



Nrf2 Deficiency Accelerates IL-17-Dependent Neutrophilic Airway Inflammation in Asthmatic Mice
 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Model
2.2. Experimental Protocols
2.3. Measurement of Airway Hyperresponsiveness (AHR)
2.4. Bronchoalveolar Lavage
2.5. Histopathology and Immunohistochemistry
2.6. Purification of Helper T (Th) Cells
2.7. RNA Isolation and Real-Time PCR
2.8. Multiplex Immunology
2.9. Western Blot Analysis
2.10. Statistical Analysis
3. Results
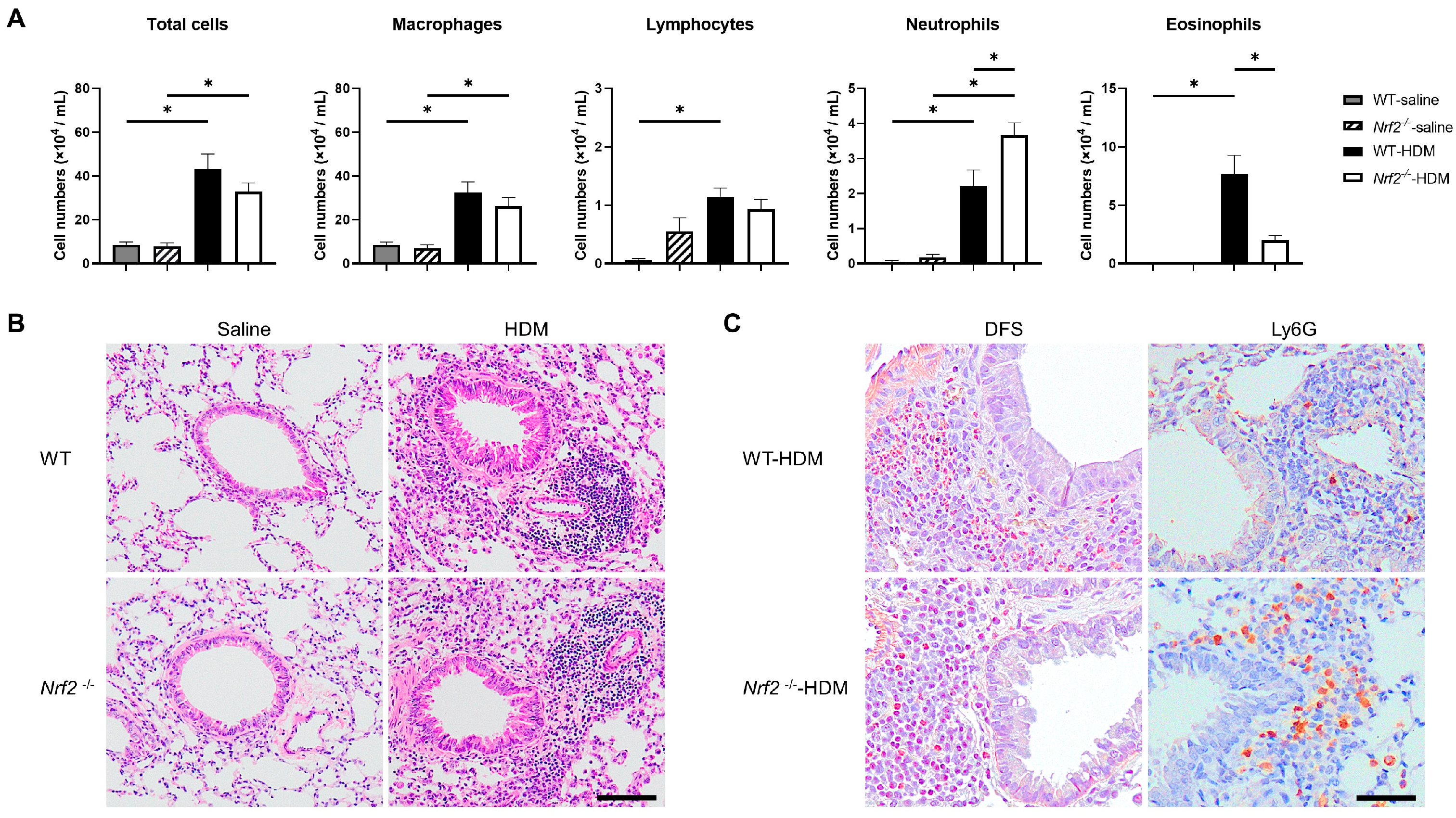
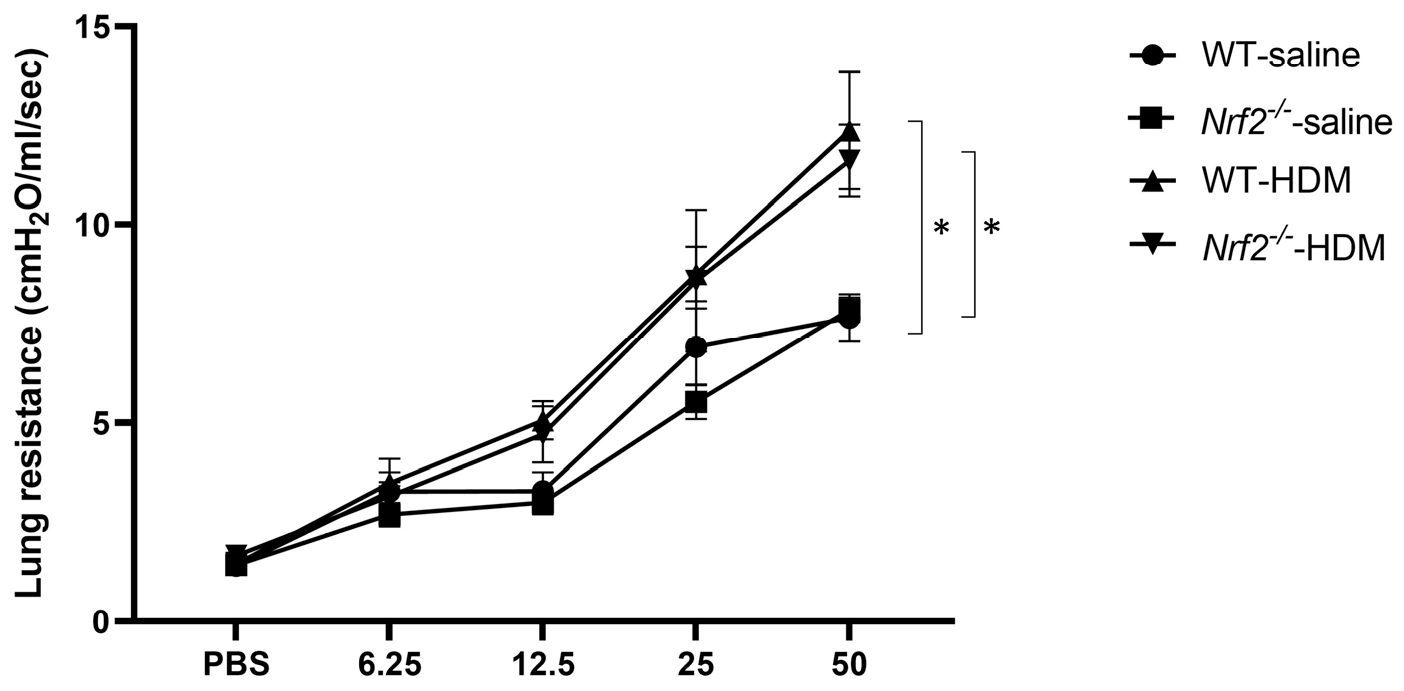
3.1. Nrf2 Deficiency Exacerbates Neutrophilic Airway Inflammation in a Mouse Model of Asthma
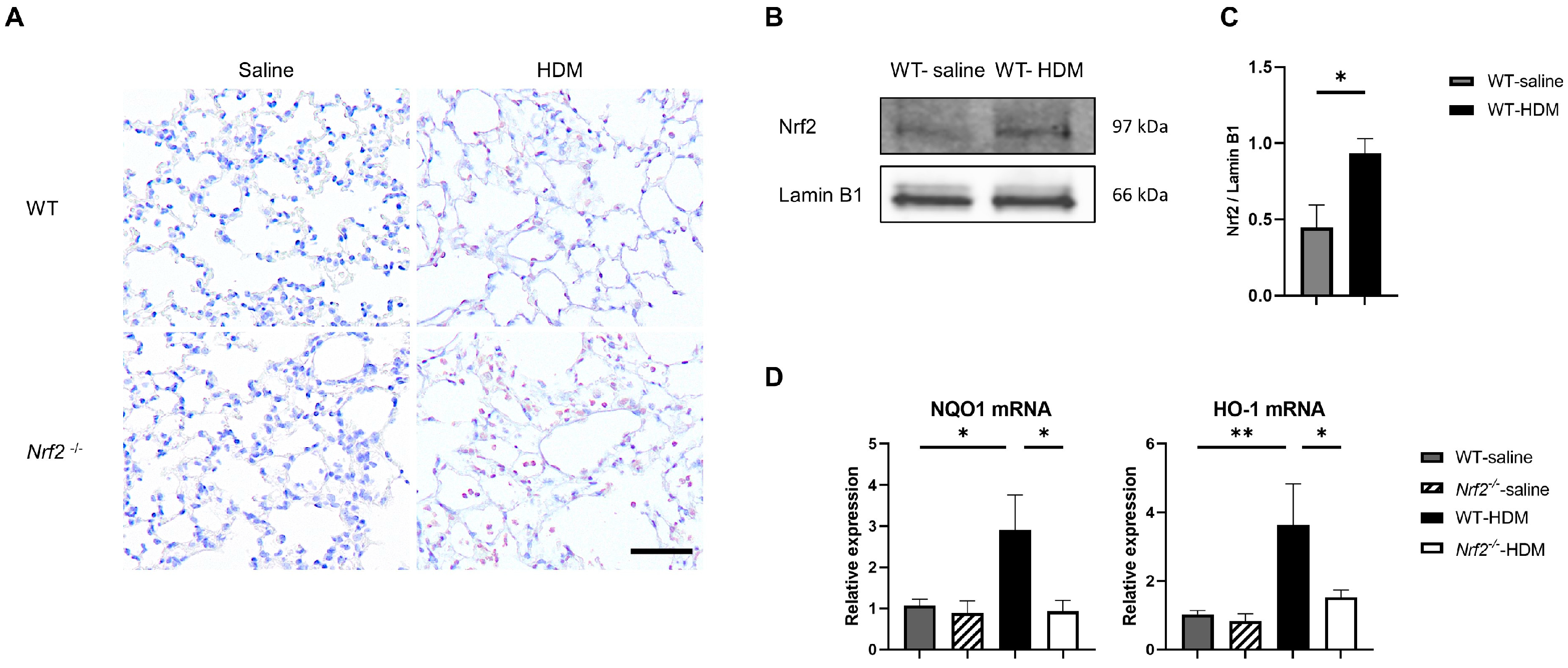
3.2. Oxidative Stress Is Exacerbated in an Nrf2-Deficient Mouse Model of Asthma
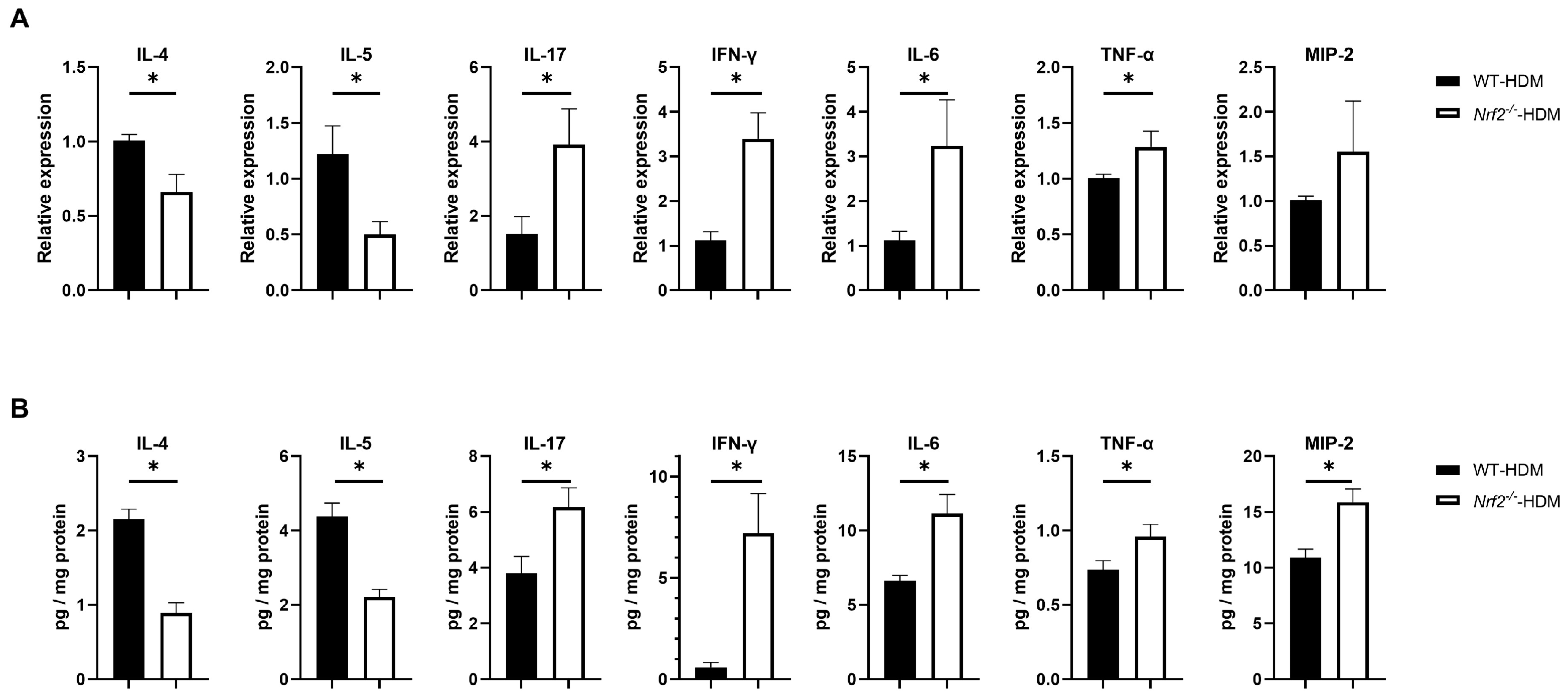
3.3. Nrf2 Deletion Alters the Expression of Inflammatory Cytokines and Chemokines
3.4. The RORγt Gene Is Overexpressed in Lung Th Cells in HDM-Stimulated Nrf2−/− Mice
3.5. Treatment with Anti-IL17 Antibody Improves Neutrophilic Airway Inflammation in HDM-Stimulated Nrf2−/− Mice
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hammad, H.; Lambrecht, B.N. The basic immunology of asthma. Cell 2021, 184, 1469–1485. [Google Scholar] [CrossRef]
- 2024 GINA Report, Global Strategy for Asthma Management and Prevention. Available online: https://ginasthma.org/reports/ (accessed on 16 May 2024).
- Heaney, L.G.; Perez de Llano, L.; Al-Ahmad, M.; Backer, V.; Busby, J.; Canonica, G.W.; Christoff, G.C.; Cosio, B.G.; FitzGerald, J.M.; Heffler, E.; et al. Eosinophilic and noneosinophilic asthma: An expert consensus framework to characterize phenotypes in a global real-life severe asthma cohort. Chest 2021, 160, 814–830. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Itoh, K.; Takahashi, S.; Sato, H.; Yanagawa, T.; Katoh, Y.; Bannai, S.; Yamamoto, M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J. Biol. Chem. 2000, 275, 16023–16029. [Google Scholar] [CrossRef] [PubMed]
- Ishii, Y.; Itoh, K.; Morishima, Y.; Kimura, T.; Kiwamoto, T.; IIzuka, T.; Hegab, A.E.; Hosoya, T.; Nomura, A.; Sakamoto, T. Transcription factor Nrf2 plays a pivotal role in protection against elastase-induced pulmonary inflammation and emphysema. J. Immunol. 2005, 175, 6968–6975. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, T.; Ishii, Y.; Itoh, K.; Kiwamoto, T.; Kimura, T.; Matsuno, Y.; Morishima, Y.; Hegab, A.E.; Homma, S.; Nomura, A. Nrf2-deficient mice are highly susceptible to cigarette smoke-induced emphysema. Genes Cells 2005, 10, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.Y.; Reddy, S.P.; Yamamoto, M.; Kleeberger, S.R. The transcription factor NRF2 protects against pulmonary fibrosis. FASEB J. 2004, 18, 1258–1260. [Google Scholar] [CrossRef]
- Kikuchi, N.; Ishii, Y.; Morishima, Y.; Yageta, Y.; Haraguchi, N.; Itoh, K.; Yamamoto, M.; Hizawa, N. Nrf2 protects against pulmonary fibrosis by regulating the lung oxidant level and Th1/Th2 balance. Respir. Res. 2010, 11, 31. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, M.; Ishii, Y.; Itoh, K.; Iizuka, T.; Morishima, Y.; Kimura, T.; Kiwamoto, T.; Matsuno, Y.; Hegab, A.E.; Nomura, A.; et al. Role of 15-deoxy delta(12,14) prostaglandin J2 and Nrf2 pathways in protection against acute lung injury. Am. J. Respir. Crit. Care Med. 2005, 171, 1260–1266. [Google Scholar] [CrossRef]
- Yageta, Y.; Ishii, Y.; Morishima, Y.; Masuko, H.; Ano, S.; Yamadori, T.; Itoh, K.; Takeuchi, K.; Yamamoto, M.; Hizawa, N. Role of Nrf2 in host defense against influenza virus in cigarette smoke-exposed mice. J. Virol. 2011, 85, 4679–4690. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, M.; Matsuyama, M.; Kawaguchi, M.; Kiwamoto, T.; Matsuno, Y.; Morishima, Y.; Yoshida, K.; Sherpa, M.; Yazaki, K.; Osawa, H.; et al. Nrf2 regulates granuloma formation and macrophage activation during mycobacterium avium infection via mediating Nramp1 and HO-1 expressions. mBio 2021, 12, e01947-20. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.; Kim, H.B.; Lee, S.Y.; Kim, B.S.; Kim, J.H.; Yu, J.; Kim, B.J.; Lee, D.H.; Seong, M.W.; Hong, S.J. Effect of active smoking on asthma symptoms, pulmonary function, and BHR in adolescents. Pediatr. Pulmonol. 2009, 44, 954–961. [Google Scholar] [CrossRef] [PubMed]
- Boulet, L.P.; Lemière, C.; Archambault, F.; Carrier, G.; Descary, M.C.; Deschesnes, F. Smoking and asthma: Clinical and radiologic features, lung function, and airway inflammation. Chest 2006, 129, 661–668. [Google Scholar] [CrossRef]
- Chalmers, G.W.; MacLeod, K.J.; Thomson, L.; Little, S.A.; McSharry, C.; Thomson, N.C. Smoking and airway inflammation in patients with mild asthma. Chest 2001, 120, 1917–1922. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, T.; Obase, Y.; Kishikawa, R.; Iwanaga, T. Influence of cigarette smoking on airway inflammation and inhaled corticosteroid treatment in patients with asthma. Allergy Asthma Proc. 2016, 37, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Min, M.G.; Song, D.J.; Miller, M.; Cho, J.Y.; McElwain, S.; Ferguson, P.; Broide, D.H. Coexposure to environmental tobacco smoke increases levels of allergen-induced airway remodeling in mice. J. Immunol. 2007, 178, 5321–5328. [Google Scholar] [CrossRef] [PubMed]
- Osborne, M.L.; Pedula, K.L.; O’Hollaren, M.; Ettinger, K.M.; Stibolt, T.; Buist, A.S.; Vollmer, W.M. Assessing future need for acute care in adult asthmatics: The Profile of Asthma Risk Study: A prospective health maintenance organization-based study. Chest 2007, 132, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Kauppi, P.; Kupiainen, H.; Lindqvist, A.; Haahtela, T.; Laitinen, T. Long-term smoking increases the need for acute care among asthma patients: A case control study. BMC Pulm. Med. 2014, 14, 119. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, J.E.; McMahon, A.D.; Chaudhuri, R.; Thompson, J.M.; Wood, S.F.; Thomson, N.C. Efficacy of low and high dose inhaled corticosteroid in smokers versus non-smokers with mild asthma. Thorax 2005, 60, 282–287. [Google Scholar] [CrossRef]
- Dijkstra, A.; Vonk, J.M.; Jongepier, H.; Koppelman, G.H.; Schouten, J.P.; ten Hacken, N. H: Timens, W: Postma, D.S. Lung function decline in asthma: Association with inhaled corticosteroids, smoking and sex. Thorax 2006, 61, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, H.; Morishima, Y.; Ishii, Y.; Yoshida, K.; Nakajima, M.; Tsunoda, Y.; Hayashi, S.Y.; Kiwamoto, T.; Matsuno, Y.; Kawaguchi, M.; et al. Sulforaphane ameliorates steroid insensitivity through an Nrf2-dependent pathway in cigarette smoke-exposed asthmatic mice. Free Radic. Biol. Med. 2018, 129, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Klein, D.K.; Silberbrandt, A.; Frøssing, L.; Hvidtfeldt, M.; von Bülow, A.; Nair, P.; Mukherjee, M.; Porsbjerg, C. Impact of former smoking exposure on airway eosinophilic activation and autoimmunity in patients with severe asthma. Eur. Respir. J. 2022, 60, 2102446. [Google Scholar] [CrossRef]
- Dong, C.; Yang, X.; Luo, W.; Fan, E.; Assam, N.P.; Kang, J.; Zhang, Y.; Huang, M.; Xu, J.; Huang, K.; et al. C–BIOPRED consortium. Influence of sex, cigarette smoking and airway inflammation on treatable traits in CBIOPRED severe asthma. Clin. Transl. Allergy 2022, 12, e12189. [Google Scholar] [CrossRef]
- Cowan, D.C.; Cowan, J.O.; Palmay, R.; Williamson, A.; Taylor, D.R. Effects of steroid therapy on inflammatory cell subtypes in asthma. Thorax 2010, 65, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Cox, G. Glucocorticoid treatment inhibits apoptosis in human neutrophils. Separation of survival and activation outcomes. J. Immunol. 1995, 154, 4719–4725. [Google Scholar] [CrossRef]
- Hyde, E.J.; Wakelin, K.A.; Daniels, N.J.; Ghosh, S.; Ronchese, F. Similar immune mechanisms control experimental airway eosinophilia elicited by different allergens and treatment protocols. BMC Immunol. 2019, 20, 18. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, T.; Guo, J.; Mitzner, W.A.; Roman, J.; Singh, A.; Fryer, A.D.; Yamamoto, M.; Kensler, T.W.; Tuder, R.M.; Georas, S.N.; et al. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J. Exp. Med. 2005, 202, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Abu Khweek, A.; Kim, E.; Joldrichsen, M.R.; Amer, A.O.; Boyaka, P.N. Insights into mucosal innate immune responses in house dust mite-mediated allergic asthma. Front Immunol. 2020, 11, 534501. [Google Scholar] [CrossRef]
- Zhao, S.; Jiang, Y.; Yang, X.; Guo, D.; Wang, Y.; Wang, J.; Wang, R.; Wang, C. Lipopolysaccharides promote a shift from Th2-derived airway eosinophilic inflammation to Th17-derived neutrophilic inflammation in an ovalbumin-sensitized murine asthma model. J. Asthma 2017, 54, 447–455. [Google Scholar] [CrossRef]
- Tan, H.T.; Hagner, S.; Ruchti, F.; Radzikowska, U.; Tan, G.; Altunbulakli, C.; Eljaszewicz, A.; Moniuszko, M.; Akdis, M.; Akdis, C.A.; et al. Tight junction, mucin, and inflammasome-related molecules are differentially expressed in eosinophilic, mixed, and neutrophilic experimental asthma in mice. Allergy 2019, 74, 294–307. [Google Scholar] [CrossRef] [PubMed]
- Nadeem, A.; Chhabra, S.K.; Masood, A.; Raj, H.G. Increased oxidative stress and altered levels of antioxidants in asthma. J. Allergy Clin. Immunol. 2003, 111, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Ammar, M.; Bahloul, N.; Amri, O.; Omri, R.; Ghozzi, H.; Kammoun, S.; Zeghal, K.; Ben Mahmoud, L. Oxidative stress in patients with asthma and its relation to uncontrolled asthma. J. Clin. Lab. Anal. 2022, 36, e24345. [Google Scholar] [CrossRef] [PubMed]
- Fernando, Y.; Wickramasinghe, P.; De Silva, U.; Alahakoon, M.; Anuradha, K.W.D.A.; Handunnetti, S. Differences in serum markers of oxidative stress in well controlled and poorly controlled asthma in Sri Lankan children: A pilot study. Allergy Asthma Clin. Immunol. 2020, 16, 66. [Google Scholar] [CrossRef] [PubMed]
- Jasemi, S.V.; Khazaei, H.; Fakhri, S.; Mohammadi-Noori, E.; Farzaei, M.H. Naringenin Improves Ovalbumin-Induced Allergic Asthma in Rats through Antioxidant and Anti-Inflammatory Effects. Evid. Based Complement. Alternat. Med. 2022, 2022, 9110798. [Google Scholar] [CrossRef] [PubMed]
- Sussan, T.E.; Gajghate, S.; Chatterjee, S.; Mandke, P.; McCormick, S.; Sudini, K.; Kumar, S.; Breysse, P.N.; Diette, G.B.; Sidhaye, V.K.; et al. Nrf2 reduces allergic asthma in mice through enhanced airway epithelial cytoprotective function. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L27–L36. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Li, W.; Feng, Y.; Xu, J.; Cao, H. Edaravone attenuates experimental asthma in mice through induction of HO-1 and the Keap1/Nrf2 pathway. Exp. Ther. Med. 2020, 19, 1407–1416. [Google Scholar] [CrossRef]
- Zhang, J.H.; Yang, X.; Chen, Y.P.; Zhang, J.F.; Li, C.Q. Nrf2 Activator RTA-408 protects against ozone-induced acute asthma exacerbation by suppressing ROS and gammadeltaT17 cells. Inflammation 2019, 42, 1843–1856. [Google Scholar] [CrossRef]
- Hellings, P.W.; Kasran, A.; Liu, Z.; Vandekerckhove, P.; Wuyts, A.; Overbergh, L.; Mathieu, C.; Ceuppens, J.L. Interleukin-17 orchestrates the granulocyte influx into airways after allergen inhalation in a mouse model of allergic asthma. Am. J. Respir. Cell Mol. Biol. 2003, 28, 42–50. [Google Scholar] [CrossRef]
- Xie, Y.; Abel, P.W.; Casale, T.B.; Tu, Y. TH17 cells and corticosteroid insensitivity in severe asthma. J. Allergy Clin. Immunol. 2022, 149, 467–479. [Google Scholar] [CrossRef]
- McKinley, L.; Alcorn, J.F.; Peterson, A.; Dupont, R.B.; Kapadia, S.; Logar, A.; Henry, A.; Irvin, C.G.; Piganelli, J.D.; Ray, A.; et al. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J. Immunol. 2008, 181, 4089–4097. [Google Scholar] [CrossRef] [PubMed]
- Ano, S.; Morishima, Y.; Ishii, Y.; Yoh, K.; Yageta, Y.; Ohtsuka, S.; Matsuyama, M.; Kawaguchi, M.; Takahashi, S.; Hizawa, N. Transcription factors GATA-3 and RORγt are important for determining the phenotype of allergic airway inflammation in a murine model of asthma. J. Immunol. 2013, 190, 1056–1065. [Google Scholar] [CrossRef] [PubMed]
- Al-Harbi, N.O.; Nadeem, A.; Ahmad, S.F.; AlThagfan, S.S.; Alqinyah, M.; Alqahtani, F.; Ibrahim, K.E.; Al-Harbi, M.M. Sulforaphane treatment reverses corticosteroid resistance in a mixed granulocytic mouse model of asthma by upregulation of antioxidants and attenuation of Th17 immune responses in the airways. Eur. J. Pharmacol. 2019, 855, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Chesné, J.; Braza, F.; Mahay, G.; Brouard, S.; Aronica, M.; Magnan, A. IL-17 in severe asthma. Where do we stand? Am. J. Respir. Crit. Care Med. 2014, 190, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.H.G. IL-17 and IL-17-producing cells in protection versus pathology. Nat. Rev. Immunol. 2023, 23, 38–54. [Google Scholar] [CrossRef]
- Ivanov, I.I.; McKenzie, B.S.; Zhou, L.; Tadokoro, C.E.; Lepelley, A.; Lafaille, J.J.; Cua, D.J.; Littman, D.R. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 2006, 126, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Weng, C.M.; Lee, M.J.; He, J.R.; Chao, M.W.; Wang, C.H.; Kuo, H.P. Diesel exhaust particles up-regulate interleukin-17A expression via ROS/NF-κB in airway epithelium. Biochem. Pharmacol. 2018, 151, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Su, B.; Tao, P.; Yang, X.; Zheng, L.; Lin, Y.; Zou, X.; Yang, H.; Wu, W.; Zhang, T.; et al. Interplay of IL-33 and IL-35 modulates Th2/Th17 responses in cigarette smoke exposure HDM-induced asthma. Inflammation 2024, 47, 173–190. [Google Scholar] [CrossRef] [PubMed]
- Brandt, E.B.; Kovacic, M.B.; Lee, G.B.; Gibson, A.M.; Acciani, T.H.; Le Cras, T.D.; Ryan, P.H.; Budelsky, A.L.; Khurana Hershey, G.K. Diesel exhaust particle induction of IL-17A contributes to severe asthma. J. Allergy Clin. Immunol. 2013, 132, 1194–1204.e2. [Google Scholar] [CrossRef]
- Liang, Y.; Shen, Y.; Kuang, L.; Zhou, G.; Zhang, L.; Zhong, X.; Zhang, J.; Liu, J. Cigarette smoke exposure promotes differentiation of CD4+ T cells toward Th17 cells by CD40-CD40L costimulatory pathway in mice. Int. J. Chron. Obstruct. Pulmon. Dis. 2018, 13, 959–968. [Google Scholar] [CrossRef]
- McGovern, T.K.; Goldberger, M.; Allard, B.; Farahnak, S.; Hamamoto, Y.; O’Sullivan, M.; Hirota, N.; Martel, G.; Rousseau, S.; Martin, J.G. Neutrophils mediate airway hyperresponsiveness after chlorine-induced airway injury in the mouse. Am. J. Respir. Cell Mol. Biol. 2015, 52, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Sun, L.; Jiang, T.; Zhang, D.; He, D.; Nie, H. TNFα promotes Th17 cell differentiation through IL-6 and IL-1β produced by monocytes in rheumatoid arthritis. J. Immunol. Res. 2014, 2014, 85352. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Ghilardi, N.; Xie, M.H.; de Sauvage, F.J.; Gurney, A.L. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J. Biol. Chem. 2003, 278, 1910–1914. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.A.; Rangasamy, T.; Bauer, S.M.; Killedar, S.; Karp, M.; Kensler, T.W.; Yamamoto, M.; Breysse, P.; Biswal, S.; Georas, S.N. Disruption of the transcription factor Nrf2 promotes pro-oxidative dendritic cells that stimulate Th2-like immunoresponsiveness upon activation by ambient particulate matter. J. Immunol. 2008, 181, 4545–4559. [Google Scholar] [CrossRef] [PubMed]
- Rockwell, C.E.; Zhang, M.; Fields, P.E.; Klaassen, C.D. Th2 skewing by activation of Nrf2 in CD4+ T cells. J. Immunol. 2012, 188, 1630–1637. [Google Scholar] [CrossRef] [PubMed]
- Hwang, E.S.; Szabo, S.J.; Schwartzberg, P.L.; Glimcher, L.H. T helper cell fate specified by kinase-mediated interaction of T-bet with GATA-3. Science 2005, 307, 430–433. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, M.; Hirose, K.; Kagami, S.; Takatori, H.; Wakashin, H.; Tamachi, T.; Watanabe, N.; Saito, Y.; Iwamoto, I.; Nakajima, H. T-bet inhibits both TH2 cell-mediated eosinophil recruitment and TH17 cell-mediated neutrophil recruitment into the airways. J. Allergy Clin. Immunol. 2007, 119, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Busse, W.W.; Holgate, S.; Kerwin, E.; Chon, Y.; Feng, J.; Lin, J.; Lin, S.L. Randomized, double-blind, placebo-controlled study of brodalumab, a human anti-IL-17 receptor monoclonal antibody, in moderate to severe asthma. Am. J. Respir. Crit. Care Med. 2013, 188, 1294–1302. [Google Scholar] [CrossRef]
- Thiruvengadam, M.; Venkidasamy, B.; Subramanian, U.; Samynathan, R.; Ali Shariati, M.; Rebezov, M.; Girish, S.; Thangavel, S.; Dhanapal, A.R.; Fedoseeva, N.; et al. Bioactive Compounds in Oxidative Stress-Mediated Diseases: Targeting the NRF2/ARE Signaling Pathway and Epigenetic Regulation. Antioxidants 2021, 10, 1859. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuramoto, K.; Morishima, Y.; Yoshida, K.; Ano, S.; Kawashima, K.; Yabuuchi, Y.; Sakai, C.; Matsumura, S.; Nishino, K.; Yazaki, K.; et al. Nrf2 Deficiency Accelerates IL-17-Dependent Neutrophilic Airway Inflammation in Asthmatic Mice. Antioxidants 2024, 13, 818. https://doi.org/10.3390/antiox13070818
Kuramoto K, Morishima Y, Yoshida K, Ano S, Kawashima K, Yabuuchi Y, Sakai C, Matsumura S, Nishino K, Yazaki K, et al. Nrf2 Deficiency Accelerates IL-17-Dependent Neutrophilic Airway Inflammation in Asthmatic Mice. Antioxidants. 2024; 13(7):818. https://doi.org/10.3390/antiox13070818
Chicago/Turabian StyleKuramoto, Kenya, Yuko Morishima, Kazufumi Yoshida, Satoshi Ano, Kai Kawashima, Yuki Yabuuchi, Chio Sakai, Sosuke Matsumura, Kengo Nishino, Kai Yazaki, and et al. 2024. "Nrf2 Deficiency Accelerates IL-17-Dependent Neutrophilic Airway Inflammation in Asthmatic Mice" Antioxidants 13, no. 7: 818. https://doi.org/10.3390/antiox13070818
APA StyleKuramoto, K., Morishima, Y., Yoshida, K., Ano, S., Kawashima, K., Yabuuchi, Y., Sakai, C., Matsumura, S., Nishino, K., Yazaki, K., Matsuyama, M., Kiwamoto, T., Ishii, Y., & Hizawa, N. (2024). Nrf2 Deficiency Accelerates IL-17-Dependent Neutrophilic Airway Inflammation in Asthmatic Mice. Antioxidants, 13(7), 818. https://doi.org/10.3390/antiox13070818