V.B. Pro-Oxidant Activity of Polyphenols and Its Implication on Cancer Chemoprevention and Chemotherapy. Biochem. Pharmacol. 2015, 98, 371–380. [Google Scholar] [CrossRef]
- Pepe, G.; Salviati, E.; Rapa, S.F.; Ostacolo, C.; Cascioferro, S.; Manfra, M.; Autore, G.; Marzocco, S.; Campiglia, P. Citrus sinensis and Vitis vinifera Protect Cardiomyocytes from Doxorubicin-Induced Oxidative Stress: Evaluation of Onconutraceutical Potential of Vegetable Smoothies. Antioxidants 2020, 9, 378. [Google Scholar] [CrossRef]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef]
- Farvid, M.S.; Chen, W.Y.; Rosner, B.A.; Tamimi, R.M.; Willett, W.C.; Eliassen, A.H. Fruit and vegetable consumption and breast cancer incidence: Repeated measures over 30 years of follow-up. Int. J. Cancer 2019, 144, 1496–1510. [Google Scholar] [CrossRef]
- Codini, M. Why Vitamin C Could Be an Excellent Complementary Remedy to Conventional Therapies for Breast Cancer. Int. J. Mol. Sci. 2020, 21, 8397. [Google Scholar] [CrossRef]
- Giménez-Bastida, J.A.; González-Sarrías, A. Dietary Bioactive Compounds and Breast Cancer. Int. J. Mol. Sci. 2023, 24, 9731. [Google Scholar] [CrossRef]
- Procházková, D.; Boušová, I.; Wilhelmová, N. Antioxidant and prooxidant properties of flavonoids. Fitoterapia 2011, 82, 513–523. [Google Scholar] [CrossRef]
- Zheng, L.F.; Dai, F.; Zhou, B.; Yang, L.; Liu, Z.L. Prooxidant activity of hydroxycinnamic acids on DNA damage in the presence of Cu(II) ions: Mechanism and structure-activity relationship. Food Chem. Toxicol. 2008, 46, 149–156. [Google Scholar] [CrossRef]
- Eghbaliferiz, S.; Iranshahi, M. Prooxidant activity of polyphenols, flavonoids, anthocyanins and carotenoids: Updated review of mechanisms and catalyzing metals. Phytother. Res. 2016, 30, 1379–1391. [Google Scholar] [CrossRef]
- Li, X.; Yang, L.; Hao, M.; Song, T.; He, Y.; Yang, M.; Zhang, J. Chlorogenic acid as an indispensible partner of caffeic acid in coffee via selective regulation of prooxidative actions of caffeic acid. Food Res. Int. 2023, 173, 113482. [Google Scholar] [CrossRef]
- Ribeiro, D.; Freitas, M.; Silva, A.M.S.; Carvalho, F.; Fernandes, E. Antioxidant and pro-oxidant activities of carotenoids and their oxidation products. Food Chem. Toxicol. 2018, 120, 681–699. [Google Scholar] [CrossRef]
- Patel, M. Targeting oxidative stress in central nervous system disorders. Trends Pharmacol. Sci. 2016, 37, 768–778. [Google Scholar] [CrossRef]
- Liu, L.; Cui, H.; Xu, Y. Quantitative estimation of oxidative stress in cancer tissue cells through gene expression data analyses. Front. Genet. 2020, 11, 494. [Google Scholar] [CrossRef]
- Tafani, M.; Sansone, L.; Limana, F.; Arcangeli, T.; De Santis, E.; Polese, M.; Fini, M.; Russo, M.A. The Interplay of Reactive Oxygen Species, Hypoxia, Inflammation, and Sirtuins in Cancer Initiation and Progression. Oxid. Med. Cell Longev. 2016, 2016, 3907147. [Google Scholar] [CrossRef]
- Li, A.N.; Li, S.; Zhang, Y.J.; Xu, X.R.; Chen, Y.M.; Li, H.B. Resources and biological activities of natural polyphenols. Nutrients 2014, 6, 6020–6047. [Google Scholar] [CrossRef]
- Marino, P.; Pepe, G.; Basilicata, M.G.; Vestuto, V.; Marzocco, S.; Autore, G.; Procino, A.; Gomez-Monterrey, I.M.; Manfra, M.; Campiglia, P. Potential Role of Natural Antioxidant Products in Oncological Diseases. Antioxidants 2023, 12, 704. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Castañeda-Arriaga, R.; Pérez-González, A.; Reina, M.; Alvarez-Idaboy, J.R.; Galano, A. Comprehensive Investigation of the Antioxidant and Pro-oxidant Effects of Phenolic Compounds: A Double-Edged Sword in the Context of Oxidative Stress? J. Phys. Chem. B 2018, 122, 6198–6214. [Google Scholar] [CrossRef]
- Quagliariello, V.; Basilicata, M.G.; Pepe, G.; De Anseris, R.; Di Mauro, A.; Scognamiglio, G.; Palma, G.; Vestuto, V.; Buccolo, S.; Luciano, A.; et al. Combination of Spirulina platensis, Ganoderma lucidum and Moringa oleifera Improves Cardiac Functions and Reduces Pro-Inflammatory Biomarkers in Preclinical Models of Short-Term Doxorubicin-Mediated Cardiotoxicity: New Frontiers in Cardioncology? J. Cardiovasc. Dev. Dis. 2022, 9, 423. [Google Scholar] [CrossRef]
- Bezerra, D.P.; Militão, G.C.G.; de Morais, M.C.; de Sousa, D.P. The Dual Antioxidant/Prooxidant Effect of Eugenol and Its Action in Cancer Development and Treatment. Nutrients 2017, 9, 1367. [Google Scholar] [CrossRef]
- Hodnick, W.F.; Milosavljević, E.B.; Nelson, J.H.; Pardini, R.S. Electrochemistry of flavonoids. Relationships between redox potentials, inhibition of mitochondrial respiration, and production of oxygen radicals by flavonoids. Biochem. Pharmacol. 1988, 37, 2607–2611. [Google Scholar] [CrossRef]
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Arancibia-Hernández, Y.L.; Hernández-Cruz, E.Y.; Pedraza-Chaverri, J. RONS and Oxidative Stress: An Overview of Basic Concepts. Oxygen 2022, 2, 437–478. [Google Scholar] [CrossRef]
- Lin, J.H.; Walter, P.; Yen, T.S. Endoplasmic reticulum stress in disease pathogenesis. Annu. Rev. Pathol. 2008, 3, 399–425. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.X.; Tanaka, L.Y.; Wosniak, J.; Laurindo, F.R. Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: Roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxid. Redox Signal 2009, 11, 2409–2427. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.G.; Venkatesan, J.; Shim, M.S. Selective Anticancer Therapy Using Pro-Oxidant Drug-Loaded Chitosan-Fucoidan Nanoparticles. Int. J. Mol. Sci. 2019, 20, 3220. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Seervi, M.; Rani, A.; Sharma, A.K.; Santhosh Kumar, T.R. ROS mediated ER stress induces Bax-Bak dependent and independent apoptosis in response to Thioridazine. Biomed. Pharmacother. 2018, 106, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Read, A.; Schröder, M. The Unfolded Protein Response: An Overview. Biology 2021, 10, 384. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Seki, T.; Maeda, H. Therapeutic strategies by modulating oxygen stress in cancer and inflammation. Adv. Drug Deliv. Rev. 2009, 61, 290–302. [Google Scholar] [CrossRef]
- de Freitas Rodrigues, C.; Ramos Boldori, J.; Valandro Soares, M.; Somacal, S.; Emanuelli, T.; Izaguirry, A.; Weber Santos Cibin, F.; Rossini Augusti, P.; Casagrande Denardin, C. Goji berry (Lycium barbarum L.) juice reduces lifespan and premature aging of Caenorhabditis elegans: Is it safe to consume it? Food Res. Int. 2021, 144, 110297. [Google Scholar] [CrossRef]
- Lu, Y.; Guo, S.; Zhang, F.; Yan, H.; Qian, D.W.; Wang, H.Q.; Jin, L.; Duan, J.A. Comparison of Functional Components and Antioxidant Activity of Lycium barbarum L. Fruits from Different Regions in China. Molecules 2019, 24, 2228. [Google Scholar] [CrossRef]
- Potterat, O. Goji (Lycium barbarum and L. chinense): Phytochemistry, pharmacology and safety in the perspective of traditional uses and recent popularity. Planta Med. 2010, 76, 7–19. [Google Scholar] [CrossRef]
- Ma, R.; Zhang, X.X.; Ni, Z.J.; Thakur, K.; Wang, W.; Yan, Y.M.; Cao, Y.L.; Zhang, J.G.; Rengasamy, K.R.R.; Wei, Z.J. Lycium barbarum (Goji) as functional food: A review of its nutrition, phytochemical structure, biological features, and food industry prospects. Crit. Rev. Food Sci. Nutr. 2023, 63, 10621–10635. [Google Scholar] [CrossRef]
- Qian, D.; Zhao, Y.; Yang, G.; Huang, L. Systematic Review of Chemical Constituents in the Genus Lycium (Solanaceae). Molecules 2017, 22, 911. [Google Scholar] [CrossRef] [PubMed]
- Chen, D. Chemical constituents from Lycium barbarum (Solanaceae) and their chemophenetic significance. Biochem. Syst. Ecol. 2021, 97, 104292. [Google Scholar] [CrossRef]
- Yang, Y.; Chang, Y.; Wu, Y.; Liu, H.; Liu, Q.; Kang, Z.; Wu, M.; Yin, H.; Duan, J. A homogeneous polysaccharide from Lycium barbarum: Structural characterizations, anti-obesity effects and impacts on gut microbiota. Int. J. Biol. Macromol. 2021, 183, 2074–2087. [Google Scholar] [CrossRef]
- Toh, D.W.K.; Xia, X.; Sutanto, C.N.; Low, J.H.M.; Poh, K.K.; Wang, J.W.; Foo, R.S.; Kim, J.E. Enhancing the cardiovascular protective effects of a healthy dietary pattern with wolfberry (Lycium barbarum): A randomized controlled trial. Am. J. Clin. Nutr. 2021, 114, 80–89. [Google Scholar] [CrossRef]
- Kaloyan, D.; Georgiev, J.; Slavov, I. Synergistic Growth Inhibitory Effects of Lycium barbarum (Goji berry) Extract with Doxorubicin against Human Breast Cancer Cells. J. Pharm. Pharmacol. Res. 2019, 3, 051–058. [Google Scholar]
- Jin, H.; Li, M.; Tian, F.; Yu, F.; Zhao, W. An Overview of Antitumour Activity of Polysaccharides. Molecules 2022, 27, 8083. [Google Scholar] [CrossRef]
- Miranda, M.R.; Vestuto, V.; Amodio, G.; Manfra, M.; Pepe, G.; Campiglia, P. Antitumor Mechanisms of Lycium barbarum Fruit: An Overview of In Vitro and In Vivo Potential. Life 2024, 14, 420. [Google Scholar] [CrossRef]
- Aquino, G.; Basilicata, M.G.; Crescenzi, C.; Vestuto, V.; Salviati, E.; Cerrato, M.; Ciaglia, T.; Sansone, F.; Pepe, G.; Campiglia, P. Optimization of microwave-assisted extraction of antioxidant compounds from spring onion leaves using Box–Behnken design. Sci. Rep. 2023, 13, 14923. [Google Scholar] [CrossRef]
- Imeneo, V.; De Bruno, A.; Piscopo, A.; Romeo, R.; Poiana, M. Valorization of ‘Rossa di Tropea’ Onion Waste through Green Recovery Techniques of Antioxidant Compounds. Sustainability 2022, 14, 4387. [Google Scholar] [CrossRef]
- Šamec, D.; Bogović, M.; Vincek, D.; Martinčić, J.; Salopek-Sondi, B. Assessing the authenticity of the white cabbage (Brassica oleracea var. capitata f. alba) cv. ‘Varaždinski’ by molecular and phytochemical markers. Food Res. Int. 2014, 60, 266–272. [Google Scholar] [CrossRef]
- Wang, H.; Zhu, Y.; Xie, D.; Zhang, H.; Zhang, Y.; Jin, P.; Du, Q. The Effect of Microwave Radiation on the Green Color Loss of Green Tea Powder. Foods 2022, 11, 2540. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Choe, J.; Bu, T.; Liu, S.; Kim, S. Comparison of Antioxidant Properties and Metabolite Profiling of Acer pseudoplatanus Leaves of Different Colors. Antioxidants 2022, 12, 65. [Google Scholar] [CrossRef]
- Moreno, M.T.; Rodríguez Mellado, J.M. Spectrophotometric and Electrochemical Assessment of the Antioxidant Capacity of Aqueous and Ethanolic Extracts of Citrus Flavedos. Oxygen 2022, 2, 99–108. [Google Scholar] [CrossRef]
- Walker, R.B.; Everette, J.D. Comparative reaction rates of various antioxidants with ABTS radical cation. J. Agric. Food Chem. 2009, 57, 1156–1161. [Google Scholar] [CrossRef]
- Umar, T.; Shalini, S.; Raza, M.K.; Gusain, S.; Kumar, J.; Ahmed, W.; Tiwari, M.; Hoda, N. New amyloid beta-disaggregating agents: Synthesis, pharmacological evaluation, crystal structure and molecular docking of N-(4-((7-chloroquinolin-4-yl)oxy)-3-ethoxybenzyl)amines. MedChemComm 2018, 9, 1891–1904. [Google Scholar] [CrossRef]
- Santoro, A.; Grimaldi, M.; Buonocore, M.; Stillitano, I.; Gloria, A.; Santin, M.; Bobba, F.; Sublimi Saponetti, M.; Ciaglia, E.; D’Ursi, A.M. New Aβ(1-42) ligands from anti-amyloid antibodies: Design, synthesis, and structural interaction. Eur. J. Med. Chem. 2022, 237, 114400. [Google Scholar] [CrossRef] [PubMed]
- Colarusso, E.; Ceccacci, S.; Monti, M.C.; Gazzillo, E.; Giordano, A.; Chini, M.G.; Ferraro, M.G.; Piccolo, M.; Ruggiero, D.; Irace, C.; et al. Identification of 2,4,5-trisubstituted-2,4-dihydro-3H-1,2,4-triazol-3-one-based small molecules as selective BRD9 binders. Eur. J. Med. Chem. 2023, 5, 115018. [Google Scholar] [CrossRef]
- Pagliara, V.; Amodio, G.; Vestuto, V.; Franceschelli, S.; Russo, N.A.; Cirillo, V.; Mottola, G.; Remondelli, P.; Moltedo, O. Myogenesis in C2C12 Cells Requires Phosphorylation of ATF6α by p38 MAPK. Biomedicines 2023, 11, 1457. [Google Scholar] [CrossRef]
- Buonocore, M.; Grimaldi, M.; Santoro, A.; Covelli, V.; Marino, C.; Napolitano, E.; Novi, S.; Tecce, M.F.; Ciaglia, E.; Montella, F.; et al. Exploiting the Features of Short Peptides to Recognize Specific Cell Surface Markers. Int. J. Mol. Sci. 2023, 24, 15610. [Google Scholar] [CrossRef] [PubMed]
- Cerqua, I.; Musella, S.; Peltner, L.K.; D’Avino, D.; Di Sarno, V.; Granato, E.; Vestuto, V.; Di Matteo, R.; Pace, S.; Ciaglia, T.; et al. Discovery and Optimization of Indoline-Based Compounds as Dual 5-LOX/sEH Inhibitors: In Vitro and In Vivo Anti-Inflammatory Characterization. J. Med. Chem. 2022, 65, 14456–14480. [Google Scholar] [CrossRef] [PubMed]
- Di Sarno, V.; Giovannelli, P.; Medina-Peris, A.; Ciaglia, T.; Di Donato, M.; Musella, S.; Lauro, G.; Vestuto, V.; Smaldone, G.; Di Matteo, F.; et al. New TRPM8 blockers exert anticancer activity over castration-resistant prostate cancer models. Eur. J. Med. Chem. 2022, 238, 114435. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, J.; Tao, W.; Zhang, X.; Gao, X.; Yong, J.; Duan, J.A. Lycium ruthenicum studies: Molecular biology, phytochemistry and pharmacology. Food Chem. 2018, 240, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Ilić, T.; Dodevska, M.; Marčetić, M.; Božić, D.; Kodranov, I.; Vidović, B. Chemical Characterization, Antioxidant and Antimicrobial Properties of Goji Berries Cultivated in Serbia. Foods 2020, 6, 1614. [Google Scholar] [CrossRef] [PubMed]
- Skenderidis, P.; Lampakis, D.; Giavasis, I.; Leontopoulos, S.; Petrotos, K.; Hadjichristodoulou, C.; Tsakalof, A. Chemical Properties, Fatty-Acid Composition, and Antioxidant Activity of Goji Berry (Lycium barbarum L. and Lycium chinense Mill.) Fruits. Antioxidants 2019, 8, 60. [Google Scholar] [CrossRef] [PubMed]
- Raj, K.; Rajni, S.; Mohan, S.T.; Shweta, S.; Amarjit, K. Comparative study of phytochemicals, antioxidant activities and chromatographic profiling of different parts of Lycium ruthenicum Murr of Trans-Himalayan region. Phytomed. Plus 2022, 2, 100339. [Google Scholar] [CrossRef]
- Xiao, X.; Ren, W.; Zhang, N.; Bing, T.; Liu, X.; Zhao, Z.; Shangguan, D. Comparative study of the chemical constituents and bioactivities of the extracts from fruits, leaves and root barks of Lycium barbarum. Molecules 2019, 24, 1585. [Google Scholar] [CrossRef] [PubMed]
- Yossa Nzeuwa, I.B.; Xia, Y.; Qiao, Z.; Feng, F.; Bian, J.; Liu, W.; Qu, W. Comparison of the origin and phenolic contents of Lycium ruthenicum Murr. by high-performance liquid chromatography fingerprinting combined with quadrupole time-of-flight mass spectrometry and chemometrics. J. Sep. Sci. 2017, 40, 1234–1243. [Google Scholar] [CrossRef]
- Alcázar Magaña, A.; Kamimura, N.; Soumyanath, A.; Stevens, J.F.; Maier, C.S. Caffeoylquinic acids: Chemistry, biosynthesis, occurrence, analytical challenges, and bioactivity. Plant J. 2021, 107, 1299–1319. [Google Scholar] [CrossRef]
- Liu, W.; Xia, M.; Bai, J.; Yang, L.; Wang, Z.; Wang, R.; Shi, Y. Chemical characterization and 5α-reductase inhibitory activity of phenolic compounds in goji berries. J. Pharm. Biomed. Anal. 2021, 201, 114119. [Google Scholar] [CrossRef]
- Oliw, E.H.; Su, C.; Skogström, T.; Benthin, G. Analysis of novel hydroperoxides and other metabolites of oleic, linoleic, and linolenic acids by liquid chromatography-mass spectrometry with ion trap MSn. Lipids 1998, 33, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Pontieri, P.; Pepe, G.; Campiglia, P.; Merciai, F.; Basilicata, M.G.; Smolensky, D.; Calcagnile, M.; Troisi, J.; Romano, R.; Del Giudice, F.; et al. Comparison of Content in Phenolic Compounds and Antioxidant Capacity in Grains of White, Red, and Black Sorghum Varieties Grown in the Mediterranean Area. ACS Food Sci. 2021, 1, 1109–1119. [Google Scholar] [CrossRef]
- Kang, J.; Price, W.E.; Ashton, J.; Tapsell, L.C.; Johnson, S. Identification and characterization of phenolic compounds in hydromethanolic extracts of sorghum wholegrains by LC-ESI-MS(n). Food Chem. 2016, 211, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Bondia-Pons, I.; Savolainen, O.; Törrönen, R.; Martinez, A.; Poutanen, K.; Hanhineva, K. Metabolic profiling of Goji berry extracts for discrimination of geographical origin by non-targeted liquid chromatography coupled to quadrupole time-of-flight mass spectrometry. Food Res. Int. 2014, 63, 132–138. [Google Scholar] [CrossRef]
- Lu, W.; Jiang, Q.; Shi, H.; Niu, Y.; Gao, B.; Yu, L.L. Partial least-squares-discriminant analysis differentiating Chinese wolfberries by UPLC-MS and flow injection mass spectrometric (FIMS) fingerprints. J. Agric. Food Chem. 2014, 62, 9073–9080. [Google Scholar] [CrossRef]
- Duan, W.; Zhang, Z.; Zhu, J.; Zhang, D.; Qian, D.; Teng, F.; Zhao, Y.; Chen, F.; Li, R.; Yang, J.J.M. Comparative Analysis of the Phenolic Profile of Lycium barbarum L. Fruits from Different Regions in China. Molecules 2022, 27, 5842. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Lin, S.; Jia, Y.; Rehman, F.; Zeng, S.; Wang, Y. Anthocyanin and spermidine derivative hexoses coordinately increase in the ripening fruit of Lycium ruthenicum. Food Chem. 2020, 311, 125874. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Gao, R.; Liu, J.; Zhang, S.; Zhao, Y.; Yu, Z. UPLC-Q-Exactive-based rats serum metabolomics for characterization of traditional Chinese medicine Natures and Flavors. J. Ethnopharmacol. 2023, 302, 115931. [Google Scholar] [CrossRef]
- Dumont, D.; Danielato, G.; Chastellier, A.; Hibrand Saint Oyant, L.; Fanciullino, A.-L.; Lugan, R.J.M. Multi-targeted metabolic profiling of carotenoids, phenolic compounds and primary metabolites in goji (Lycium spp.) berry and tomato (Solanum lycopersicum) reveals inter and intra genus biomarkers. Metabolites 2020, 10, 422. [Google Scholar] [CrossRef]
- Carolina, L.; Ramirez, A.; Aguilera, R.J. Differential nuclear staining assay for high-throughput screening to identify cytotoxic compounds. Curr. Cell. Biochem. 2011, 1, 1. [Google Scholar]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2001, 6, 128. [Google Scholar] [CrossRef]
- Suchiya, K. Switching from Apoptosis to Pyroptosis: Gasdermin-Elicited Inflammation and Antitumor Immunity. Int. J. Mol. Sci. 2021, 22, 426. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Du, J.; Li, D.; Yang, P.; Kou, Y.; Li, C.; Zhou, Q.; Lu, Y.; Hasegawa, T.; Li, M. Oxidative stress induced pyroptosis leads to osteogenic dysfunction of MG63 cells. J. Mol. Histol. 2020, 51, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Ping, Z.; Tou, F.; Zhan, Y.; Cai, X.; Lang, H.; Fan, H.; Jun, M.; Liang, S. Oxidative Stress and Pyroptosis in Doxorubicin-Induced Heart Failure and Atrial Fibrillation. Oxid. Med. Cell Longev. 2023, 2023, 4938287. [Google Scholar] [CrossRef]
- Carvalho, C.; Santos, R.X.; Cardoso, S.; Correia, S.; Oliveira, P.J.; Santos, M.S.; Moreira, P.I. Doxorubicin: The good, the bad and the ugly effect. Curr. Med. Chem. 2009, 16, 3267–3285. [Google Scholar] [CrossRef]
- Beriault, D.R.; Werstuck, G.H. Detection and quantification of endoplasmic reticulum stress in living cells using the fluorescent compound, Thioflavin T. Biochim. Biophys. Acta 2013, 1833, 2293–2301. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, H.D. Long and short (timeframe) of endoplasmic reticulum stress-induced cell death. FEBS J. 2016, 283, 3718–3722. [Google Scholar] [CrossRef]
- Badiola, N.; Penas, C.; Miñano-Molina, A.; Barneda-Zahonero, B.; Fadó, R.; Sánchez-Opazo, G.; Comella, J.X.; Sabriá, J.; Zhu, C.; Blomgren, K.; et al. Induction of ER stress in response to oxygen-glucose deprivation of cortical cultures involves the activation of the PERK and IRE-1 pathways and of caspase-12. Cell Death Dis. 2011, 2, e149. [Google Scholar] [CrossRef]
- Morishima, N.; Nakanishi, K.; Tsuchiya, K.; Shibata, T.; Seiwa, E. Translocation of Bim to the endoplasmic reticulum (ER) mediates ER stress signaling for activation of caspase-12 during ER stress-induced apoptosis. J. Biol. Chem. 2004, 279, 50375–50381. [Google Scholar] [CrossRef]
- Lebeaupin, C.; Proics, E.; de Bieville, C. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis. 2005, 6, e1879. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cao, T.; Luo, C.; Cai, J.; Zhou, X.; Xiao, X.; Liu, S. Crosstalk between ER stress, NLRP3 inflammasome, and inflammation. Appl. Microbiol. Biotechnol. 2020, 104, 6129–6140. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.; Yang, Y.; Jin, T.; Jiang, W.; Zhou, R. Orchestration of NLRP3 inflammasome activation by ion fluxes. Trends Immunol. 2018, 39, 393–406. [Google Scholar] [CrossRef]
- Gomez-Suaga, P.; Bravo-San Pedro, J.M.; Gonzalez-Polo, R.A.; Fuentes, J.M.; Niso-Santano, M. ER-mitochondria signaling in Parkinson’s disease. Cell Death Dis. 2018, 9, 337. [Google Scholar] [CrossRef] [PubMed]
- Belvedere, R.; Morretta, E.; Novizio, N.; Morello, S.; Bruno, O.; Brullo, C.; Petrella, A. The Pyrazolyl-Urea Gege3 Inhibits the Activity of ANXA1 in the Angiogenesis Induced by the Pancreatic Cancer Derived EVs. Biomolecules 2021, 11, 1758. [Google Scholar] [CrossRef] [PubMed]
- Senthil, K.; Aranganathan, S.; Nalini, N. Evidence of oxidative stress in the circulation of ovarian cancer patients. Clin. Chim. Acta 2004, 339, 27–32. [Google Scholar] [CrossRef]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef]
- Shin, J.; Song, M.H.; Oh, J.W.; Keum, Y.S.; Saini, R.K. Pro-Oxidant Actions of Carotenoids in Triggering Apoptosis of Cancer Cells: A Review of Emerging Evidence. Antioxidants 2020, 9, 532. [Google Scholar] [CrossRef]
- Martin-Cordero, C.; Leon-Gonzalez, A.J.; Calderon-Montano, J.M.; Burgos-Moron, E.; Lopez-Lazaro, M. Pro-oxidant natural products as anticancer agents. Curr. Drug Targets 2012, 13, 1006–1028. [Google Scholar] [CrossRef]
- D’angelo, S.; D’Angelo, S.; Martino, E.; Ilisso, C.P.; Bagarolo, M.L.; Porcelli, M.; Cacciapuoti, G. Pro-oxidant and pro-apoptotic activity of polyphenol extract from Annurca apple and its underlying mechanisms in human breast cancer cells. Int. J. Oncol. 2017, 51, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Kepinska, M.; Kizek, R.; Milnerowicz, H. Metallothionein and Superoxide Dismutase—Antioxidative Protein Status in Fullerene-Doxorubicin Delivery to MCF-7 Human Breast Cancer Cells. Int. J. Mol. Sci. 2018, 19, 3253. [Google Scholar] [CrossRef] [PubMed]
- Irazabal, M.V.; Torres, V.E. Reactive Oxygen Species and Redox Signaling in Chronic Kidney Disease. Cells 2020, 9, 1342. [Google Scholar] [CrossRef] [PubMed]
- Courage, S.D.; Haihui, Z.; Vera, G.; David, A.-D.; Yuqing, D. Anti- and pro-oxidant properties of polyphenols and their role in modulating glutathione synthesis, activity and cellular redox potential: Potential synergies for disease management. Adv. Redox Res. 2024, 11, 100099. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Martucciello, S.; Masullo, M.; Cerulli, A.; Piacente, S. Natural Products Targeting ER Stress, and the Functional Link to Mitochondria. Int. J. Mol. Sci. 2020, 21, 1905. [Google Scholar] [CrossRef] [PubMed]
- Vestuto, V.; Di Sarno, V.; Musella, S.; Di Dona, G.; Moltedo, O.; Gomez-Monterrey, I.M.; Bertamino, A.; Ostacolo, C.; Campiglia, P.; Ciaglia, T. New Frontiers on ER Stress Modulation: Are TRP Channels the Leading Actors? Int. J. Mol. Sci. 2023, 24, 185. [Google Scholar] [CrossRef] [PubMed]
- Georgiev, K.D.; Slavov, I.J.; Iliev, I.A. Antioxidant Activity and Antiproliferative Effects of Lycium barbarum’s (Goji berry) Fractions on Breast Cancer Cell Lines. Folia Med. 2019, 61, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Zhang, J.; Liu, L.; Xu, B.; Han, H.; Dai, W.; Pei, X.; Fu, X.; Hou, S. A novel anticancer property of Lycium barbarum polysaccharide in triggering ferroptosis of breast cancer cells. J. Zhejiang Univ. Sci. B 2022, 23, 286–299. [Google Scholar] [CrossRef]
- Wawruszak, A.; Czerwonka, A.; Okła, K.; Rzeski, W. Anticancer effect of ethanol Lycium barbarum (Goji berry) extract on human breast cancer T47D cell line. Nat. Prod. Res. 2016, 30, 1993–1996. [Google Scholar] [CrossRef]
- Claudine, M.; Gary, W.; Christine, M.; Augustin, S.; Christian, R. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. Am. J. Clin. Nutr. 2005, 81, 230S–242S. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Xia, H.; Tang, H.; Yang, L.; Sun, G. Tissue distribution of Lycium barbarum polysaccharides in rat tissue by fluorescein isothiocyanate labeling. Food Sci. Hum. Wellness 2022, 11, 837–844. [Google Scholar] [CrossRef]
- Xia, H.; Yang, C.; Zhou, B.; Tang, H.; Yang, L.; Liao, W.; Sun, G. Pharmacokinetics and Excretion Study of Lycium barbarum Polysaccharides in Rats by FITC-Fluorescence Labeling. Foods 2021, 10, 2851. [Google Scholar] [CrossRef] [PubMed]
- Ávila-Gálvez, M.Á.; González-Sarrías, A.; Martínez-Díaz, F.; Abellán, B.; Martínez-Torrano, A.J.; Fernández-López, A.J.; Giménez-Bastida, J.A.; Espín, J.C. Disposition of Dietary Polyphenols in Breast Cancer Patients’ Tumors, and Their Associated Anticancer Activity: The Particular Case of Curcumin. Mol. Nutr. Food Res. 2021, 65, e2100163. [Google Scholar] [CrossRef] [PubMed]
- Xin, Y.F.; Zhou, G.L.; Deng, Z.Y.; Chen, Y.X.; Wu, Y.G.; Xu, P.S. Protective effect of Lycium barbarum on doxorubicin-induced cardiotoxicity. Phytother. Res. 2007, 21, 1020–1024. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Luo, S.; Luo, X.; Hu, M.; Ma, F.; Wang, Y. Fraction from Lycium barbarum Polysaccharides Reduces Immunotoxicity and Enhances Antitumor Activity of Doxorubicin in Mice. Integr. Cancer Ther. 2018, 17, 860–866. [Google Scholar] [CrossRef]
- Sun, L.; Zuo, C.; Liu, X.; Guo, Y.; Wang, X.; Dong, Z.; Han, M. Combined Photothermal Therapy and Lycium barbarum Polysaccharide for Topical Administration to Improve the Efficacy of Doxorubicin in the Treatment of Breast Cancer. Pharmaceutics 2022, 14, 2677. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miranda, M.R.; Basilicata, M.G.; Vestuto, V.; Aquino, G.; Marino, P.; Salviati, E.; Ciaglia, T.; Domínguez-Rodríguez, G.; Moltedo, O.; Campiglia, P.; et al. Anticancer Therapies Based on Oxidative Damage: Lycium barbarum Inhibits the Proliferation of MCF-7 Cells by Activating Pyroptosis through Endoplasmic Reticulum Stress. Antioxidants 2024, 13, 708. https://doi.org/10.3390/antiox13060708
Miranda MR, Basilicata MG, Vestuto V, Aquino G, Marino P, Salviati E, Ciaglia T, Domínguez-Rodríguez G, Moltedo O, Campiglia P, et al. Anticancer Therapies Based on Oxidative Damage: Lycium barbarum Inhibits the Proliferation of MCF-7 Cells by Activating Pyroptosis through Endoplasmic Reticulum Stress. Antioxidants. 2024; 13(6):708. https://doi.org/10.3390/antiox13060708
Chicago/Turabian StyleMiranda, Maria Rosaria, Manuela Giovanna Basilicata, Vincenzo Vestuto, Giovanna Aquino, Pasquale Marino, Emanuela Salviati, Tania Ciaglia, Gloria Domínguez-Rodríguez, Ornella Moltedo, Pietro Campiglia, and et al. 2024. "Anticancer Therapies Based on Oxidative Damage: Lycium barbarum Inhibits the Proliferation of MCF-7 Cells by Activating Pyroptosis through Endoplasmic Reticulum Stress" Antioxidants 13, no. 6: 708. https://doi.org/10.3390/antiox13060708
APA StyleMiranda, M. R., Basilicata, M. G., Vestuto, V., Aquino, G., Marino, P., Salviati, E., Ciaglia, T., Domínguez-Rodríguez, G., Moltedo, O., Campiglia, P., Pepe, G., & Manfra, M. (2024). Anticancer Therapies Based on Oxidative Damage: Lycium barbarum Inhibits the Proliferation of MCF-7 Cells by Activating Pyroptosis through Endoplasmic Reticulum Stress. Antioxidants, 13(6), 708. https://doi.org/10.3390/antiox13060708