
Metabolomics Combined with Physiology and Transcriptomics Reveal the Response of Samsoniella hepiali to Key Metabolic Pathways and Its Degradation Mechanism during Subculture



Abstract

1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Observation of Apparent Morphology and Determination of Growth Rate, Dry Weight, Fresh Weight, and Sporulation
2.3. Antioxidative Enzyme Activity and Malondialdehyde (MDA) Content
2.4. Untargeted Metabolomics Analysis
2.5. Transcriptome Analysis
2.6. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR) Verification
3. Results
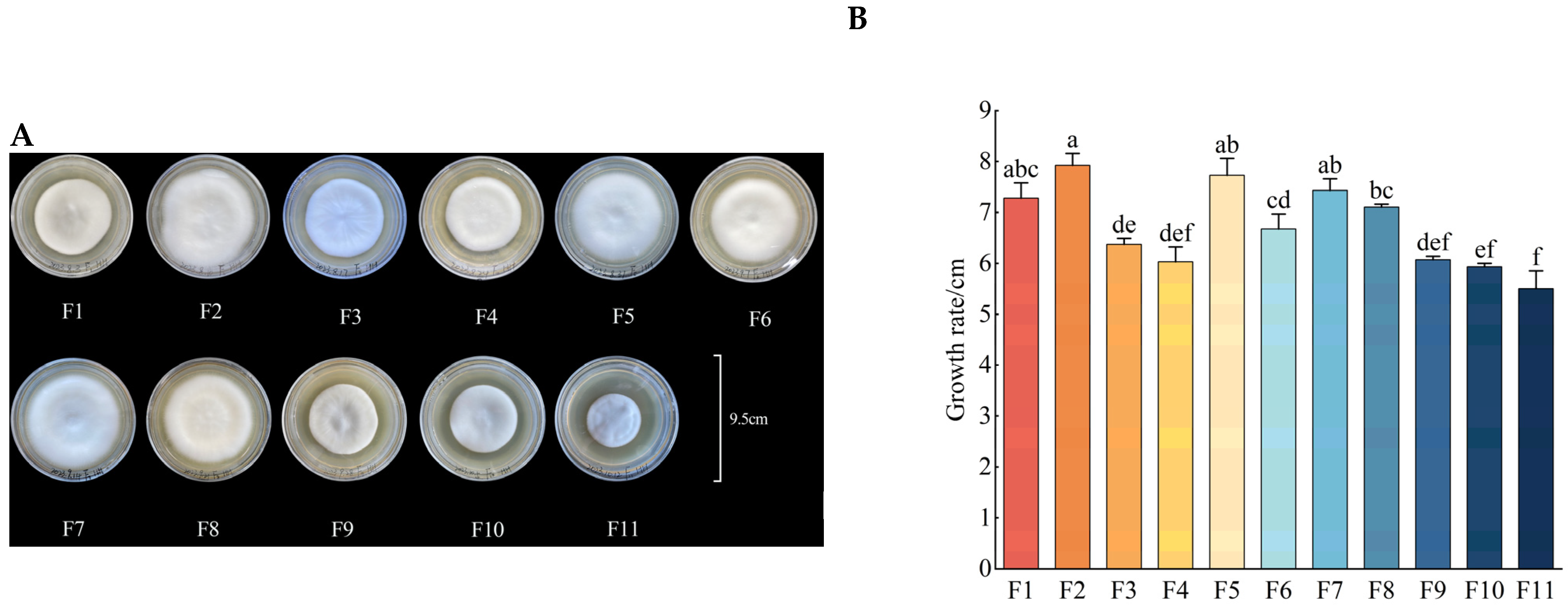
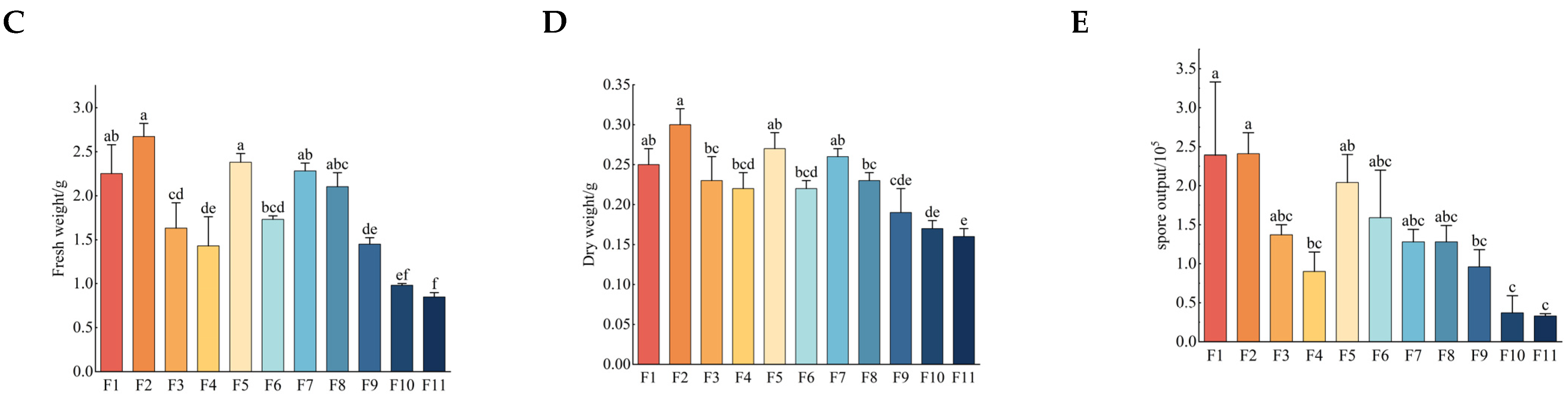
3.1. Growth Rate, Fresh Weight, Dry Weight and Sporulation
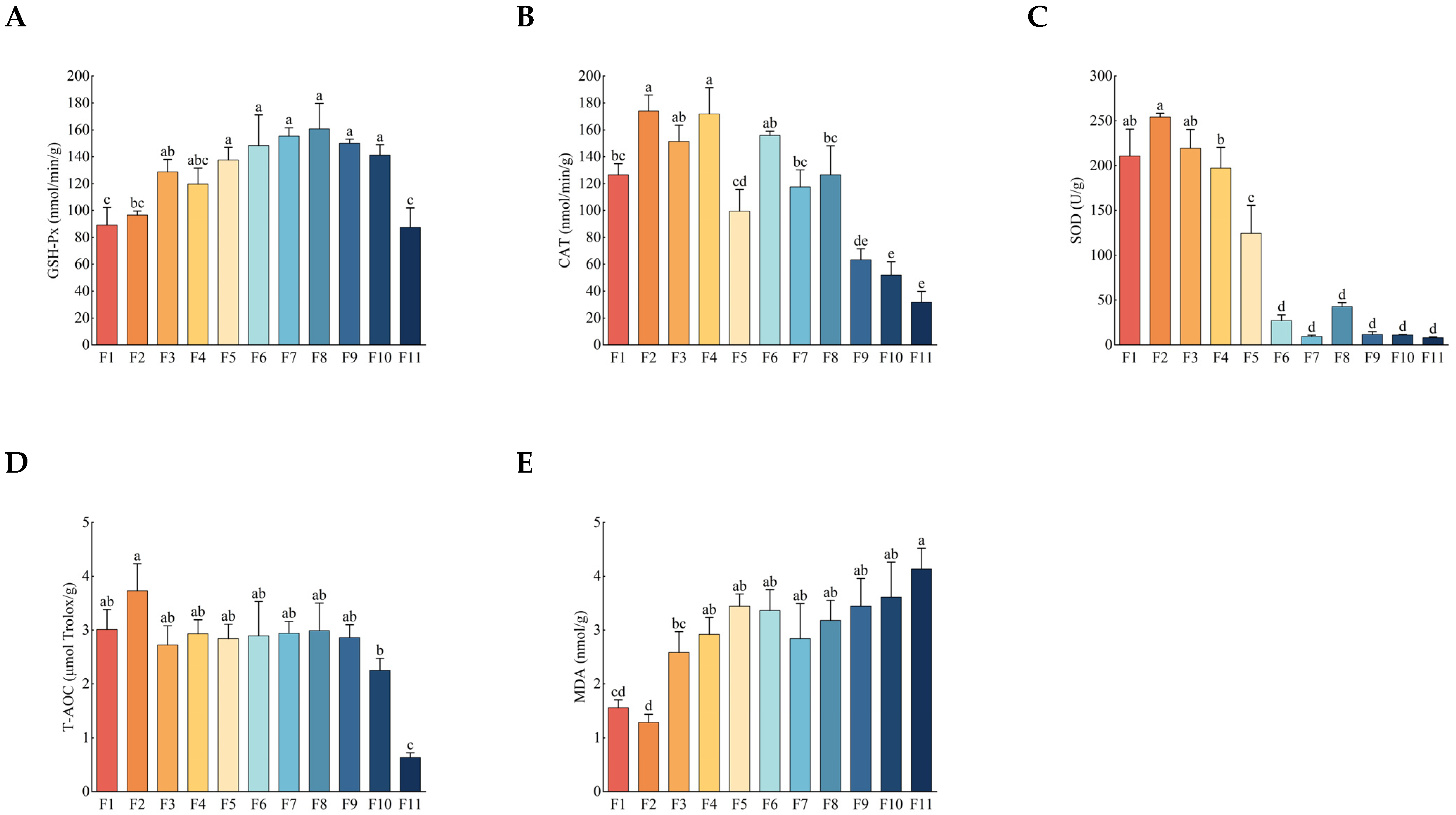
3.2. Antioxidant Activity and Malondialdehyde (MDA) Content
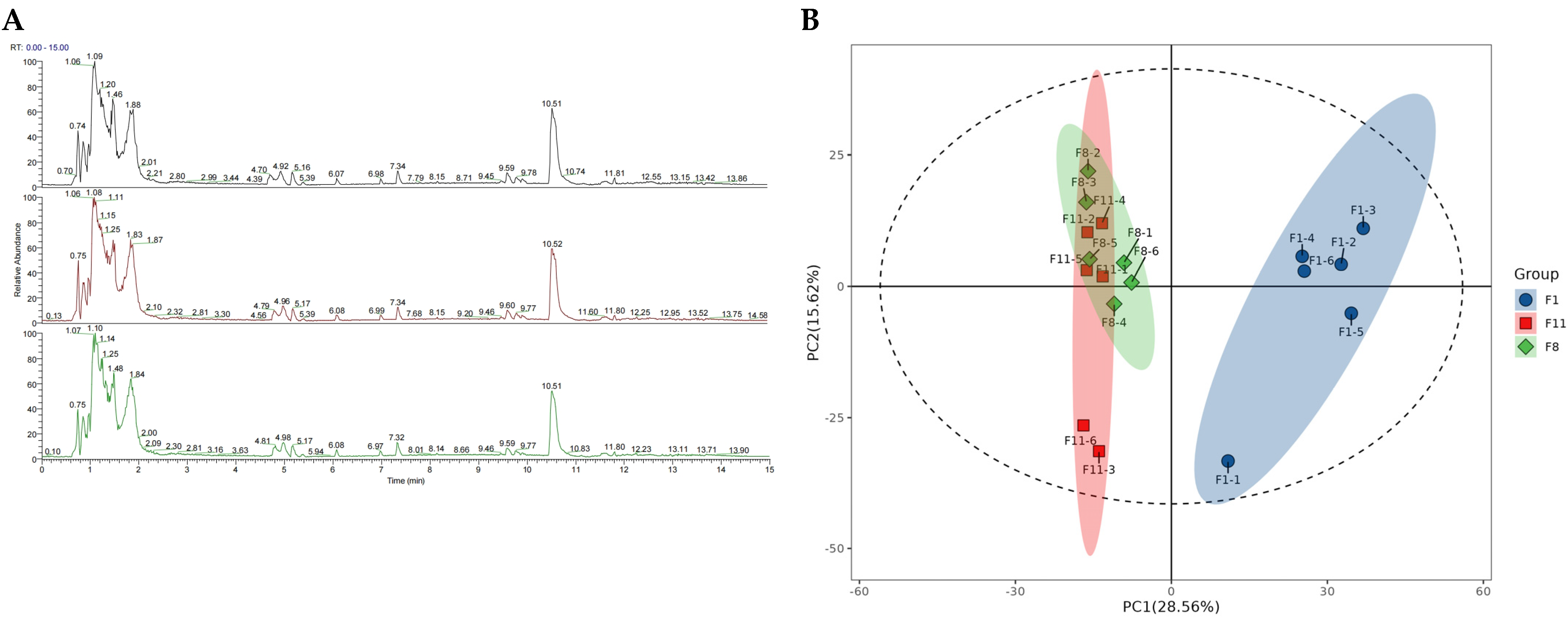
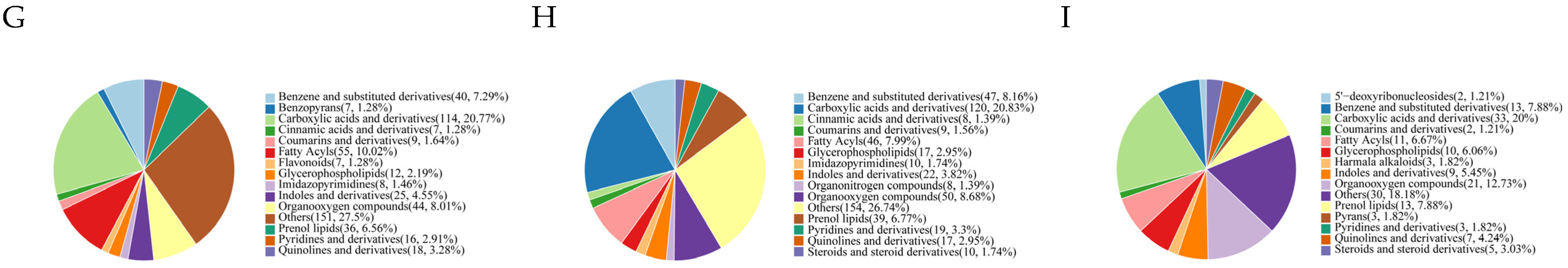
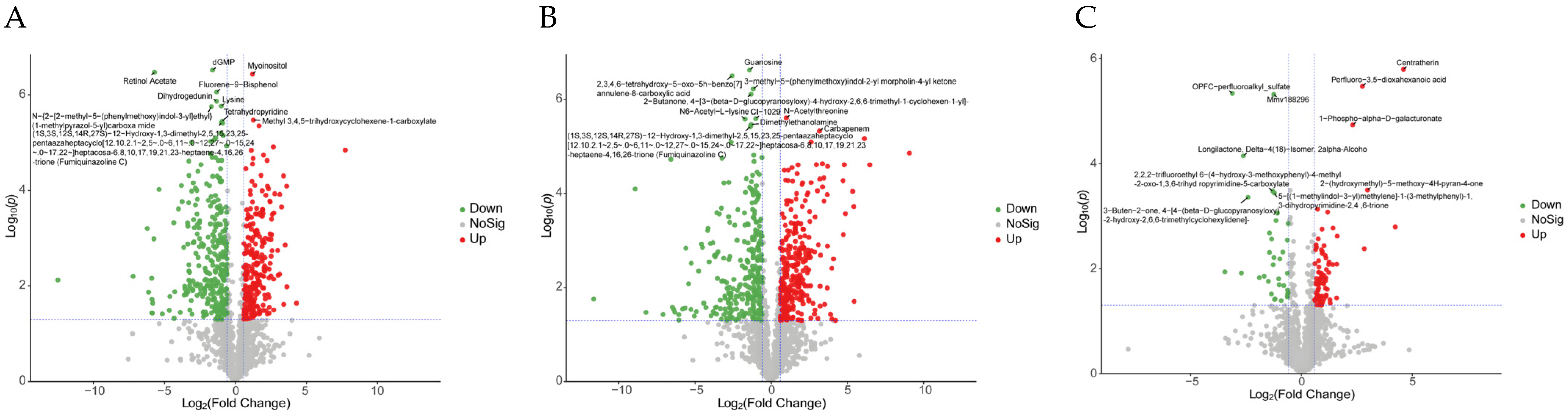
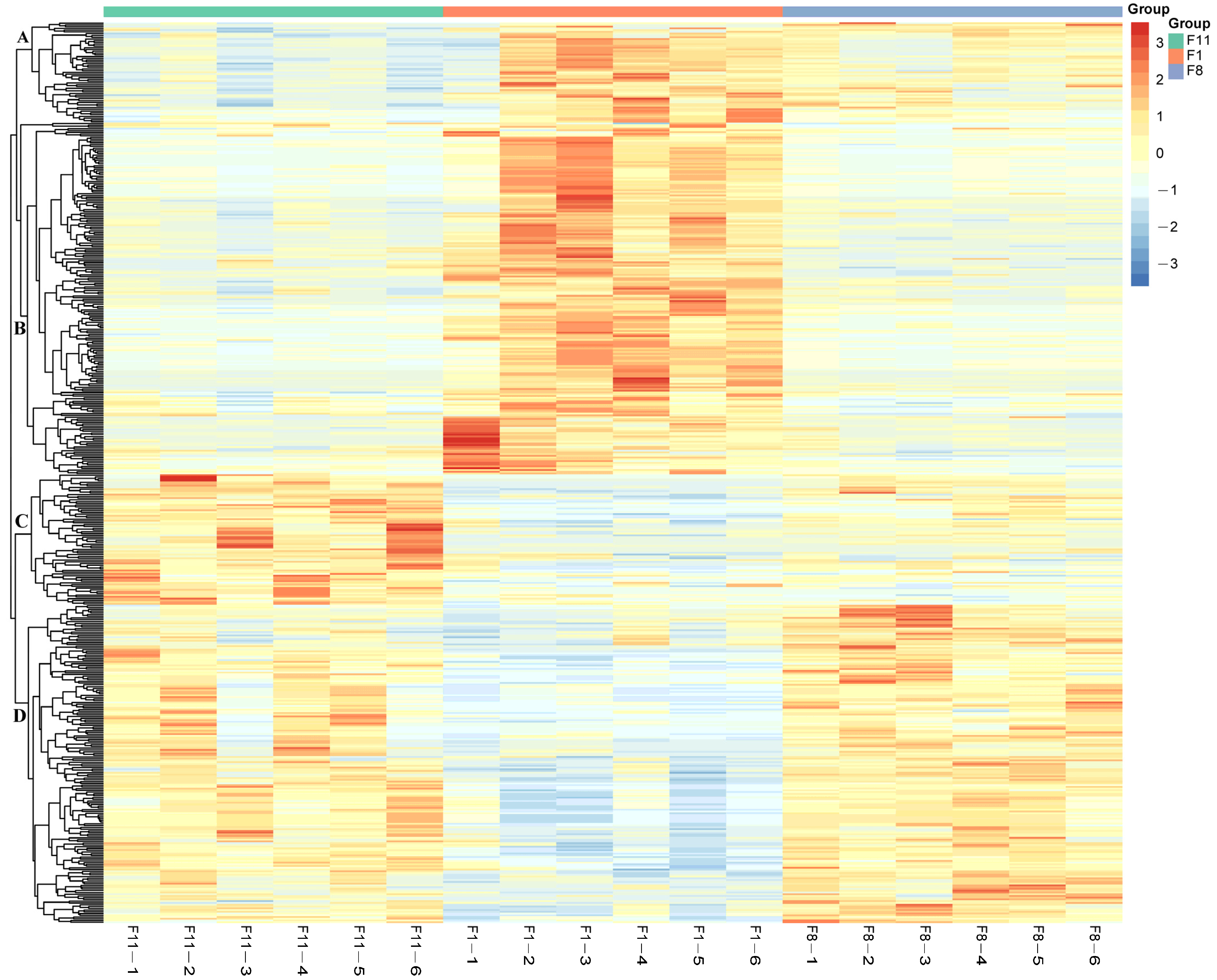
3.3. Metabolomic Analysis
3.3.1. Metabolic Analysis of Different Generations of Subcultured Strains
3.3.2. Identification of Differentially Accumulated Metabolites (DAMs) in Different
Subculture Strains
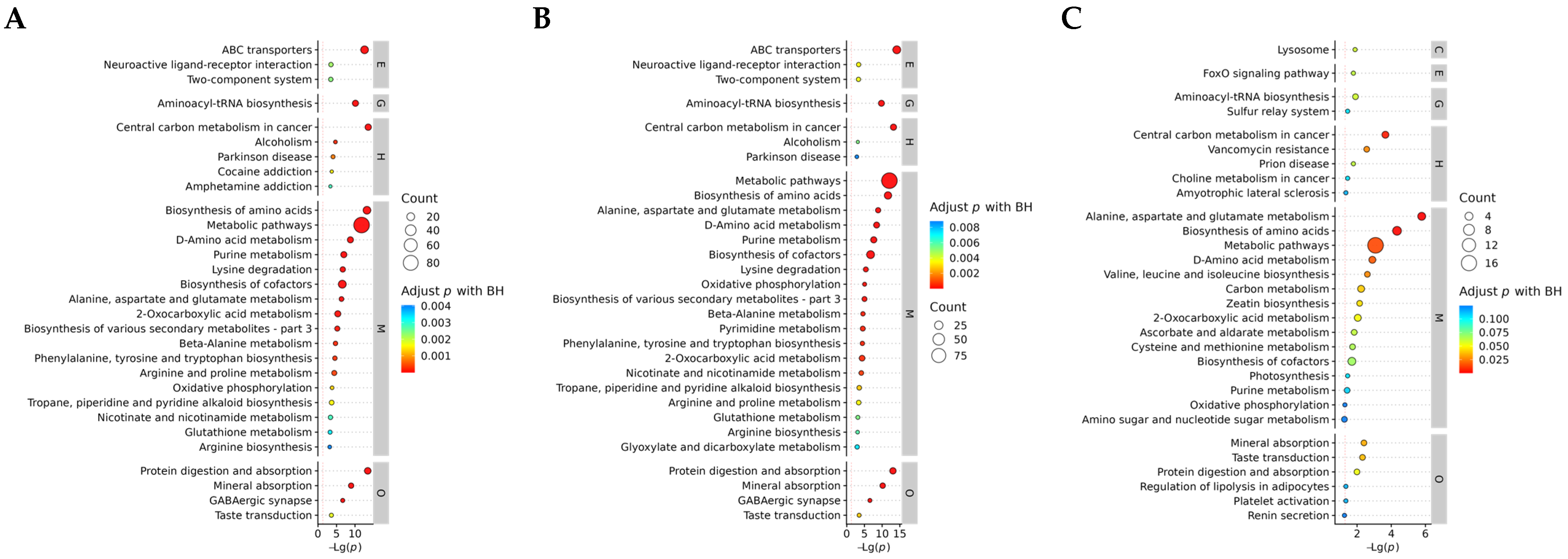
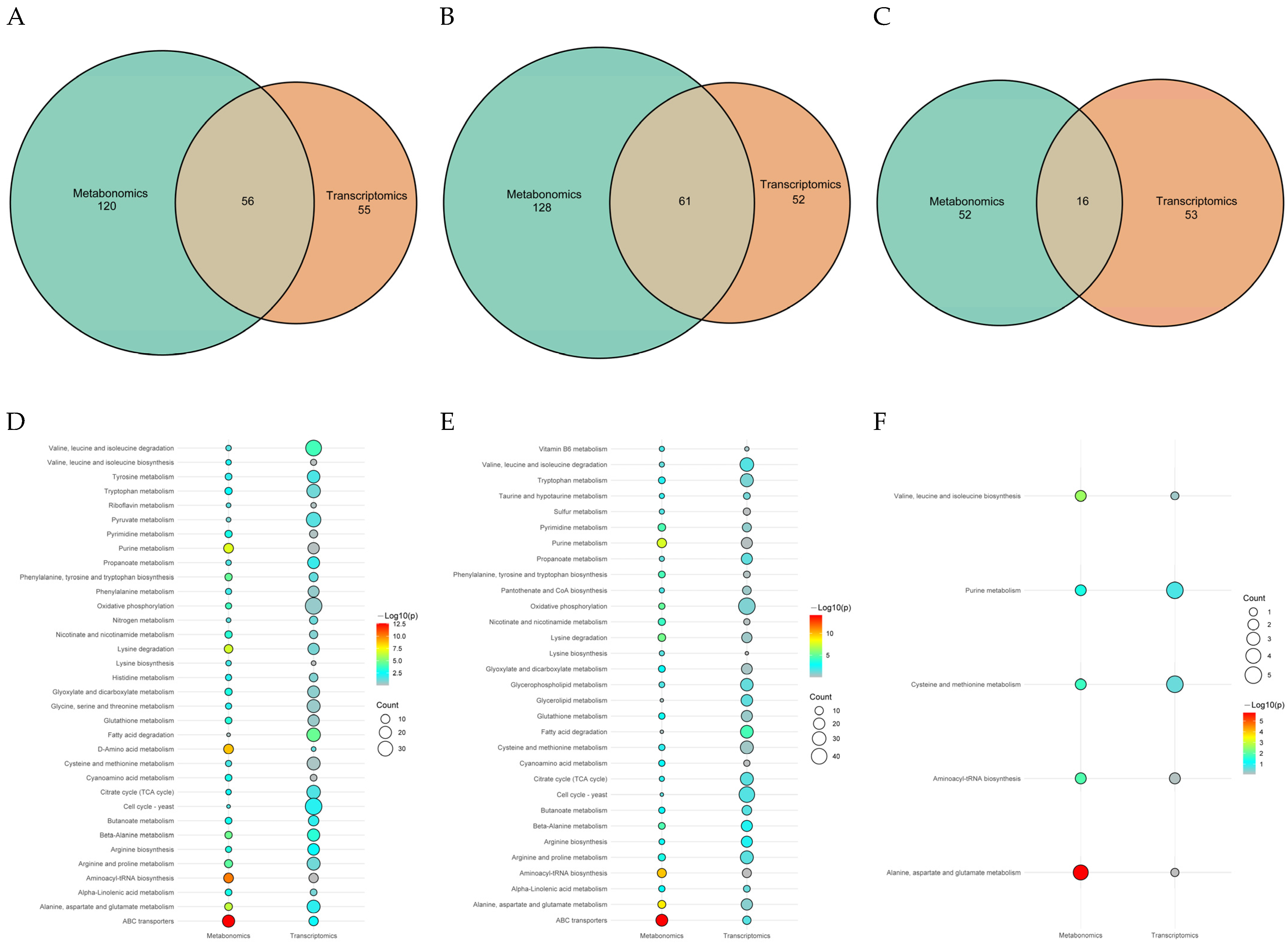
3.3.3. KEGG Enrichment Analysis of DAMs
3.4. Transcriptomic Analysis
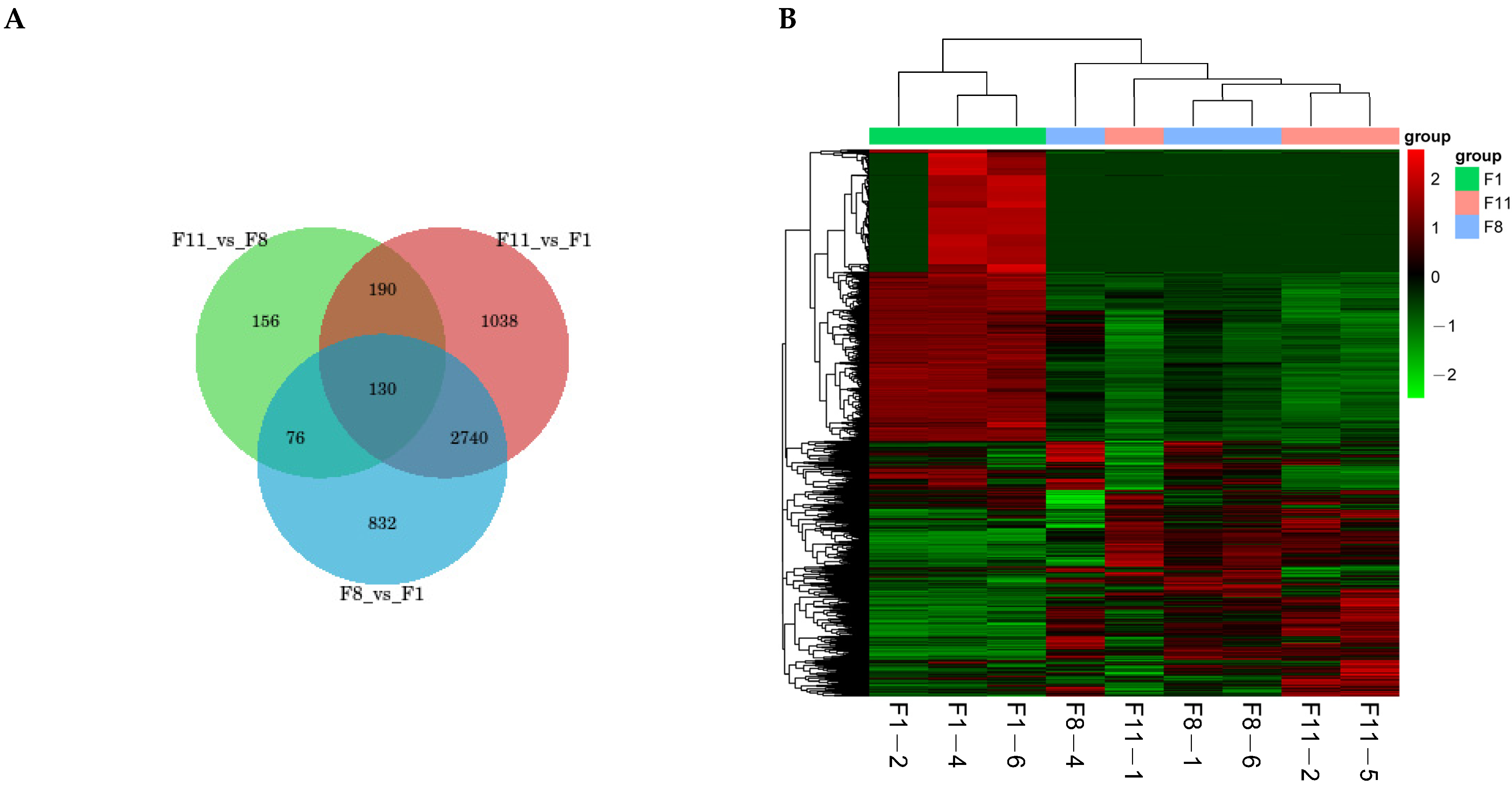
3.4.1. Transcriptome Analysis of Different Subculture Strains
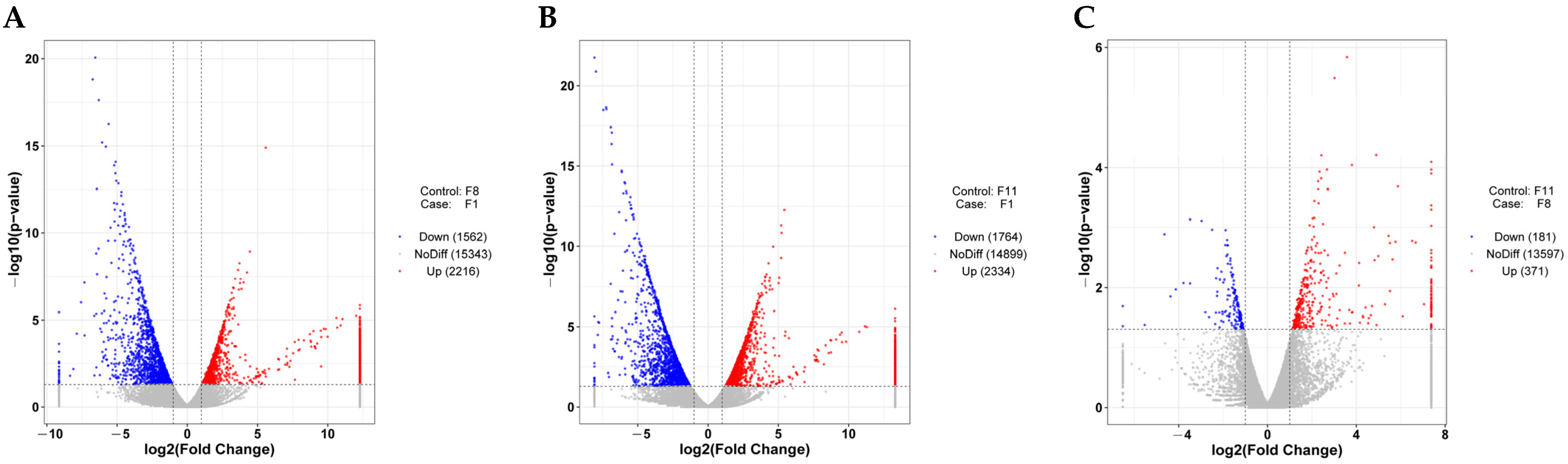
3.4.2. Differentially Expressed Genes (DEGs) of Different Subcultured Strains
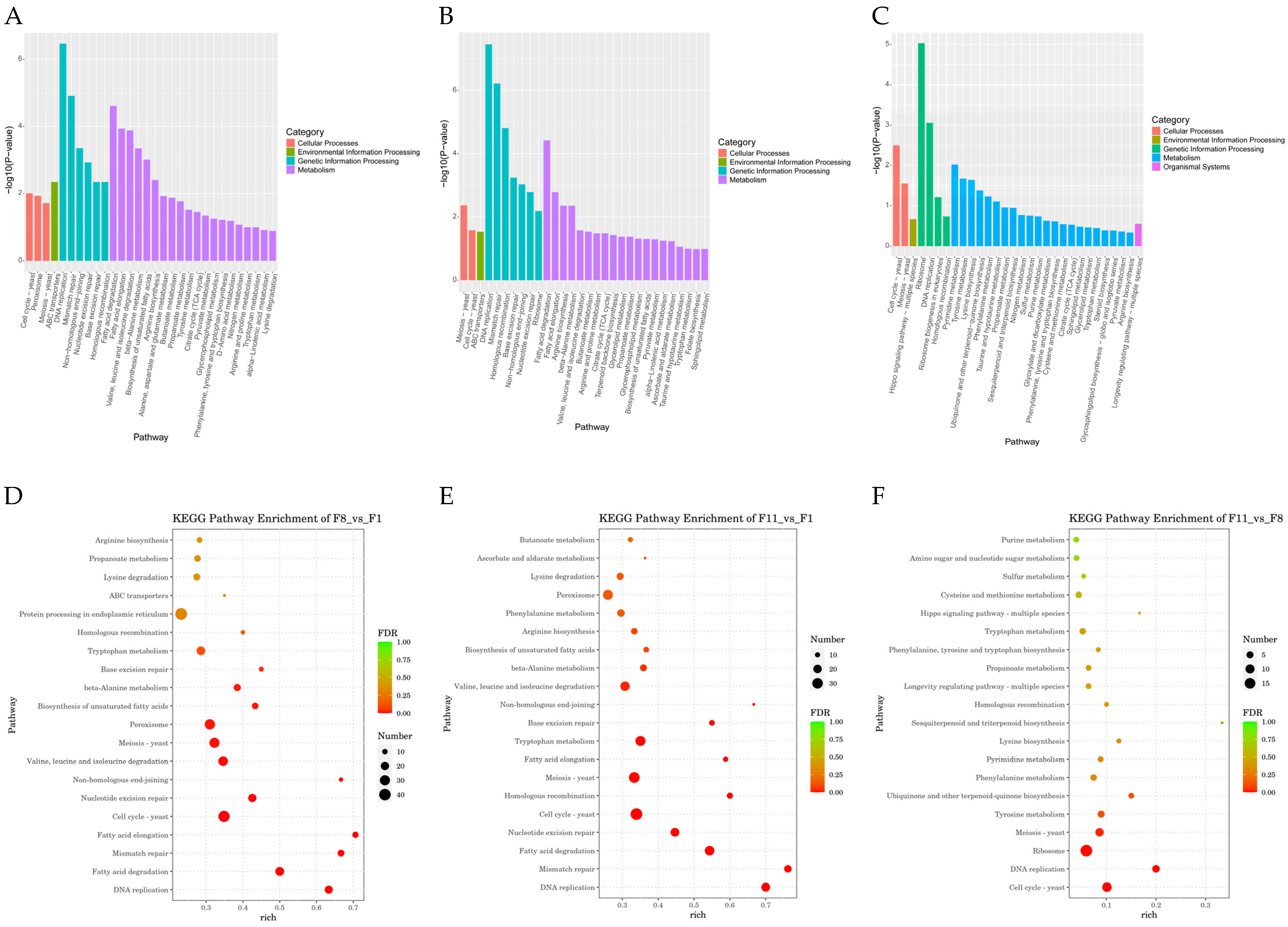
3.4.3. GO Analysis and KEGG Pathway Analysis
3.5. Joint Analysis of the Transcriptome and Metabolome
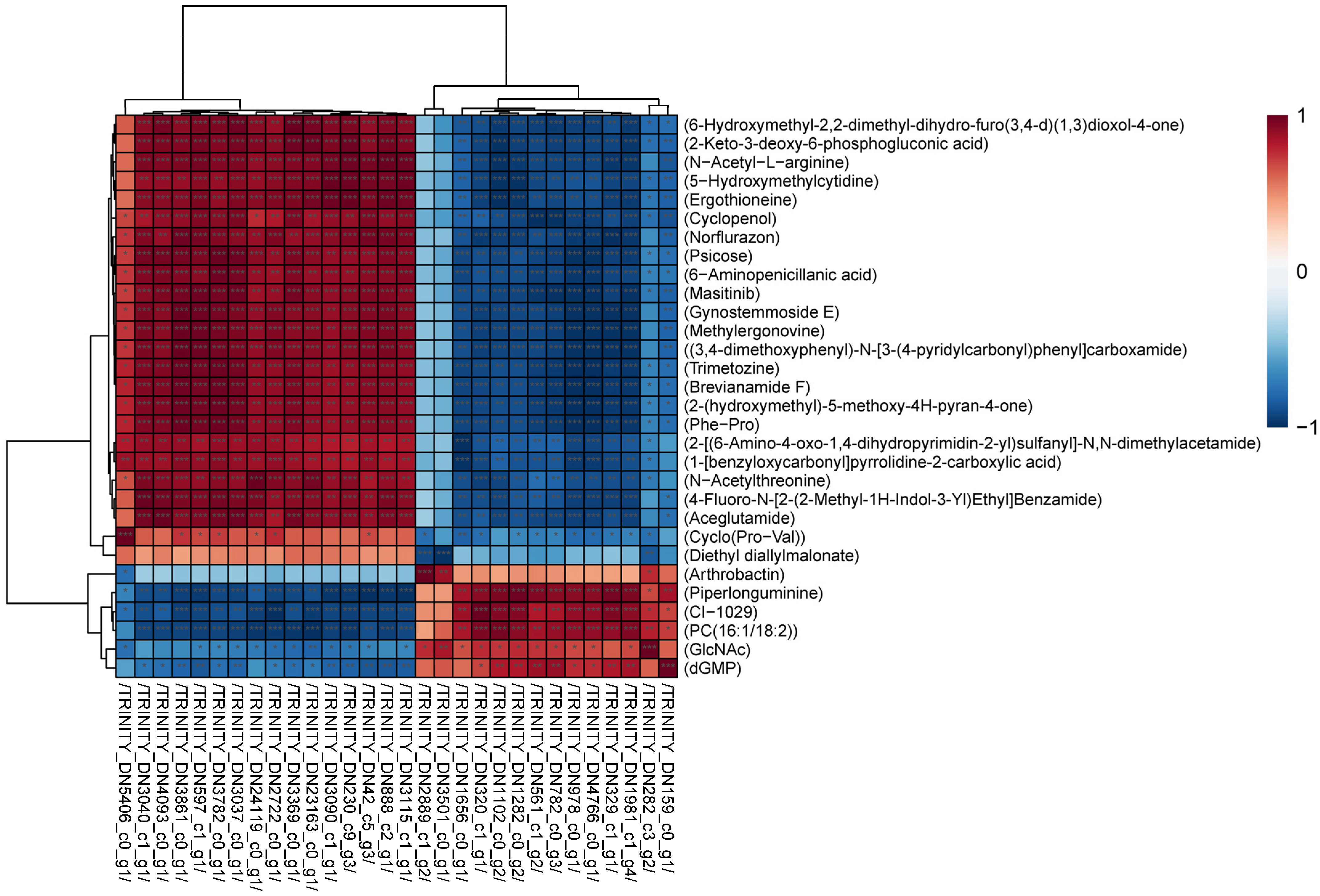
3.5.1. Correlations between DEGs and DAMs
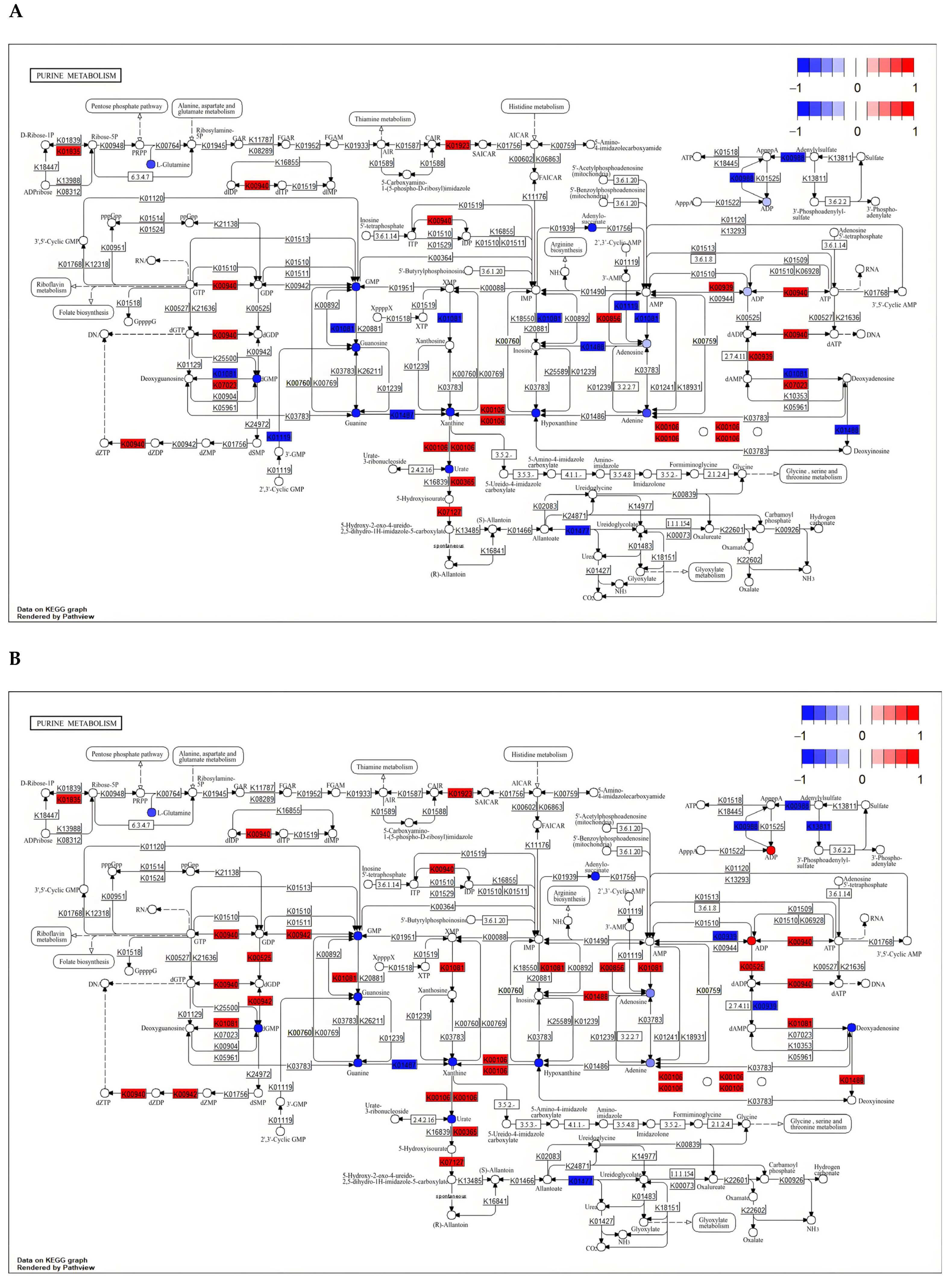
3.5.2. Critical Pathway Analysis
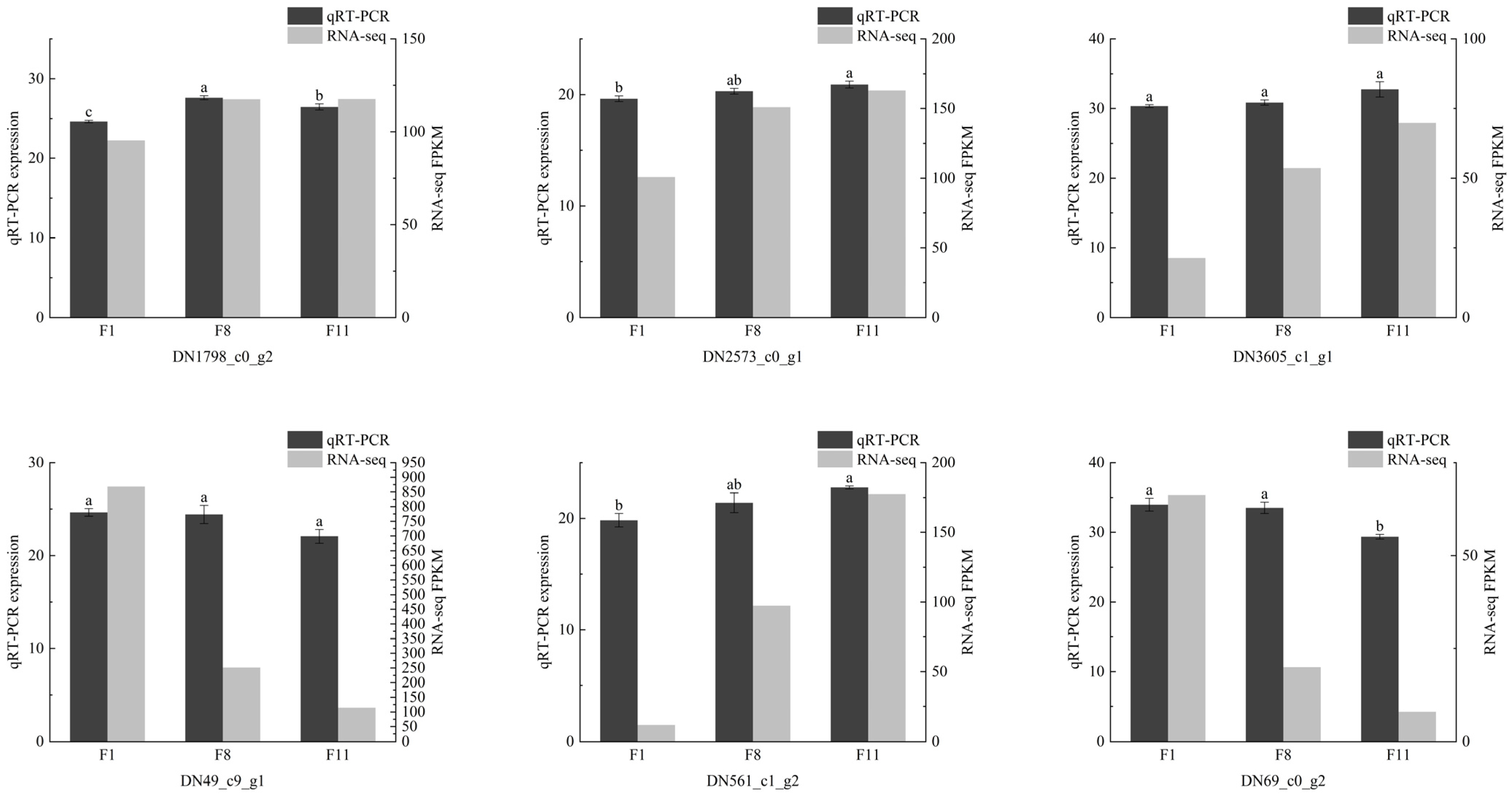
3.6. Confirmation of DEGs via qRT-PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cairns, T.; Zheng, X.; Zheng, P.; Sun, J.; Meyer, V. Turning inside out: Filamentous fungal secretion and its applications in biotechnology, agriculture, and the clinic. J. Fungi 2021, 7, 535. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhao, Q.; He, Q. Application of CRISPR in Filamentous Fungi and Macrofungi: From Component Function to Development Potentiality. ACS Synth. Biol. 2023, 12, 1908–1923. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Gao, J.; Wang, W.; Wang, K.; Xu, G.; Huang, H.; Ji, X. CRISPR/Cas9-based genome editing in the filamentous fungus Fusarium fujikuroi and its application in strain engineering for gibberellic acid production. ACS Synth. Biol. 2019, 8, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.; Lin, Y.; Wan, Y.; Chen, K.; Huang, J.; Chang, P. Degenerated virulence and irregular development of Fusarium oxysporum f. sp. niveum induced by successive subculture. J. Fungi 2020, 6, 382. [Google Scholar]
- Lou, H.; Lin, J.; Guo, L.; Wang, X.; Tian, S.; Liu, C.; Zhao, Y.; Zhao, R. Advances in research on Cordyceps militaris degeneration. Appl. Microbiol. Biotechnol. 2019, 103, 7835–7841. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; He, L.; Wang, Z.; Chen, J. The Influences of Subculture of Cordyceps militaris to Colonial Morphologyand Fruit-body Yield. J. Shenyang Agric. Univ. 2006, 37, 538–541. [Google Scholar]
- Zhao, F.; Wang, Q.; An, X.; Tan, Q.; Yun, J.; Zhang, Y. Oxidative damage from repeated tissue isolation for subculturing causes degeneration in Volvariella volvacea. Front. Microbiol. 2023, 14, 1210496. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Bai, Y.; Zhang, Y.; Fan, Y.; Guo, W. Effects of Subculturing on the Growth, Development and Pathogenicity of Setosphaeria turcica. J. Maize Sci. 2013, 21, 134–136+140. [Google Scholar]
- Zhao, F.; Liu, X.; Chen, C.; Cheng, Z.; Wang, W.; Yun, J. Successive mycelial subculturing decreased lignocellulase activity and increased ROS accumulation in Volvariella volvacea. Front. Microbiol. 2022, 13, 997485. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Ha, M.; Li, P.; Jin, J. Research Progress on the Mechanism of Cordyceps militaris Strain Degradation. J. Jilin Agric. Sci. Technol. Univ. 2023, 32, 12–16. [Google Scholar]
- Chen, X.; Zhang, Z.; Liu, X.; Cui, B.; Miao, W.; Cheng, W.; Zhao, F. Characteristics analysis reveals the progress of Volvariella volvacea mycelium subculture degeneration. Front. Microbiol. 2019, 10, 412945. [Google Scholar] [CrossRef]
- Wellham, P.; Hafeez, A.; Gregori, A.; Brock, M.; Kim, D.; Chandler, D.; Moor, C. Culture degeneration reduces sex-related gene expression, alters metabolite production and reduces insect pathogenic response in Cordyceps militaris. Microorganisms 2021, 9, 1559. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Xu, J.; Hou, H.; Wu, S.; Xiong, K.; He, L.; Li, C.; Wu, X. Transcriptome analysis of Samsoniella hepiali induced by salicylic acid and crucial genes digging for metabolic pathways of cordycepic acid. Acta Microbiol. Sin. 2022, 62, 3751–3767. [Google Scholar]
- Zha, Y.; Zhang, Z.; Li, X.; Xu, H.; Liu, X.; Li, Y. Optimization of Polyphenols Extraction from Cordyceps sinensis mycelium by Response Surface Methodology. Chin. Qinghai J. Anim. Vet. Sci. 2019, 49, 10–17. [Google Scholar]
- An, C.; Ma, S.; Chang, F.; Shangguan, Y.; Xue, W. Overview of Research and Development Strategy of Cordyceps sinensis related Mycelium Fermentation Products. Shaanxi J. Agric. Sci. 2016, 62, 119–122. [Google Scholar]
- Danner, C.; Mach, R.; Mach-Aigner, A. The phenomenon of strain degeneration in biotechnologically relevant fungi. Appl. Microbiol. Biotechnol. 2023, 107, 4745–4758. [Google Scholar] [CrossRef] [PubMed]
- Yin, J. Study on the Degradative Mechanism of Cordyceps militaris in Subculture. Master’s Thesis, Jiangsu University of Science and Technology, Zhenjiang, China, 2018. [Google Scholar]
- Li, L. Study on the Degradation Mechanism of Filamentous Fungi. Ph.D. Thesis, Institute of Plant Physiology and Ecology Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, China, 2012. [Google Scholar]
- Sobota-Grzeszyk, A.; Kuźmicki, M.; Szamatowicz, J. Myoinositol in the prevention of gestational diabetes mellitus: Is it sensible? J. Diabetes Res. 2019, 2019, 915253. [Google Scholar] [CrossRef] [PubMed]
- El-Gamal, M.; Brahim, I.; Hisham, N.; Aladdin, R.; Mohammed, H.; Bahaaeldin, A. Recent updates of carbapenem antibiotics. Eur. J. Med. Chem. 2017, 131, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Schneider, E.; Hofmeister, O.; Kälble, S.; Seifert, R. Apoptotic and anti-proliferative effect of guanosine and guanosine derivatives in HuT-78 T lymphoma cells. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2020, 393, 1251–1267. [Google Scholar] [CrossRef]
- Bettio, L.; Gil-Mohapel, J.; Rodrigues, A. Guanosine and its role in neuropathologies. Purinergic Signal. 2016, 12, 411–426. [Google Scholar] [CrossRef]
- Lobo, J.; Castro, E.; Gouvea, D.; Fernandes, C.; Almeida, F.; Amorim, L.; Burth, P.; Rocha, L.; Santos, M.; Harmerski, L.; et al. Antiproliferative activity of Eremanthus crotonoides extracts and centratherin demonstrated in brain tumor cell lines. Rev. Bras. Farmacogn. 2012, 22, 1295–1300. [Google Scholar] [CrossRef]
- Yin, J.; Xin, X.; Weng, Y.; Gui, Z. Transcriptome-wide analysis reveals the progress of Cordyceps militaris subculture degeneration. PLoS ONE 2017, 12, e0186279. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, S.; Li, J.; Zhang, L.; Zhang, L.; Lei, Z. Influence of Subculture on Virulence to Frankliniella occidentalis and Conidial Production of the Entomopathogenic Fungus Beauveria bassiana. Sci. Agric. Sin. 2016, 49, 2977–2987. [Google Scholar]
- Park, H.; Kim, K. Selection of Lecanicillium strains with high virulence against developmental stages of Bemisia tabaci. Mycobiology 2010, 38, 210–214. [Google Scholar] [CrossRef]
- Papa, K.E.; Srb, A.M.; Federer, W.T. Inheritance of growth rate in Neurospora crassa: Reverse selection in an improved strain. Heredity 1967, 22, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W. Intestinal Toxicity Effect of Heavy Metal Cadmium on Procambarus clarkii. Master’s Thesis, Shanxi University, Taiyuan, China, 2023. [Google Scholar]
- Zhang, Y.; Li, H.; Yang, R.; Wang, C. Study on antioxidant enzymatic activities of Trichosporon asahii. Indian J. Microbiol. 2016, 56, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhou, G.; Qin, H.; Guan, Y.; Wang, T.; Ni, W.; Xie, H.; Xing, Y.; Tian, G.; Lyu, M.; et al. Metabolomics combined with physiology and transcriptomics reveal key metabolic pathway responses in apple plants exposure to different selenium concentrations. J. Hazard. Mater. 2024, 464, 132953. [Google Scholar] [CrossRef]
- Zhang, S.; Shao, L.; Sun, Z.; Huang, Y.; Liu, N. An atmospheric pollutant (inorganic nitrogen) alters the response of evergreen broad-leaved tree species to extreme drought. Ecotoxicol. Environ. Saf. 2020, 187, 109750. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W. Study on Biological Characteristics of Initial Degradation of Cordyceps militaris during Successive Transfer Culture. Master’s Thesis, Shenyang Agricultural University, Shenyang, China, 2021. [Google Scholar]
- Wang, C.; Lv, D.; Li, L. Study on the pathogenesis and degradation mechanism of entomogenous fungi. In Proceedings of the Mycological Society of China, Wuhan, China, 5–7 November 2008; pp. 16–23. [Google Scholar]
- Zhang, J.; Wang, N.; Chen, W.; Zhang, W.; Zhang, H.; Yu, H.; Yi, Y. Integrated metabolomics and transcriptomics reveal metabolites difference between wild and cultivated Ophiocordyceps sinensis. Food Res. Int. 2023, 163, 112275. [Google Scholar] [CrossRef]
- Sun, S.; Deng, C.; Zhang, L.; Hu, K. Molecular analysis and biochemical characteristics of degenerated strains of Cordyceps militaris. Arch. Microbiol. 2017, 199, 939–944. [Google Scholar] [CrossRef]
- Cai, H.; Gan, X.; Jin, Z.; Hao, G. Carboxylic Acid Derivatives in Herbicide Development. J. Agric. Food Chem. 2023, 26, 9973–9993. [Google Scholar] [CrossRef]
- Sullivan, L.; Luengo, A.; Danai, L.; Bush, L.; Diehl, F.; Hosios, A.; Lau, A.; Elmiligy, S.; Malstrom, S.; Lewis, C.; et al. Aspartate is an endogenous metabolic limitation for tumour growth. Nat. Cell Biol. 2018, 20, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Brosnan, M.; Brosnan, J. Histidine metabolism and function. J. Nutr. 2020, 150 (Suppl. 1), 2570S–2575S. [Google Scholar] [CrossRef]
- Lambros, M.; Tran, T.; Fei, Q.; Nicolaou, M. Citric acid: A multifunctional pharmaceutical excipient. Pharmaceutics 2022, 14, 972. [Google Scholar] [CrossRef]
- Wang, X. Studies on Cordyceps militaris Degeneration and Regulation Mechanism of Cordycepin Metabolism. Master’s Thesis, Ludong University, Yantai, China, 2023. [Google Scholar]
- Xie, X.; Cheng, T.; Wang, G.; Duan, J.; Xia, Q. ABC and MATE Transporters of Plant and Their Roles in Membrane Trans-port of Secondary Metabolites. Plant Physiol. J. 2011, 47, 752–758. [Google Scholar]
- Li, X.; Hu, X.; Zhang, T.; Cheng, J.; Yu, Z.; Ren, F.; Shi, W. Study on the syntheticprocess of corgycepin. Chin. J. Med. Chem. 2023, 33, 656–659. [Google Scholar]
- Li, T.; Hou, Y.; Gou, W.; Shang, H.; Xu, F.; Li, Y.; Hou, W.; Zhou, F. Visual analysis of amino acid radiation protection research based on CiteSpace. Sci. Technol. Food Ind. 2024, 1–15. [Google Scholar] [CrossRef]
- Zhang, L.; Nie, H.; Wen, J.; Liao, H.; Ling, C.; Xu, F.; Dong, X. Multi-Omics Analysis Revealed Biological Differences of Pathogenic Fusarium Species of Panax notoginseng. J. Nucl. Agric. Sci. 2024, 38, 443–454. [Google Scholar]
- Lowe, R.; Shirley, N.; Bleackley, M.; Dolan, S.; Shafee, T. Transcriptomics technologies. PLoS Comput. Biol. 2017, 13, e1005457. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Liu, X.; Wu, Z.; Teng, N. Transcriptome and metabolome analyses provide insights into the Stomium degeneration mechanism in lily. Int. J. Mol. Sci. 2021, 22, 12124. [Google Scholar] [CrossRef]
- Choi, D.; Min, M.; Kim, M.; Lee, R.; Kwon, S.; Bae, S. Hrq1 facilitates nucleotide excision repair of DNA damage induced by 4-nitroquinoline-1-oxide and cisplatin in Saccharomyces cerevisiae. J. Microbiol. 2014, 52, 292–298. [Google Scholar] [CrossRef]
- Rogers, C.; Sanders, E.; Nguyen, P.; Smith-Kinnaman, W.; Mosley, A.; Bochman, M. The genetic and physical interactomes of the Saccharomyces cerevisiae Hrq1 helicase. G3 Genes Genomes Genet. 2020, 10, 4347–4357. [Google Scholar] [CrossRef] [PubMed]
- Sertic, S.; Quadri, R.; Lazzaro, F.; Muzi-Falconi, M. EXO1: A tightly regulated nuclease. DNA Repair 2020, 93, 102929. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Hu, B.; Jiang, M.; Deng, W.; Zheng, P.; Fu, B. Bioinformatic analysis of the expression and clinical significance of the DNA replication regulator MCM complex in bladder cancer. Int. J. Gen. Med. 2022, 15, 5465–5485. [Google Scholar] [CrossRef] [PubMed]
- Bochman, M.; Schwacha, A. The Mcm complex: Unwinding the mechanism of a replicative helicase. Microbiol. Mol. Biol. Rev. 2009, 73, 652–683. [Google Scholar] [CrossRef] [PubMed]
- Petry, S. Mechanisms of mitotic spindle assembly. Annu. Rev. Biochem. 2016, 85, 659–683. [Google Scholar] [CrossRef]
- Wada, A.; Sawada, Y.; Sugino, H.; Nakamura, M. Angioedema and Fatty Acids. Int. J. Mol. Sci. 2021, 22, 9000. [Google Scholar] [CrossRef] [PubMed]
- Falomir-Lockhart, L.; Cavazzutti, G.; Giménez, E.; Toscani, A. Fatty acid signaling mechanisms in neural cells: Fatty acid receptors. Front. Cell. Neurosci. 2019, 13, 162. [Google Scholar] [CrossRef] [PubMed]
- Pavoncello, V.; Barras, F.; Bouveret, E. Degradation of exogenous fatty acids in Escherichia coli. Biomolecules 2022, 12, 1019. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Wang, Y.; Yuan, M.; Huang, Z.; Zou, Q.; Pu, Y.; Zhao, B.; Cai, Z. Role of mismatch repair in aging. Int. J. Biol. Sci. 2021, 17, 3923. [Google Scholar] [CrossRef]
- Ten, S. The Role and Mechanism of Ixr1 of Saccharomyces cerevisiae in DNA Replication Stress. Ph.D. Thesis, Jilin University, Changchun, China, 2023. [Google Scholar]
- Alimova, I.; Ng, J.; Harris, P.; Birks, D.; Donson, A.; Taylor, M.; Foreman, N.; Venkataraman, S.; Vibhakar, R. MPS1 kinase as a potential therapeutic target in medulloblastoma. Oncol. Rep. 2016, 36, 2633–2640. [Google Scholar] [CrossRef]
- Lee, J.; Cheong, H.; Koh, Y.; Ahn, K.; Shin, H.; Yoon, S. MCM7 polymorphisms associated with the AML relapse and overall survival. Ann. Hematol. 2017, 96, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Zeng, T.; Guan, Y.; Li, Y.; Wu, Q.; Tang, X.; Zeng, X.; Ling, H.; Zou, J. The DNA replication regulator MCM6: An emerging cancer biomarker and target. Clin. Chim. Acta 2021, 517, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Pan, S.; Cui, J.; Bai, J.; Jiang, P.; He, Y. Functional analysis of the correlation between ABCB11 gene mutation and primary intrahepatic stone. Mol. Med. Rep. 2019, 19, 195–204. [Google Scholar] [CrossRef]
- Gordo-Gilart, R.; Andueza, S.; Hierro, L.; Jara, P.; Alvarez, L. Functional rescue of trafficking-impaired ABCB4 mutants by chemical chaperones. PLoS ONE 2016, 11, e0150098. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; Wang, J.; Yang, J.; Yang, G. Abnormal expression of ABCD3 is an independent prognostic factor for colorectal cancer. Oncol. Lett. 2020, 19, 3567–3577. [Google Scholar] [CrossRef]
- Tian, P.; Ning, J.; Zou, Q.; Lu, Y.; Guo, B.; Shi, M. Effect of 5-Aza-2′-deoxycytidine on the expression of Sfrp1 in NRK-52E cells cultured with high glucose. Basic Clin. Med. 2022, 42, 1194–1199. [Google Scholar]
- Ren, G.; Kim, J. 1786-P: ACSL1 Contributes to Thermogenesis and Differentiation of Brown Adipose Tissue. Diabetes 2019, 68 (Suppl. S1), 1786-P. [Google Scholar] [CrossRef]
- Li, T.; Li, X.; Meng, H.; Chen, L.; Meng, F. ACSL1 affects triglyceride levels through the PPARγ pathway. Int. J. Med. Sci. 2020, 17, 720. [Google Scholar] [CrossRef]
- Li, Y.; Yang, M.; Tan, J.; Shen, C.; Deng, S.; Fu, X.; Gao, S.; Li, H.; Zhang, X.; Cai, W. Targeting ACSL1 promotes cardiomyocyte proliferation and cardiac regeneration. Life Sci. 2022, 294, 120371. [Google Scholar] [CrossRef]
- Bowman, T.; O’Keeffe, K.; D’Aquila, T.; Yan, Q.; Griffin, J.; Killion, E.; Salter, D.; Mashek, D.; Buhman, K.; Greenberg, A. Acyl CoA synthetase 5 (ACSL5) ablation in mice increases energy expenditure and insulin sensitivity and delays fat absorption. Mol. Metab. 2016, 5, 210–220. [Google Scholar] [CrossRef]
- Reinartz, A.; Haynes, C.; Knuechel, R.; Merrill, A.; Gassler, N. Lipid-Induced Up-Regulation of Acyl-CoA Synthetase 5 Promotes Apoptosis in Human Hepatoctes. Biophys. J. 2010, 98, 466a. [Google Scholar] [CrossRef]
- Zhang, S.; Xiong, J.; Lou, W.; Ning, Z.; Zhang, D.; Yang, J. Antifungal effect of triglycerol monolaurate synthesized by Lipozyme 435-mediated esterification. J. Microbiol. Biotechnol. 2020, 30, 561. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.; Pearce, E. Amino assets: How amino acids support immunity. Cell Metab. 2020, 32, 154–175. [Google Scholar] [CrossRef]
- Dato, S.; Hoxha, E.; Crocco, P.; Iannone, F.; Passarino, G.; Rose, G. Amino acids and amino acid sensing: Implication for aging and diseases. Biogerontology 2019, 20, 17–31. [Google Scholar] [CrossRef]
- Li, J.; Yang, D.; Oldham, J.; Linderholm, A.; Zhang, J.; Liu, J.; Kenyon, N.; Chen, C. Therapeutic targeting of argininosuccinate synthase 1 (ASS1)-deficient pulmonary fibrosis. Mol. Ther. 2021, 29, 1487–1500. [Google Scholar] [CrossRef]
- Jerkins, A.; Steele, R. Dietary sulfur amino acid modulation of cysteine sulfinic acid decarboxylase. Am. J. Physiol.-Endocrinol. Metab. 1991, 261, E551–E555. [Google Scholar] [CrossRef]
- Huang, Z.; Xie, N.; Illes, P.; Virgilio, F.; Ulrich, H.; Semyanov, A.; Verkhratsky, A.; Sperlagh, B.; Yu, S.; Huang, C.; et al. From purines to purinergic signalling: Molecular functions and human diseases. Signal Transduct. Target. Ther. 2021, 6, 162. [Google Scholar] [CrossRef]
- Chitty, J.; Fraser, J. Purine acquisition and synthesis by human fungal pathogens. Microorganisms 2017, 5, 33. [Google Scholar] [CrossRef]
- Cai, F.; Xu, H.; Zha, D.; Wang, X.; Li, P.; Yu, S.; Yao, Y.; Chang, X.; Chen, J.; Lu, Y.; et al. AK2 promotes the migration and invasion of lung adenocarcinoma by activating TGF-β/smad pathway in vitro and in vivo. Front. Pharmacol. 2021, 12, 714365. [Google Scholar] [CrossRef]
- Lee, H.; Pyo, J.; Oh, Y.; Kim, H.; Hong, S.; Jeon, Y.; Kim, H.; Cho, D.; Woo, H.; Song, S.; et al. AK2 activates a novel apoptotic pathway through formation of a complex with FADD and caspase-10. Nat. Cell Biol. 2007, 9, 1303–1310. [Google Scholar] [CrossRef]
- Conte, F.; Ashikov, A.; Mijdam, R.; Ven, E.; Scherpenzeel, M.; Veizaj, R.; Mahalleh-Yousefi, S.; Post, M.; Huijben, K.; Panneman, D.; et al. In Vitro Skeletal Muscle Model of PGM1 Deficiency Reveals Altered Energy Homeostasis. Int. J. Mol. Sci. 2023, 24, 8247. [Google Scholar] [CrossRef] [PubMed]
- Nemmar, A.; Karaca, T.; Beegam, S.; Yuvaraju, P.; Yasin, J.; Ali, B. Lung oxidative stress, DNA damage, apoptosis, and fibrosis in adenine-induced chronic kidney disease in mice. Front. Physiol. 2017, 8, 286097. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z. Effects of Water Stress on the Growth and Development of Ophiocordyceps sinensis. Master’s Thesis, Lanzhou Jiaotong University, Lanzhou, China, 2022. [Google Scholar]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed]
- Thévenot, E.; Roux, A.; Xu, Y.; Ezan, E.; Junot, C. Analysis of the human adult urinary metabolome variations with age, body mass index, and gender by implementing a comprehensive workflow for univariate and OPLS statistical analyses. J. Proteome Res. 2015, 14, 3322–3335. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Raw Reads | Clean Reads | Raw Bases (bp) | Clean Bases (bp) | Error Rate (%) | Q20 (%) | Q30 (%) | GC Content (%) |
|---|---|---|---|---|---|---|---|---|
| F11_1 | 59,297,640 | 58,657,916 | 8,953,943,640 | 8,842,076,476 | 0.001886 | 99.01 | 97.02 | 56.32 |
| F11_2 | 61,246,102 | 60,561,152 | 9,248,161,402 | 9,122,703,173 | 0.001779 | 99.01 | 97.04 | 56.25 |
| F11_5 | 52,317,986 | 51,644,146 | 7,900,015,886 | 7,783,025,546 | 0.001787 | 98.94 | 96.85 | 56.44 |
| F1_2 | 70,055,490 | 69,094,666 | 10,578,378,990 | 10,412,699,146 | 0.007917 | 98.79 | 96.44 | 56.04 |
| F1_4 | 50,425,330 | 49,984,240 | 7,614,224,830 | 7,537,756,194 | 0.002278 | 99.15 | 97.37 | 55.80 |
| F1_6 | 52,412,658 | 51,744,950 | 7,914,311,358 | 7,796,427,041 | 0.002247 | 98.88 | 96.68 | 55.57 |
| F8_1 | 65,251,706 | 64,482,778 | 9,853,007,606 | 9,715,403,210 | 0.001802 | 98.95 | 96.89 | 56.04 |
| F8_4 | 67,137,558 | 66,349,958 | 10,137,771,258 | 10,001,417,170 | 0.001818 | 99.00 | 97.03 | 55.72 |
| F8_6 | 55,659,734 | 55,055,572 | 8,404,619,834 | 8,297,541,185 | 0.001751 | 99.03 | 97.07 | 56.17 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, H.; Cao, Z.; Wang, T.; Tang, C.; Li, Y.; Li, X. Metabolomics Combined with Physiology and Transcriptomics Reveal the Response of Samsoniella hepiali to Key Metabolic Pathways and Its Degradation Mechanism during Subculture. Antioxidants 2024, 13, 780. https://doi.org/10.3390/antiox13070780
He H, Cao Z, Wang T, Tang C, Li Y, Li X. Metabolomics Combined with Physiology and Transcriptomics Reveal the Response of Samsoniella hepiali to Key Metabolic Pathways and Its Degradation Mechanism during Subculture. Antioxidants. 2024; 13(7):780. https://doi.org/10.3390/antiox13070780
Chicago/Turabian StyleHe, Hui, Zhengfei Cao, Tao Wang, Chuyu Tang, Yuling Li, and Xiuzhang Li. 2024. "Metabolomics Combined with Physiology and Transcriptomics Reveal the Response of Samsoniella hepiali to Key Metabolic Pathways and Its Degradation Mechanism during Subculture" Antioxidants 13, no. 7: 780. https://doi.org/10.3390/antiox13070780
APA StyleHe, H., Cao, Z., Wang, T., Tang, C., Li, Y., & Li, X. (2024). Metabolomics Combined with Physiology and Transcriptomics Reveal the Response of Samsoniella hepiali to Key Metabolic Pathways and Its Degradation Mechanism during Subculture. Antioxidants, 13(7), 780. https://doi.org/10.3390/antiox13070780