

N-acetyl Cysteine Overdose Induced Acute Toxicity and Hepatic Microvesicular Steatosis by Disrupting GSH and Interfering Lipid Metabolisms in Normal Mice

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Animals and Experimental Designs
2.3. Serum Biochemical Analyses
2.4. H&E Staining of Multiple Tissue Sections
2.5. Measurements of Hepatic TG and FFA
2.6. Measurements of Hepatic GSH and CAT Levels
2.7. Measurements of Hepatic MDA and ATP Levels
2.8. Measurements of Hepatic mRNA Levels of Lipid Metabolism Genes by Real-Time PCR
2.9. Western Blotting
2.10. Statistical Analysis
3. Results
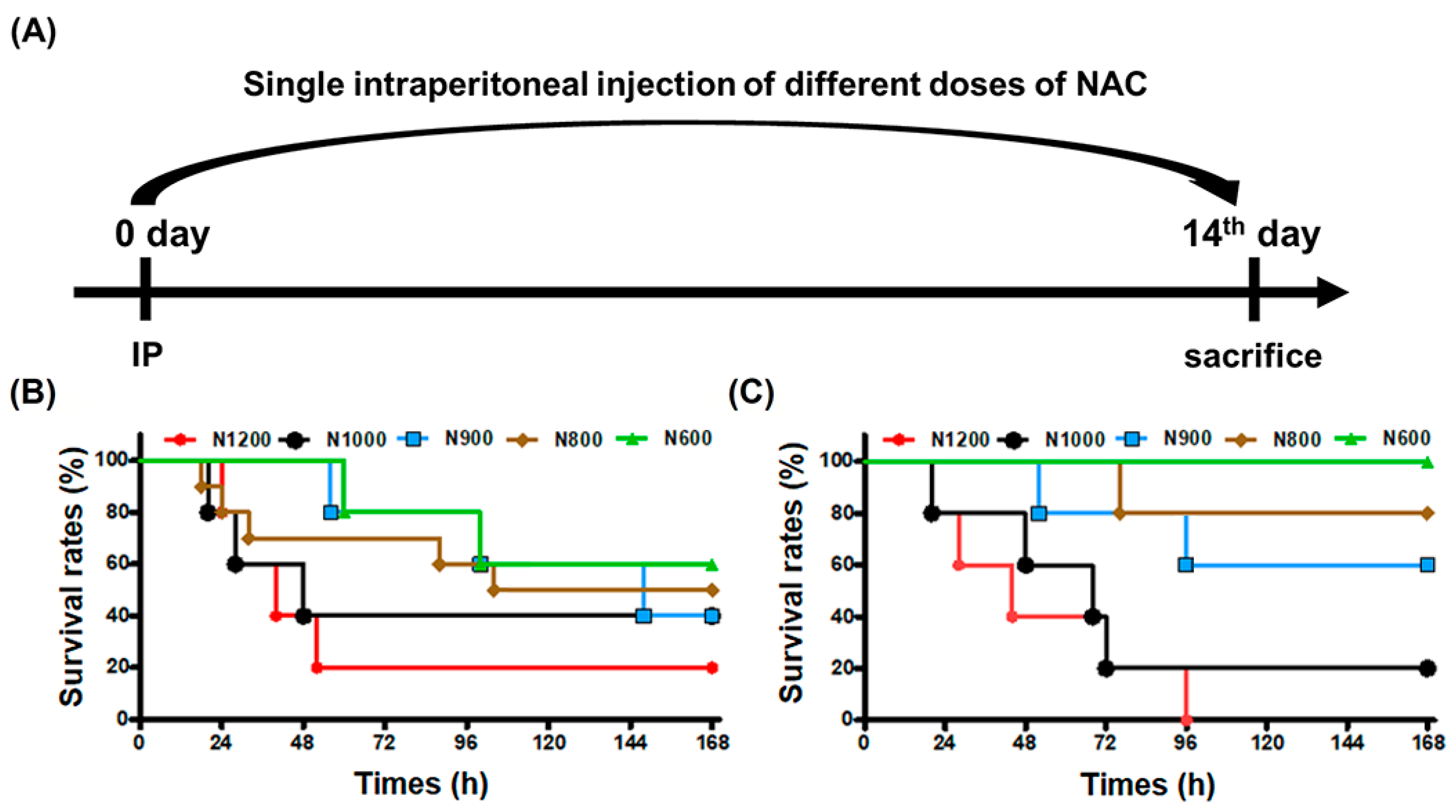
3.1. LD50 of Male and Female Mice following IP of NAC
3.2. Histopathological Findings and Serum Biochemical Values of Surviving Male and Female Mice after High-Dose NAC Treatment
3.3. Toxic Mechanisms of NAC Determined by Hepatic and Renal Biochemical Indices in Sera
3.4. Toxic Mechanisms of NAC Determined by Major Organ Analyses
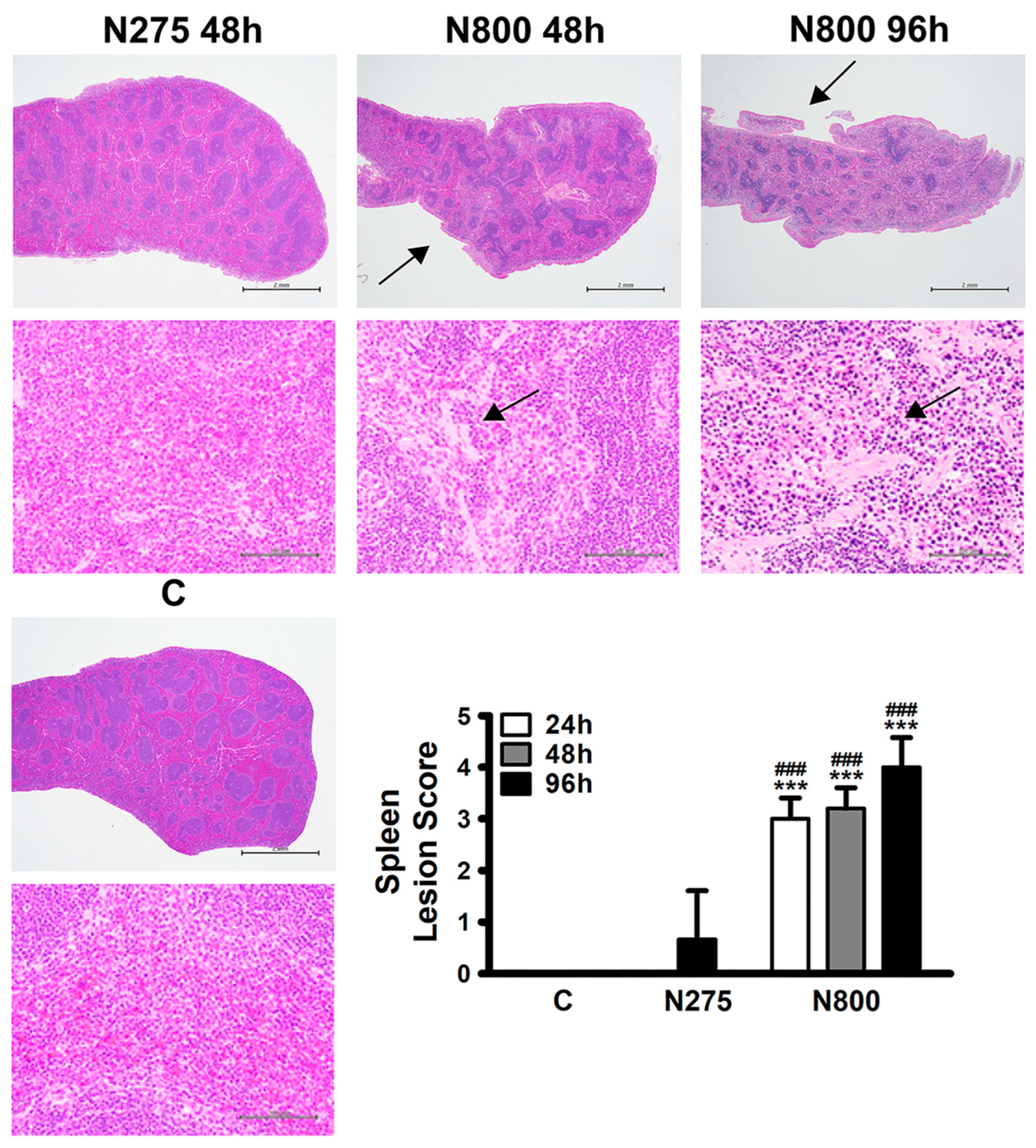
3.5. Toxic Mechanisms of NAC Determined by Hepatic, Renal, and Splenic Histopathology
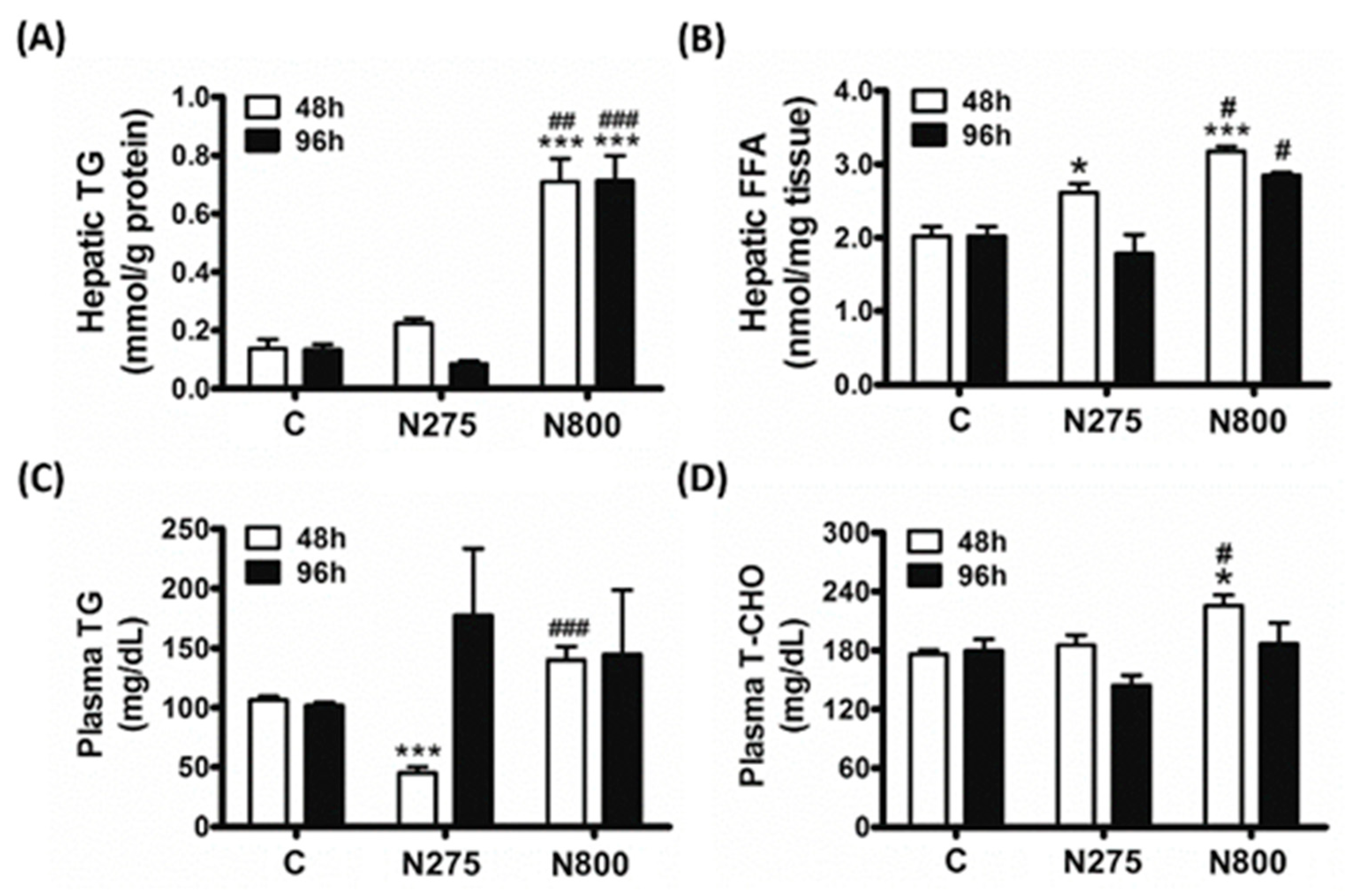
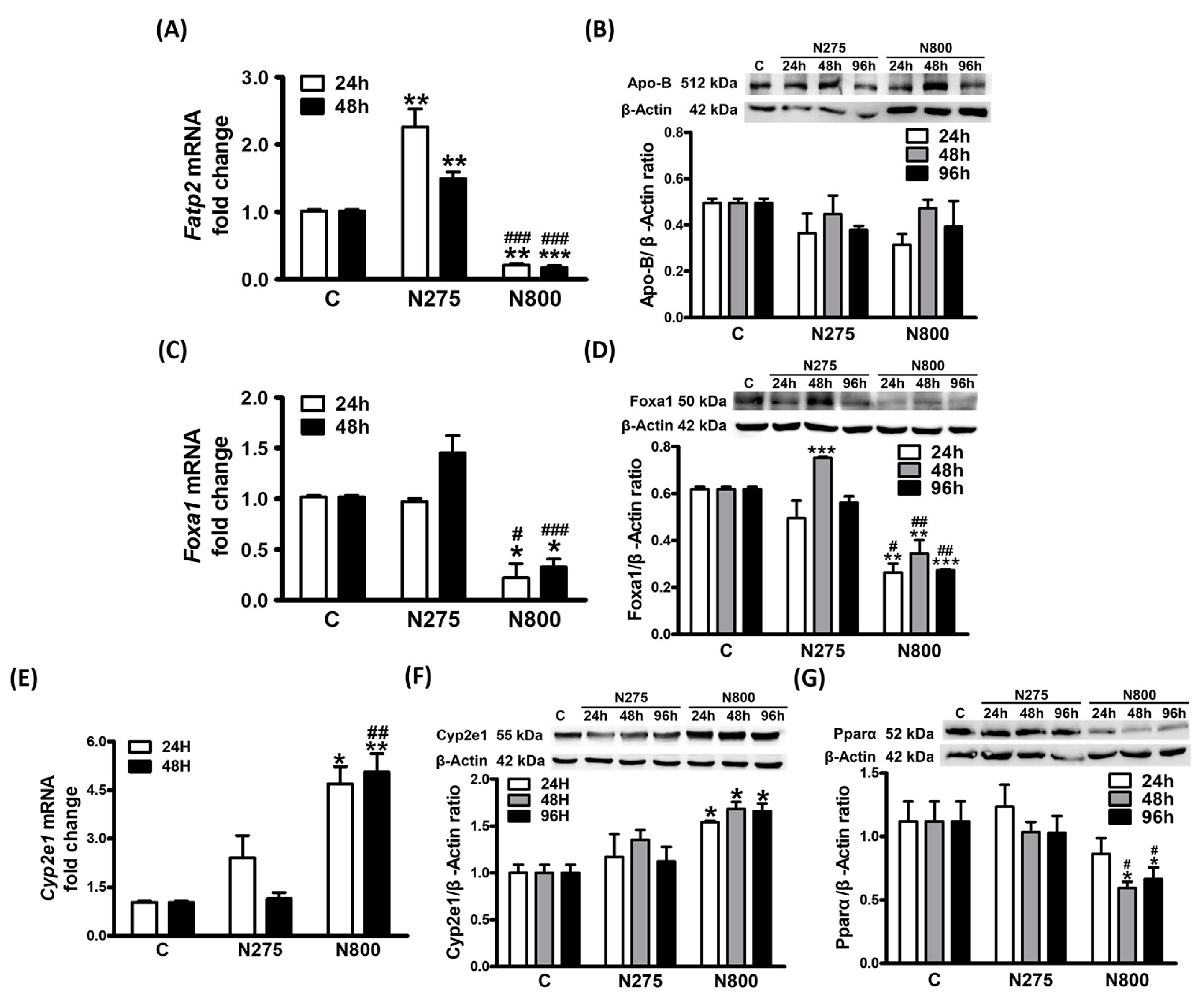
3.6. Effects of NAC on Lipid Metabolism
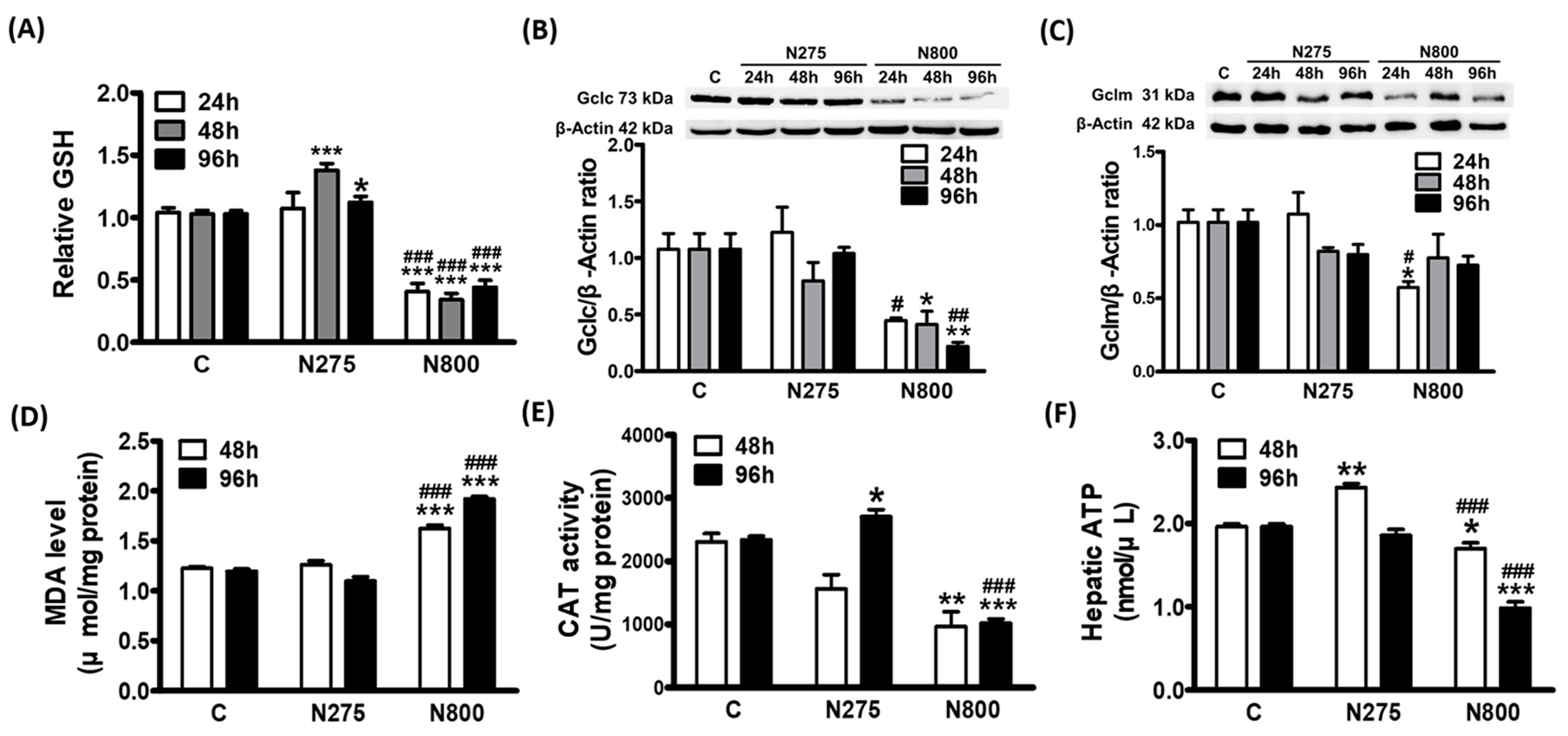
3.7. Effects of NAC on Hepatic GSH and ATP Syntheses
3.8. Effects of NAC on Hepatic Lipid Metabolism
3.9. Effects of NAC on NAFL-Related Gene Expressions: Foxa1, Cyp2e1, and Pparα
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jaeschke, H. Acetaminophen Hepatotoxicity: Not as Simple as One Might Think! Introductory Comments on the Special Issue-Recent Advances in Acetaminophen Hepatotoxicity. Livers 2022, 2, 105–107. [Google Scholar] [CrossRef] [PubMed]
- Dludla, P.V.; Nkambule, B.B.; Mazibuko-Mbeje, S.E.; Nyambuya, T.M.; Marcheggiani, F.; Cirilli, I.; Ziqubu, K.; Shabalala, S.C.; Johnson, R.; Louw, J.; et al. N-Acetyl Cysteine Targets Hepatic Lipid Accumulation to Curb Oxidative Stress and Inflammation in NAFLD: A Comprehensive Analysis of the Literature. Antioxidants 2020, 9, 1283. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.P.; Cotrim, H.P.; Stefano, J.T.; Siqueira, A.C.G.; Salgado, A.L.A.; Parise, E.R. N-Acetylcysteine and/or Ursodeoxycholic Acid Associated with Metformin in Non-Alcoholic Steatohepatitis: An Open-Label Multicenter Randomized Controlled Trial. Arq. Gastroenterol. 2019, 56, 184–190. [Google Scholar] [CrossRef] [PubMed]
- El-Lakkany, N.M.; Seif El-Din, S.H.; Sabra, A.A.; Hammam, O.A.; Ebeid, F.A. Co-administration of metformin and N-acetylcysteine with dietary control improves the biochemical and histological manifestations in rats with non-alcoholic fatty liver. Res. Pharm. Sci. 2016, 11, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, H.; Perl, A.; Smith, D.; Lewis, T.; Kon, Z.; Goldenberg, R.; Yarta, K.; Staniloae, C.; Williams, M. Therapeutic blockade of inflammation in severe COVID-19 infection with intravenous N-acetylcysteine. Clin. Immunol. 2020, 219, 108544. [Google Scholar] [CrossRef] [PubMed]
- Atkuri, K.R.; Mantovani, J.J.; Herzenberg, L.A.; Herzenberg, L.A. N-Acetylcysteine--a safe antidote for cysteine/glutathione deficiency. Curr. Opin. Pharmacol. 2007, 7, 355–359. [Google Scholar] [CrossRef]
- Nakhaee, S.; Dastjerdi, M.; Roumi, H.; Mehrpour, O.; Farrokhfall, K. N-acetylcysteine dose-dependently improves the analgesic effect of acetaminophen on the rat hot plate test. BMC Pharmacol. Toxicol. 2021, 22, 4. [Google Scholar] [CrossRef] [PubMed]
- Tardiolo, G.; Bramanti, P.; Mazzon, E. Overview on the Effects of N-Acetylcysteine in Neurodegenerative Diseases. Molecules 2018, 23, 3305. [Google Scholar] [CrossRef] [PubMed]
- Ooi, S.L.; Green, R.; Pak, S.C. N-Acetylcysteine for the Treatment of Psychiatric Disorders: A Review of Current Evidence. BioMed Res. Int. 2018, 2018, 2469486. [Google Scholar] [CrossRef]
- Elsayed, A.; Elkomy, A.; Elkammar, R.; Youssef, G.; Abdelhiee, E.Y.; Abdo, W.; Fadl, S.E.; Soliman, A.; Aboubakr, M. Synergistic protective effects of lycopene and N-acetylcysteine against cisplatin-induced hepatorenal toxicity in rats. Sci. Rep. 2021, 11, 13979. [Google Scholar] [CrossRef]
- Tenório, M.; Graciliano, N.G.; Moura, F.A.; Oliveira, A.C.M.; Goulart, M.O.F. N-Acetylcysteine (NAC): Impacts on Human Health. Antioxidants 2021, 10, 967. [Google Scholar] [CrossRef]
- Wu, G.; Fang, Y.Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione metabolism and its implications for health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [CrossRef]
- Licata, A.; Minissale, M.G.; Stankevičiūtė, S.; Sanabria-Cabrera, J.; Lucena, M.I.; Andrade, R.J.; Almasio, P.L. N-Acetylcysteine for Preventing Acetaminophen-Induced Liver Injury: A Comprehensive Review. Front. Pharmacol. 2022, 13, 828565. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Shi, J.; Hu, Z.; Hang, C. Inhibitory effect on cerebral inflammatory response following traumatic brain injury in rats: A potential neuroprotective mechanism of N-acetylcysteine. Mediat. Inflamm. 2008, 2008, 716458. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, M.; He, X.; Zhang, B.; Peng, W.; Guo, L. N-acetyl cysteine inhibits the lipopolysaccharide-induced inflammatory response in bone marrow mesenchymal stem cells by suppressing the TXNIP/NLRP3/IL-1β signaling pathway. Mol. Med. Rep. 2020, 22, 3299–3306. [Google Scholar] [CrossRef] [PubMed]
- Szakmany, T.; Hauser, B.; Radermacher, P. N-acetylcysteine for sepsis and systemic inflammatory response in adults. Cochrane Database Syst. Rev. 2012, 2012, Cd006616. [Google Scholar] [CrossRef]
- Walayat, S.; Shoaib, H.; Asghar, M.; Kim, M.; Dhillon, S. Role of N-acetylcysteine in non-acetaminophen-related acute liver failure: An updated meta-analysis and systematic review. Ann. Gastroenterol. 2021, 34, 235–240. [Google Scholar] [CrossRef]
- Pei, Y.; Liu, H.; Yang, Y.; Yang, Y.; Jiao, Y.; Tay, F.R.; Chen, J. Biological Activities and Potential Oral Applications of N-Acetylcysteine: Progress and Prospects. Oxid. Med. Cell. Longev. 2018, 2018, 2835787. [Google Scholar] [CrossRef] [PubMed]
- Bavarsad Shahripour, R.; Harrigan, M.R.; Alexandrov, A.V. N-acetylcysteine (NAC) in neurological disorders: Mechanisms of action and therapeutic opportunities. Brain Behav. 2014, 4, 108–122. [Google Scholar] [CrossRef]
- Bonanomi, L.; Gazzaniga, A. Toxicological, pharmacokinetic and metabolic studies on acetylcysteine. Eur. J. Respir. Dis. Suppl. 1980, 111, 45–51. [Google Scholar]
- Lewis, R.J., Sr. Sax’s Dangerous Properties of Industrial Materials, 11th ed.; Wiley-Interscience, John Wiley & Sons: Hoboken, NJ, USA, 2004. [Google Scholar]
- Liou, G.G.; Hsieh, C.C.; Lee, Y.J.; Li, P.H.; Tsai, M.S.; Li, C.T.; Wang, S.H. N-Acetyl Cysteine Overdose Inducing Hepatic Steatosis and Systemic Inflammation in Both Propacetamol-Induced Hepatotoxic and Normal Mice. Antioxidants 2021, 10, 442. [Google Scholar] [CrossRef] [PubMed]
- Spence, E.E.M.; Shwetz, S.; Ryan, L.; Anton, N.; Joffe, A.R. Non-Intentional N-Acetylcysteine Overdose Associated with Cerebral Edema and Brain Death. Case Rep. Gastroenterol. 2023, 17, 96–103. [Google Scholar] [CrossRef]
- Al Shoyaib, A.; Archie, S.R.; Karamyan, V.T. Intraperitoneal Route of Drug Administration: Should it Be Used in Experimental Animal Studies? Pharm. Res. 2019, 37, 12. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, J.H.; Baron, D.N.; Moss, D.W.; Walker, P.G. Standardization of clinical enzyme assays: A reference method for aspartate and alanine transaminases. J. Clin. Pathol. 1972, 25, 940–944. [Google Scholar] [CrossRef]
- Rahman, I.; Kode, A.; Biswas, S.K. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat. Protoc. 2006, 1, 3159–3165. [Google Scholar] [CrossRef]
- Tsai, M.S.; Chien, C.C.; Lin, T.H.; Liu, C.C.; Liu, R.H.; Su, H.L.; Chiu, Y.T.; Wang, S.H. Galangin Prevents Acute Hepatorenal Toxicity in Novel Propacetamol-Induced Acetaminophen-Overdosed Mice. J. Med. Food 2015, 18, 1187–1197. [Google Scholar] [CrossRef]
- Janero, D.R. Malondialdehyde and thiobarbituric acid-reactivity as diagnostic indices of lipid peroxidation and peroxidative tissue injury. Free Radic. Biol. Med. 1990, 9, 515–540. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.S.; Wang, Y.H.; Lai, Y.Y.; Tsou, H.K.; Liou, G.G.; Ko, J.L.; Wang, S.H. Kaempferol protects against propacetamol-induced acute liver injury through CYP2E1 inactivation, UGT1A1 activation, and attenuation of oxidative stress, inflammation and apoptosis in mice. Toxicol. Lett. 2018, 290, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Yin, H.; Liu, M.; Xu, G.; Zhou, X.; Ge, P.; Yang, H.; Mao, Y. Impaired albumin function: A novel potential indicator for liver function damage? Ann. Med. 2019, 51, 333–344. [Google Scholar] [CrossRef]
- Cheng, T.; Wang, X.; Han, Y.; Hao, J.; Hu, H.; Hao, L. The level of serum albumin is associated with renal prognosis and renal function decline in patients with chronic kidney disease. BMC Nephrol. 2023, 24, 57. [Google Scholar] [CrossRef]
- Lyngdoh, T.; Marques-Vidal, P.; Paccaud, F.; Preisig, M.; Waeber, G.; Bochud, M.; Vollenweider, P. Elevated serum uric acid is associated with high circulating inflammatory cytokines in the population-based Colaus study. PLoS ONE 2011, 6, e19901. [Google Scholar] [CrossRef]
- Li, N.; Yang, X.; Wu, J.; Wang, Y.; Wang, Z.; Mu, H. Correlation between the increase in serum uric acid and the rapid decline in kidney function in adults with normal kidney function: A retrospective study in Urumqi, China. BMC Nephrol. 2023, 24, 103. [Google Scholar] [CrossRef]
- Moe, S.M. Disorders involving calcium, phosphorus, and magnesium. Prim. Care 2008, 35, 215–237, v–vi. [Google Scholar] [CrossRef]
- Benet, M.; Moya, M.; Donato, M.T.; Lahoz, A.; Hervás, D.; Guzmán, C.; Gómez-Lechón, M.J.; Castell, J.V.; Jover, R. A simple transcriptomic signature able to predict drug-induced hepatic steatosis. Arch. Toxicol. 2014, 88, 967–982. [Google Scholar] [CrossRef]
- Todisco, S.; Santarsiero, A.; Convertini, P.; De Stefano, G.; Gilio, M.; Iacobazzi, V.; Infantino, V. PPAR Alpha as a Metabolic Modulator of the Liver: Role in the Pathogenesis of Nonalcoholic Steatohepatitis (NASH). Biology 2022, 11, 792. [Google Scholar] [CrossRef]
- Aubert, J.; Begriche, K.; Knockaert, L.; Robin, M.A.; Fromenty, B. Increased expression of cytochrome P450 2E1 in nonalcoholic fatty liver disease: Mechanisms and pathophysiological role. Clin. Res. Hepatol. Gastroenterol. 2011, 35, 630–637. [Google Scholar] [CrossRef]
- Morris, M.E.; Lee, H.J.; Predko, L.M. Gender differences in the membrane transport of endogenous and exogenous compounds. Pharmacol. Rev. 2003, 55, 229–240. [Google Scholar] [CrossRef]
- Tanaka, E.; Hisawa, S. Clinically significant pharmacokinetic drug interactions with psychoactive drugs: Antidepressants and antipsychotics and the cytochrome P450 system. J. Clin. Pharm. Ther. 1999, 24, 7–16. [Google Scholar] [CrossRef]
- Chan, K.; Han, X.D.; Kan, Y.W. An important function of Nrf2 in combating oxidative stress: Detoxification of acetaminophen. Proc. Natl. Acad. Sci. USA 2001, 98, 4611–4616. [Google Scholar] [CrossRef]
- Dai, G.; He, L.; Chou, N.; Wan, Y.J. Acetaminophen metabolism does not contribute to gender difference in its hepatotoxicity in mouse. Toxicol. Sci. 2006, 92, 33–41. [Google Scholar] [CrossRef]
- Du, K.; Williams, C.D.; McGill, M.R.; Jaeschke, H. Lower susceptibility of female mice to acetaminophen hepatotoxicity: Role of mitochondrial glutathione, oxidant stress and c-jun N-terminal kinase. Toxicol. Appl. Pharmacol. 2014, 281, 58–66. [Google Scholar] [CrossRef]
- McConnachie, L.A.; Mohar, I.; Hudson, F.N.; Ware, C.B.; Ladiges, W.C.; Fernandez, C.; Chatterton-Kirchmeier, S.; White, C.C.; Pierce, R.H.; Kavanagh, T.J. Glutamate cysteine ligase modifier subunit deficiency and gender as determinants of acetaminophen-induced hepatotoxicity in mice. Toxicol. Sci. 2007, 99, 628–636. [Google Scholar] [CrossRef]
- Montano, M.M.; Deng, H.; Liu, M.; Sun, X.; Singal, R. Transcriptional regulation by the estrogen receptor of antioxidative stress enzymes and its functional implications. Oncogene 2004, 23, 2442–2453. [Google Scholar] [CrossRef]
- Botta, D.; Shi, S.; White, C.C.; Dabrowski, M.J.; Keener, C.L.; Srinouanprachanh, S.L.; Farin, F.M.; Ware, C.B.; Ladiges, W.C.; Pierce, R.H.; et al. Acetaminophen-induced liver injury is attenuated in male glutamate-cysteine ligase transgenic mice. J. Biol. Chem. 2006, 281, 28865–28875. [Google Scholar] [CrossRef]
- Chandrasekaran, V.R.; Periasamy, S.; Liu, L.L.; Liu, M.Y. 17β-Estradiol protects against acetaminophen-overdose-induced acute oxidative hepatic damage and increases the survival rate in mice. Steroids 2011, 76, 118–124. [Google Scholar] [CrossRef]
- Arfsten, D.P.; Johnson, E.W.; Wilfong, E.R.; Jung, A.E.; Bobb, A.J. Distribution of radio-labeled N-Acetyl-L-Cysteine in Sprague-Dawley rats and its effect on glutathione metabolism following single and repeat dosing by oral gavage. Cutan. Ocul. Toxicol. 2007, 26, 113–134. [Google Scholar] [CrossRef]
- McLellan, L.I.; Lewis, A.D.; Hall, D.J.; Ansell, J.D.; Wolf, C.R. Uptake and distribution of N-acetylcysteine in mice: Tissue-specific effects on glutathione concentrations. Carcinogenesis 1995, 16, 2099–2106. [Google Scholar] [CrossRef]
- Schumacher, J.D.; Guo, G.L. Mechanistic review of drug-induced steatohepatitis. Toxicol. Appl. Pharmacol. 2015, 289, 40–47. [Google Scholar] [CrossRef]
- Aboubakr, M.; Farag, A.; Elfadadny, A.; Alkafafy, M.; Soliman, A.; Elbadawy, M. Antioxidant and anti-apoptotic potency of allicin and lycopene against methotrexate-induced cardiac injury in rats. Environ. Sci. Pollut. Res. Int. 2023, 30, 88724–88733. [Google Scholar] [CrossRef]
- Townsend, D.M.; Tew, K.D.; Tapiero, H. The importance of glutathione in human disease. Biomed. Pharmacother. 2003, 57, 145–155. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, Y.; Miller, M.L.; Shen, D.; Shertzer, H.G.; Stringer, K.F.; Wang, B.; Schneider, S.N.; Nebert, D.W.; Dalton, T.P. Hepatocyte-specific Gclc deletion leads to rapid onset of steatosis with mitochondrial injury and liver failure. Hepatology 2007, 45, 1118–1128. [Google Scholar] [CrossRef]
- Valverde, M.; Rojas, E.; Kala, S.V.; Kala, G.; Lieberman, M.W. Survival and cell death in cells constitutively unable to synthesize glutathione. Mutat. Res. 2006, 594, 172–180. [Google Scholar] [CrossRef]
- Lin, C.C.; Yin, M.C. Effects of cysteine-containing compounds on biosynthesis of triacylglycerol and cholesterol and anti-oxidative protection in liver from mice consuming a high-fat diet. Br. J. Nutr. 2008, 99, 37–43. [Google Scholar] [CrossRef]
- Ma, Y.; Gao, M.; Liu, D. N-acetylcysteine Protects Mice from High Fat Diet-induced Metabolic Disorders. Pharm. Res. 2016, 33, 2033–2042. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, K.; Wang, N.; Zhang, H. N-acetylcysteine induces apoptosis via the mitochondria-dependent pathway but not via endoplasmic reticulum stress in H9c2 cells. Mol. Med. Rep. 2017, 16, 6626–6633. [Google Scholar] [CrossRef]
- Begriche, K.; Massart, J.; Robin, M.A.; Borgne-Sanchez, A.; Fromenty, B. Drug-induced toxicity on mitochondria and lipid metabolism: Mechanistic diversity and deleterious consequences for the liver. J. Hepatol. 2011, 54, 773–794. [Google Scholar] [CrossRef]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef]
- Pan, J.; Zhou, W.; Xu, R.; Xing, L.; Ji, G.; Dang, Y. Natural PPARs agonists for the treatment of nonalcoholic fatty liver disease. Biomed. Pharmacother. 2022, 151, 113127. [Google Scholar] [CrossRef]
- Mansouri, R.M.; Baugé, E.; Staels, B.; Gervois, P. Systemic and Distal Repercussions of Liver-Specific Peroxisome Proliferator-Activated Receptor-α Control of the Acute-Phase Response. Endocrinology 2008, 149, 3215–3223. [Google Scholar] [CrossRef]
- Chew, C.H.; Chew, G.S.; Najimudin, N.; Tengku-Muhammad, T.S. Interleukin-6 inhibits human peroxisome proliferator activated receptor alpha gene expression via CCAAT/enhancer-binding proteins in hepatocytes. Int. J. Biochem. Cell Biol. 2007, 39, 1975–1986. [Google Scholar] [CrossRef]
- Wang, Y.; Nakajima, T.; Gonzalez, F.J.; Tanaka, N. PPARs as Metabolic Regulators in the Liver: Lessons from Liver-Specific PPAR-Null Mice. Int. J. Mol. Sci. 2020, 21, 2061. [Google Scholar] [CrossRef] [PubMed]
- Skat-Rørdam, J.; Højland Ipsen, D.; Lykkesfeldt, J.; Tveden-Nyborg, P. A role of peroxisome proliferator-activated receptor γ in non-alcoholic fatty liver disease. Basic Clin. Pharmacol. Toxicol. 2019, 124, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Moya, M.; Benet, M.; Guzmán, C.; Tolosa, L.; García-Monzón, C.; Pareja, E.; Castell, J.V.; Jover, R. Foxa1 reduces lipid accumulation in human hepatocytes and is down-regulated in nonalcoholic fatty liver. PLoS ONE 2012, 7, e30014. [Google Scholar] [CrossRef]
- Nagaya, T.; Tanaka, N.; Suzuki, T.; Sano, K.; Horiuchi, A.; Komatsu, M.; Nakajima, T.; Nishizawa, T.; Joshita, S.; Umemura, T.; et al. Down-regulation of SREBP-1c is associated with the development of burned-out NASH. J. Hepatol. 2010, 53, 724–731. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C Male | NAC Male | C Female | NAC Female | |
|---|---|---|---|---|
| Liver | ||||
| AST (U/L) | 221.1 ± 40.5 | 185.7 ± 54.5 | 137.9 ± 42.2 | 262.3 ± 140.6 |
| ALT (U/L) | 88.0 ± 28.9 | 58.8 ± 29.3 | 36.9 ± 4.5 | 47.6 ± 17.1 |
| ALP (U/L) | 647.1 ± 25.8 | 441.8 ± 65.4 *** | 435.6 ± 33.7 | 395.5 ± 44.3 |
| T-BIL (μg/dL) | 53.1 ± 15.9 | 26.7 ± 8.3 * | 38.5 ± 2.6 | 13.9 ± 11.9 ** |
| TP (g/dL) | 4.7 ± 0.2 | 4.6 ± 0.2 | 4.8 ± 0.1 | 4.4 ± 0.1 *** |
| ALB (g/dL) | 3.1 ± 0.1 | 2.7 ± 0.2 ** | 3.2 ± 0.1 | 3.0 ± 0.1 ** |
| GLU (mg/dL) | 186.5 ± 9.5 | 195.5 ± 26.9 | 181.8 ± 18.8 | 187.3 ± 35.4 |
| Kidney | ||||
| BUN (mg/dL) | 33.6 ± 4.6 | 27.1 ± 4.2 * | 28.7 ± 1.6 | 26.8 ± 3.0 |
| CREA (mg/dL) | 0.14 ± 0.01 | 0.09 ± 0.03 * | 0.13 ± 0.02 | 0.14 ± 0.03 |
| UA (mg/dL) | 1.9 ± 0.2 | 2.8 ± 0.5 * | 1.9 ± 0.4 | 2.4 ± 0.6 |
| Lipid | ||||
| TG (mg/dL) | 113.7 ± 4.1 | 72.9 ± 10.0 ** | 79.5 ± 15.9 | 43.4 ± 8.3 *** |
| T-CHO (mg/dL) | 176.6 ± 11.7 | 148.7 ± 10.5 ** | 125.2 ± 6.6 | 105.2 ± 6.9 *** |
| HDL (mg/dL) | 131.4 ± 9.1 | 115.9 ± 9.2 * | 94.7 ± 6.4 | 75.8 ± 5.4 *** |
| LDL (mg/dL) | 29.6 ± 3.4 | 17.6 ± 3.9 *** | 15.3 ± 2.5 | 14.6 ± 2.2 |
| T-CHO/HDL | 1.344 ± 0.089 | 1.283 ± 0.091 | 1.322 ± 0.070 | 1.388 ± 0.091 |
| LDL/HDL | 0.225 ± 0.026 | 0.152 ± 0.034 * | 0.162 ± 0.026 | 0.193 ± 0.029 |
| Ca Mg P | ||||
| Ca (mg/dL) | 9.9 ± 0.2 | 9.5 ± 0.4 | 9.4 ± 0.2 | 8.6 ± 0.3 *** |
| Mg (mg/dL) | 2.9 ± 0.3 | 2.7 ± 0.3 | 2.5 ± 0.1 | 2.7 ± 0.3 |
| P (mg/dL) | 9.2 ± 1.4 | 7.9 ± 1.5 | 5.9 ± 0.9 | 9.0 ± 1.5 ** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, M.-S.; Liou, G.-G.; Liao, J.-W.; Lai, P.-Y.; Yang, D.-J.; Wu, S.-H.; Wang, S.-H. N-acetyl Cysteine Overdose Induced Acute Toxicity and Hepatic Microvesicular Steatosis by Disrupting GSH and Interfering Lipid Metabolisms in Normal Mice. Antioxidants 2024, 13, 832. https://doi.org/10.3390/antiox13070832
Tsai M-S, Liou G-G, Liao J-W, Lai P-Y, Yang D-J, Wu S-H, Wang S-H. N-acetyl Cysteine Overdose Induced Acute Toxicity and Hepatic Microvesicular Steatosis by Disrupting GSH and Interfering Lipid Metabolisms in Normal Mice. Antioxidants. 2024; 13(7):832. https://doi.org/10.3390/antiox13070832
Chicago/Turabian StyleTsai, Ming-Shiun, Gunn-Guang Liou, Jiunn-Wang Liao, Pin-Yen Lai, Di-Jie Yang, Szu-Hua Wu, and Sue-Hong Wang. 2024. "N-acetyl Cysteine Overdose Induced Acute Toxicity and Hepatic Microvesicular Steatosis by Disrupting GSH and Interfering Lipid Metabolisms in Normal Mice" Antioxidants 13, no. 7: 832. https://doi.org/10.3390/antiox13070832
APA StyleTsai, M.-S., Liou, G.-G., Liao, J.-W., Lai, P.-Y., Yang, D.-J., Wu, S.-H., & Wang, S.-H. (2024). N-acetyl Cysteine Overdose Induced Acute Toxicity and Hepatic Microvesicular Steatosis by Disrupting GSH and Interfering Lipid Metabolisms in Normal Mice. Antioxidants, 13(7), 832. https://doi.org/10.3390/antiox13070832