
Oxidative Cysteine Post Translational Modifications Drive the Redox Code Underlying Neurodegeneration and Amyotrophic Lateral Sclerosis
 , , ,
, , ,
Abstract
:1. Introduction
2. Redoxome and Cysteine Sensitive Proteins
Free Radicals in the Redox Balance Underlying Neurodegeneration
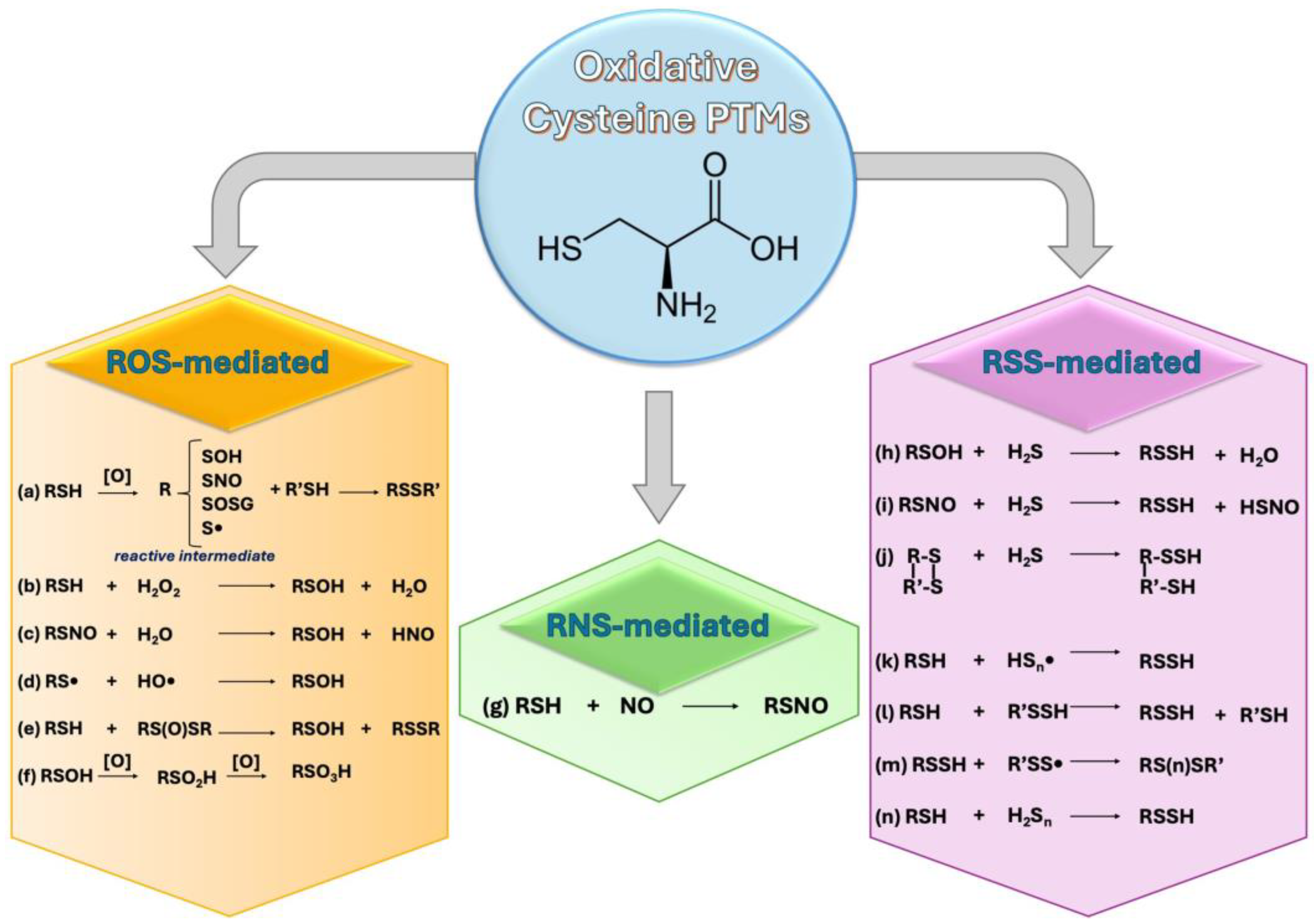
3. Free Radicals and Cys-Sensitive Proteins Crosstalk: Cys-PTMs
3.1. ROS and Cys-PTMs
- S-Thiolation
- Disulfide formation
- Sulfenylation
- Sulfinylation and Sulfonation
3.2. RNS and Cys-PTMs
- S-Nitrosylation
3.3. RSS and Cys-PTMs
- S-sulfhydration (persulfidation)
- Polysulfides
3.4. Other Cys-PTMs
3.4.1. Ox-Cys-PTMs
- S-glutathionylation
- S-Carbonylation
3.4.2. Lipid Modifications
- S-Acylation
- S-palmitoylation
- S-Prenylation
3.4.3. Molecules PTMS
- S-homocysteinylation
- Non-canonical ubiquitination
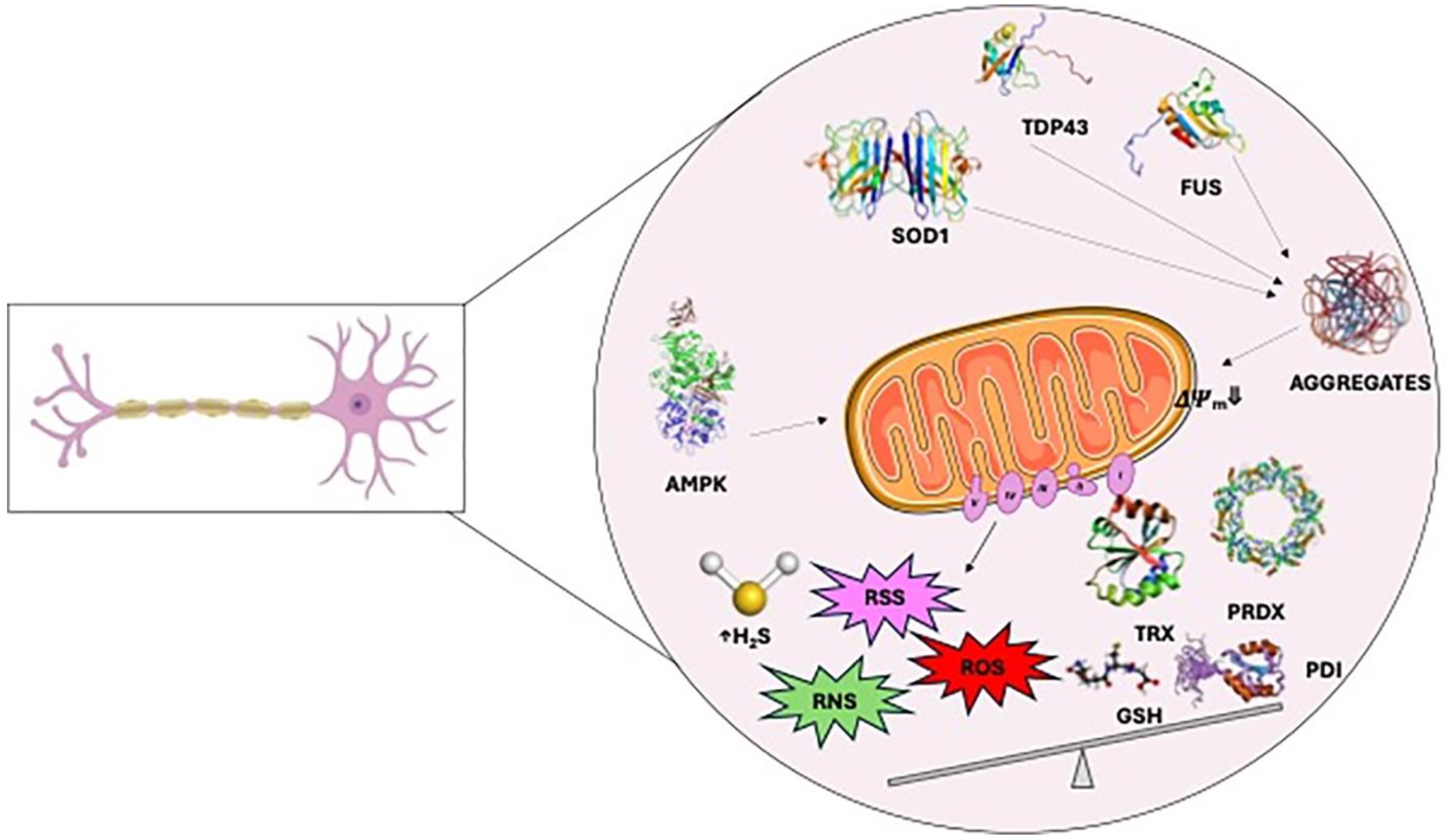
4. Redox Dysregulation in Amyotrophic Lateral Sclerosis
4.1. Cysteine Modifications in ALS
4.1.1. Cysteine Sensitive Proteins and Redox Unbalance
- Glutathione system
- Peroxiredoxins
- Hepcidin
4.1.2. Cysteine Sensitive Proteins and Protein Aggregation
- SOD1
- TDP43
- FUS
4.1.3. Cysteine Sensitive Proteins and Redox Signaling
- Protein Disulfide Isomerases
- AMPK
- FGF-2
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jennings, E.Q.; Fritz, K.S.; Galligan, J.J. Biochemical genesis of enzymatic and non-enzymatic post-translational modifications. Mol. Aspects Med. 2022, 86, 101053. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.T.; Garneau-Tsodikova, S.; Gatto, G.J. Protein Posttranslational Modifications: The Chemistry of Proteome Diversifications. Angew. Chemie Int. Ed. 2005, 44, 7342–7372. [Google Scholar] [CrossRef]
- Ramazi, S.; Zahiri, J. Post-translational modifications in proteins: Resources, tools and prediction methods. Database 2021, 2021, baab012. [Google Scholar] [CrossRef] [PubMed]
- Schaffert, L.-N.; Carter, W.G. Do Post-Translational Modifications Influence Protein Aggregation in Neurodegenerative Diseases: A Systematic Review. Brain Sci. 2020, 10, 232. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Banaclocha, M. N-Acetyl-Cysteine: Modulating the Cysteine Redox Proteome in Neurodegenerative Diseases. Antioxidants 2022, 11, 416. [Google Scholar] [CrossRef]
- Chung, H.S.; Wang, S.-B.; Venkatraman, V.; Murray, C.I.; Van Eyk, J.E. Cysteine Oxidative Posttranslational Modifications. Circ. Res. 2013, 112, 382–392. [Google Scholar] [CrossRef]
- Martínez Banaclocha, M.A. Cellular Cysteine Network (CYSTEINET): Pharmacological Intervention in Brain Aging and Neurodegenerative Diseases. In Frontiers in Clinical Drug Research-Central Nervous System; Bentham Science Publishers: Kingston, RI, USA, 2016; pp. 105–172. [Google Scholar]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Redox Mechanisms in Neurodegeneration: From Disease Outcomes to Therapeutic Opportunities. Antioxid. Redox Signal. 2019, 30, 1450–1499. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Jones, D.P. Redefining Oxidative Stress. Antioxid. Redox Signal. 2006, 8, 1865–1879. [Google Scholar] [CrossRef]
- Floyd, R.A.; Carney, J.M. Free radical damage to protein and DNA: Mechanisms involved and relevant observations on brain undergoing oxidative stress. Ann. Neurol. 1992, 32, S22–S27. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.T. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 2017, 39, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Greco, V.; Longone, P.; Spalloni, A.; Pieroni, L.; Urbani, A. Crosstalk between Oxidative Stress and Mitochondrial Damage: Focus on Amyotrophic Lateral Sclerosis. In Mitochondria in Health and in Sickness; Springer: Berlin/Heidelberg, Germany, 2019; pp. 71–82. [Google Scholar]
- Martínez-Banaclocha, M. Cysteine Network (CYSTEINET) Dysregulation in Parkinson’s Disease: Role of N-acetylcysteine. Curr. Drug Metab. 2016, 17, 368–385. [Google Scholar] [CrossRef] [PubMed]
- Bouldin, S.D.; Darch, M.A.; Hart, P.J.; Outten, C.E. Redox properties of the disulfide bond of human Cu,Zn superoxide dismutase and the effects of human glutaredoxin 1. Biochem. J. 2012, 446, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Weismiller, H.A.; Holub, T.J.; Krzesinski, B.J.; Margittai, M. A thiol-based intramolecular redox switch in four-repeat tau controls fibril assembly and disassembly. J. Biol. Chem. 2021, 297, 101021. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.J.; Hwang, A.W.; Unger, T.; Trojanowski, J.Q.; Lee, V.M.Y. Redox signalling directly regulates TDP-43 via cysteine oxidation and disulphide cross-linking. EMBO J. 2012, 31, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Kiss, R.; Zhu, M.; Jójárt, B.; Czajlik, A.; Solti, K.; Fórizs, B.; Nagy, É.; Zsila, F.; Beke-Somfai, T.; Tóth, G. Structural features of human DJ-1 in distinct Cys106 oxidative states and their relevance to its loss of function in disease. Biochim. Biophys. Acta-Gen. Subj. 2017, 1861, 2619–2629. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Yao, D.; Shi, Y.; Kabakoff, J.; Wu, W.; Reicher, J.; Ma, Y.; Moosmann, B.; Masliah, E.; Lipton, S.A.; et al. Oxidation of the cysteine-rich regions of parkin perturbs its E3 ligase activity and contributes to protein aggregation. Mol. Neurodegener. 2011, 6, 34. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shang, H.; Qiu, X.; Fujiwara, N.; Cui, L.; Li, X.-M.; Gao, T.-M.; Kong, J. Oxidative Modification of Cysteine 111 Promotes Disulfide Bond-Independent Aggregation of SOD1. Neurochem. Res. 2012, 37, 835–845. [Google Scholar] [CrossRef]
- Pieragostino, D.; Del Boccio, P.; Di Ioia, M.; Pieroni, L.; Greco, V.; De Luca, G.; D’Aguanno, S.; Rossi, C.; Franciotta, D.; Centonze, D.; et al. Oxidative modifications of cerebral transthyretin are associated with multiple sclerosis. Proteomics 2013, 13, 1002–1009. [Google Scholar] [CrossRef]
- Qu, J.; Nakamura, T.; Holland, E.A.; McKercher, S.R.; Lipton, S.A. S-nitrosylation of Cdk5. Prion 2012, 6, 364–370. [Google Scholar] [CrossRef]
- Choi, M.S.; Nakamura, T.; Cho, S.-J.; Han, X.; Holland, E.A.; Qu, J.; Petsko, G.A.; Yates, J.R.; Liddington, R.C.; Lipton, S.A. Transnitrosylation from DJ-1 to PTEN Attenuates Neuronal Cell Death in Parkinson’s Disease Models. J. Neurosci. 2014, 34, 15123–15131. [Google Scholar] [CrossRef]
- Ito, G.; Ariga, H.; Nakagawa, Y.; Iwatsubo, T. Roles of distinct cysteine residues in S-nitrosylation and dimerization of DJ-1. Biochem. Biophys. Res. Commun. 2006, 339, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.-H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-Nitrosylation of Drp1 Mediates β-Amyloid-Related Mitochondrial Fission and Neuronal Injury. Science 2009, 324, 102–105. [Google Scholar] [CrossRef]
- Haun, F.; Nakamura, T.; Shiu, A.D.; Cho, D.-H.; Tsunemi, T.; Holland, E.A.; La Spada, A.R.; Lipton, S.A. S-Nitrosylation of Dynamin-Related Protein 1 Mediates Mutant Huntingtin-Induced Mitochondrial Fragmentation and Neuronal Injury in Huntington’s Disease. Antioxid. Redox Signal. 2013, 19, 1173–1184. [Google Scholar] [CrossRef] [PubMed]
- Bae, B.-I.; Hara, M.R.; Cascio, M.B.; Wellington, C.L.; Hayden, M.R.; Ross, C.A.; Ha, H.C.; Li, X.-J.; Snyder, S.H.; Sawa, A. Mutant Huntingtin: Nuclear translocation and cytotoxicity mediated by GAPDH. Proc. Natl. Acad. Sci. USA 2006, 103, 3405–3409. [Google Scholar] [CrossRef]
- Hara, M.R.; Thomas, B.; Cascio, M.B.; Bae, B.-I.; Hester, L.D.; Dawson, V.L.; Dawson, T.M.; Sawa, A.; Snyder, S.H. Neuroprotection by pharmacologic blockade of the GAPDH death cascade. Proc. Natl. Acad. Sci. USA 2006, 103, 3887–3889. [Google Scholar] [CrossRef]
- Sen, N.; Hara, M.R.; Ahmad, A.S.; Cascio, M.B.; Kamiya, A.; Ehmsen, J.T.; Aggrawal, N.; Hester, L.; Doré, S.; Snyder, S.H.; et al. GOSPEL: A Neuroprotective Protein that Binds to GAPDH upon S-Nitrosylation. Neuron 2009, 63, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.; Gu, Z.; Nakamura, T.; Shi, Z.-Q.; Ma, Y.; Gaston, B.; Palmer, L.A.; Rockenstein, E.M.; Zhang, Z.; Masliah, E.; et al. Nitrosative stress linked to sporadic Parkinson’s disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc. Natl. Acad. Sci. USA 2004, 101, 10810–10814. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, X.; Li, C.; Guan, T.; Shang, H.; Cui, L.; Li, X.; Kong, J. S-nitrosylated protein disulfide isomerase contributes to mutant SOD1 aggregates in amyotrophic lateral sclerosis. J. Neurochem. 2013, 124, 45–58. [Google Scholar] [CrossRef]
- Fang, J.; Nakamura, T.; Cho, D.-H.; Gu, Z.; Lipton, S.A. S-nitrosylation of peroxiredoxin 2 promotes oxidative stress-induced neuronal cell death in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 18742–18747. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Jangir, D.K.; Verma, G.; Shekhar, S.; Hanpude, P.; Kumar, S.; Kumari, R.; Singh, N.; Bhavesh, N.S.; Jana, N.R.; et al. S-nitrosylation of UCHL1 induces its structural instability and promotes α-synuclein aggregation. Sci. Rep. 2017, 7, 44558. [Google Scholar] [CrossRef]
- Tsang, A.H.K.; Lee, Y.-I.; Ko, H.S.; Savitt, J.M.; Pletnikova, O.; Troncoso, J.C.; Dawson, V.L.; Dawson, T.M.; Chung, K.K.K. S-nitrosylation of XIAP compromises neuronal survival in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 4900–4905. [Google Scholar] [CrossRef]
- Sen, T.; Saha, P.; Jiang, T.; Sen, N. Sulfhydration of AKT triggers Tau-phosphorylation by activating glycogen synthase kinase 3β in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2020, 117, 4418–4427. [Google Scholar] [CrossRef]
- Giovinazzo, D.; Bursac, B.; Sbodio, J.I.; Nalluru, S.; Vignane, T.; Snowman, A.M.; Albacarys, L.M.; Sedlak, T.W.; Torregrossa, R.; Whiteman, M.; et al. Hydrogen sulfide is neuroprotective in Alzheimer’s disease by sulfhydrating GSK3β and inhibiting Tau hyperphosphorylation. Proc. Natl. Acad. Sci. USA 2021, 118, e2017225118. [Google Scholar] [CrossRef] [PubMed]
- Vandiver, M.S.; Paul, B.D.; Xu, R.; Karuppagounder, S.; Rao, F.; Snowman, A.M.; Seok Ko, H.; Il Lee, Y.; Dawson, V.L.; Dawson, T.M.; et al. Sulfhydration mediates neuroprotective actions of parkin. Nat. Commun. 2013, 4, 1626. [Google Scholar] [CrossRef]
- Greco, V.; Neri, C.; Pieragostino, D.; Spalloni, A.; Persichilli, S.; Gastaldi, M.; Mercuri, N.B.; Longone, P.; Urbani, A. Investigating Different Forms of Hydrogen Sulfide in Cerebrospinal Fluid of Various Neurological Disorders. Metabolites 2021, 11, 152. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, M.; Wang, C.; Zhang, S.; Gao, Q.; Wang, L.; Ma, L. NaSH increases SIRT1 activity and autophagy flux through sulfhydration to protect SH-SY5Y cells induced by MPP~+. Cell Cycle 2020, 19, 2216–2225. [Google Scholar] [CrossRef]
- Alvarez, B.; Salinas, G. Basic concepts of thiol chemistry and biology. In Redox Chemistry and Biology of Thiols; Elsevier: Amsterdam, The Netherlands, 2022; pp. 1–18. [Google Scholar]
- Messens, J.; Collet, J.-F. Thiol–Disulfide Exchange in Signaling: Disulfide Bonds As a Switch. Antioxid. Redox Signal. 2013, 18, 1594–1596. [Google Scholar] [CrossRef]
- EREL, Ö.; ERDOĞAN, S. Thiol-disulfide homeostasis: An integrated approach with biochemical and clinical aspects. TURKISH J. Med. Sci. 2020, 50, 1728–1738. [Google Scholar] [CrossRef]
- McBean, G.J.; Aslan, M.; Griffiths, H.R.; Torrão, R.C. Thiol redox homeostasis in neurodegenerative disease. Redox Biol. 2015, 5, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Arnesano, F.; Banci, L.; Bertini, I.; Martinelli, M.; Furukawa, Y.; O’Halloran, T.V. The Unusually Stable Quaternary Structure of Human Cu,Zn-Superoxide Dismutase 1 Is Controlled by Both Metal Occupancy and Disulfide Status. J. Biol. Chem. 2004, 279, 47998–48003. [Google Scholar] [CrossRef] [PubMed]
- Roos, G.; Messens, J. Protein sulfenic acid formation: From cellular damage to redox regulation. Free Radic. Biol. Med. 2011, 51, 314–326. [Google Scholar] [CrossRef] [PubMed]
- Lo Conte, M.; Carroll, K.S. The Redox Biochemistry of Protein Sulfenylation and Sulfinylation. J. Biol. Chem. 2013, 288, 26480–26488. [Google Scholar] [CrossRef] [PubMed]
- Beedle, A.E.M.; Lynham, S.; Garcia-Manyes, S. Protein S-sulfenylation is a fleeting molecular switch that regulates non-enzymatic oxidative folding. Nat. Commun. 2016, 7, 12490. [Google Scholar] [CrossRef]
- Peskin, A.V.; Dickerhof, N.; Poynton, R.A.; Paton, L.N.; Pace, P.E.; Hampton, M.B.; Winterbourn, C.C. Hyperoxidation of Peroxiredoxins 2 and 3. J. Biol. Chem. 2013, 288, 14170–14177. [Google Scholar] [CrossRef] [PubMed]
- Biteau, B.; Labarre, J.; Toledano, M.B. ATP-dependent reduction of cysteine–sulphinic acid by S. cerevisiae sulphiredoxin. Nature 2003, 425, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.-S.; Kang, S.W.; Woo, H.A.; Hwang, S.C.; Chae, H.Z.; Kim, K.; Rhee, S.G. Inactivation of Human Peroxiredoxin I during Catalysis as the Result of the Oxidation of the Catalytic Site Cysteine to Cysteine-sulfinic Acid. J. Biol. Chem. 2002, 277, 38029–38036. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Bian, K. Nitric oxide NO–biogeneration regulation and relevence to human diseases. Front. Biosci. 2003, 8, 997. [Google Scholar] [CrossRef]
- Radi, R. Peroxynitrite, a Stealthy Biological Oxidant. J. Biol. Chem. 2013, 288, 26464–26472. [Google Scholar] [CrossRef]
- Kirsch, M.; Korth, H.-G.; Sustmann, R.; Groot, H. de The Pathobiochemistry of Nitrogen Dioxide. Biol. Chem. 2002, 383, 383. [Google Scholar] [CrossRef]
- Anand, P.; Stamler, J.S. Enzymatic mechanisms regulating protein S-nitrosylation: Implications in health and disease. J. Mol. Med. 2012, 90, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Steenbergen, C.; Murphy, E. S-Nitrosylation: NO-Related Redox Signaling to Protect Against Oxidative Stress. Antioxid. Redox Signal. 2006, 8, 1693–1705. [Google Scholar] [CrossRef] [PubMed]
- Seth, D.; Stamler, J.S. The SNO-proteome: Causation and classifications. Curr. Opin. Chem. Biol. 2011, 15, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Koriyama, Y.; Furukawa, A. S-Nitrosylation Regulates Cell Survival and Death in the Central Nervous System. Neurochem. Res. 2018, 43, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Fernando, V.; Zheng, X.; Walia, Y.; Sharma, V.; Letson, J.; Furuta, S. S-Nitrosylation: An Emerging Paradigm of Redox Signaling. Antioxidants 2019, 8, 404. [Google Scholar] [CrossRef] [PubMed]
- Hess, D.T.; Matsumoto, A.; Kim, S.-O.; Marshall, H.E.; Stamler, J.S. Protein S-nitrosylation: Purview and parameters. Nat. Rev. Mol. Cell Biol. 2005, 6, 150–166. [Google Scholar] [CrossRef] [PubMed]
- Bouillaud, F.; Blachier, F. Mitochondria and Sulfide: A Very Old Story of Poisoning, Feeding, and Signaling? Antioxid. Redox Signal. 2011, 15, 379–391. [Google Scholar] [CrossRef]
- Iciek, M.; Bilska-Wilkosz, A.; Kozdrowicki, M.; Górny, M. Reactive sulfur species and their significance in health and disease. Biosci. Rep. 2022, 42, BSR20221006. [Google Scholar] [CrossRef]
- Libiad, M.; Yadav, P.K.; Vitvitsky, V.; Martinov, M.; Banerjee, R. Organization of the Human Mitochondrial Hydrogen Sulfide Oxidation Pathway. J. Biol. Chem. 2014, 289, 30901–30910. [Google Scholar] [CrossRef] [PubMed]
- Kabil, O.; Banerjee, R. Enzymology of H2S Biogenesis, Decay and Signaling. Antioxid. Redox Signal. 2014, 20, 770–782. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, N.; Tanaka, M.; Yoshida, M.; Ogasawara, Y.; Togawa, T.; Ishii, K.; Kimura, H. 3-Mercaptopyruvate Sulfurtransferase Produces Hydrogen Sulfide and Bound Sulfane Sulfur in the Brain. Antioxid. Redox Signal. 2009, 11, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, N.; Koike, S.; Tanaka, M.; Ishigami-Yuasa, M.; Kimura, Y.; Ogasawara, Y.; Fukui, K.; Nagahara, N.; Kimura, H. A novel pathway for the production of hydrogen sulfide from D-cysteine in mammalian cells. Nat. Commun. 2013, 4, 1366. [Google Scholar] [CrossRef]
- Mustafa, A.K.; Gadalla, M.M.; Sen, N.; Kim, S.; Mu, W.; Gazi, S.K.; Barrow, R.K.; Yang, G.; Wang, R.; Snyder, S.H. H2S Signals through Protein S-Sulfhydration. Sci. Signal. 2009, 2, ra72. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H. H2S signalling through protein sulfhydration and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 499–507. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H. H 2 S: A Novel Gasotransmitter that Signals by Sulfhydration. Trends Biochem. Sci. 2015, 40, 687–700. [Google Scholar] [CrossRef]
- Zhang, D.; Du, J.; Tang, C.; Huang, Y.; Jin, H. H2S-Induced Sulfhydration: Biological Function and Detection Methodology. Front. Pharmacol. 2017, 8, 608. [Google Scholar] [CrossRef] [PubMed]
- Petrovic, D.; Kouroussis, E.; Vignane, T.; Filipovic, M.R. The Role of Protein Persulfidation in Brain Aging and Neurodegeneration. Front. Aging Neurosci. 2021, 13, 674135. [Google Scholar] [CrossRef] [PubMed]
- Cuevasanta, E.; Lange, M.; Bonanata, J.; Coitiño, E.L.; Ferrer-Sueta, G.; Filipovic, M.R.; Alvarez, B. Reaction of Hydrogen Sulfide with Disulfide and Sulfenic Acid to Form the Strongly Nucleophilic Persulfide. J. Biol. Chem. 2015, 290, 26866–26880. [Google Scholar] [CrossRef]
- Szabo, C. Hydrogen Sulfide, an Endogenous Stimulator of Mitochondrial Function in Cancer Cells. Cells 2021, 10, 220. [Google Scholar] [CrossRef]
- Cirino, G.; Szabo, C.; Papapetropoulos, A. Physiological roles of hydrogen sulfide in mammalian cells, tissues, and organs. Physiol. Rev. 2023, 103, 31–276. [Google Scholar] [CrossRef] [PubMed]
- Davoli, A.; Greco, V.; Spalloni, A.; Guatteo, E.; Neri, C.; Rizzo, G.R.; Cordella, A.; Romigi, A.; Cortese, C.; Bernardini, S.; et al. Evidence of hydrogen sulfide involvement in amyotrophic lateral sclerosis. Ann. Neurol. 2015, 77, 697–709. [Google Scholar] [CrossRef]
- Spalloni, A.; Greco, V.; Ciriminna, G.; Corasolla Carregari, V.; Marini, F.; Pieroni, L.; Mercuri, N.B.; Urbani, A.; Longone, P. Impact of Pharmacological Inhibition of Hydrogen Sulphide Production in the SOD1G93A-ALS Mouse Model. Int. J. Mol. Sci. 2019, 20, 2550. [Google Scholar] [CrossRef] [PubMed]
- Kamoun, P.; Belardinelli, M.; Chabli, A.; Lallouchi, K.; Chadefaux-Vekemans, B. Endogenous hydrogen sulfide overproduction in Down syndrome. Am. J. Med. Genet. Part A 2003, 116, 310–311. [Google Scholar] [CrossRef]
- Belardinelli, M.-C.; Chabli, A.; Chadefaux-Vekemans, B.; Kamoun, P. Urinary Sulfur Compounds in Down Syndrome. Clin. Chem. 2001, 47, 1500–1501. [Google Scholar] [CrossRef] [PubMed]
- Barayeu, U.; Sawa, T.; Nishida, M.; Wei, F.; Motohashi, H.; Akaike, T. Supersulfide biology and translational medicine for disease control. Br. J. Pharmacol. 2023, 1–16. [Google Scholar] [CrossRef]
- Wu, Z.; Barayeu, U.; Schilling, D.; Dick, T.P.; Pratt, D.A. Emergence of (hydro)persulfides as suppressors of lipid peroxidation and ferroptotic cell death. Curr. Opin. Chem. Biol. 2023, 76, 102353. [Google Scholar] [CrossRef]
- Nagy, P.; Winterbourn, C.C. Rapid Reaction of Hydrogen Sulfide with the Neutrophil Oxidant Hypochlorous Acid to Generate Polysulfides. Chem. Res. Toxicol. 2010, 23, 1541–1543. [Google Scholar] [CrossRef]
- Kimura, Y.; Mikami, Y.; Osumi, K.; Tsugane, M.; Oka, J.; Kimura, H. Polysulfides are possible H2S-derived signaling molecules in rat brain. FASEB J. 2013, 27, 2451–2457. [Google Scholar] [CrossRef]
- Kimura, H. Hydrogen polysulfide (H2Sn) signaling along with hydrogen sulfide (H2S) and nitric oxide (NO). J. Neural Transm. 2016, 123, 1235–1245. [Google Scholar] [CrossRef] [PubMed]
- Greiner, R.; Pálinkás, Z.; Bäsell, K.; Becher, D.; Antelmann, H.; Nagy, P.; Dick, T.P. Polysulfides Link H2S to Protein Thiol Oxidation. Antioxid. Redox Signal. 2013, 19, 1749–1765. [Google Scholar] [CrossRef]
- Olson, K.R. H2S and polysulfide metabolism: Conventional and unconventional pathways. Biochem. Pharmacol. 2018, 149, 77–90. [Google Scholar] [CrossRef]
- Bogdándi, V.; Ditrói, T.; Bátai, I.Z.; Sándor, Z.; Minnion, M.; Vasas, A.; Galambos, K.; Buglyó, P.; Pintér, E.; Feelisch, M.; et al. Nitrosopersulfide (SSNO−) Is a Unique Cysteine Polysulfidating Agent with Reduction-Resistant Bioactivity. Antioxid. Redox Signal. 2020, 33, 1277–1294. [Google Scholar] [CrossRef]
- Alcock, L.J.; Perkins, M.V.; Chalker, J.M. Chemical methods for mapping cysteine oxidation. Chem. Soc. Rev. 2018, 47, 231–268. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Zhang, L.; Zhang, L.; Wang, Z.; Wang, X.; Li, C.; Chen, Y.; Shang, S.; Li, L. CysModDB: A comprehensive platform with the integration of manually curated resources and analysis tools for cysteine posttranslational modifications. Brief. Bioinform. 2022, 23, 23. [Google Scholar] [CrossRef] [PubMed]
- Gallogly, M.M.; Mieyal, J.J. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr. Opin. Pharmacol. 2007, 7, 381–391. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Rossi, R.; Giustarini, D.; Colombo, R.; Milzani, A. S-glutathionylation in protein redox regulation. Free Radic. Biol. Med. 2007, 43, 883–898. [Google Scholar] [CrossRef]
- Hurd, T.R.; Costa, N.J.; Dahm, C.C.; Beer, S.M.; Brown, S.E.; Filipovska, A.; Murphy, M.P. Glutathionylation of Mitochondrial Proteins. Antioxid. Redox Signal. 2005, 7, 999–1010. [Google Scholar] [CrossRef]
- Applegate, M.A.B.; Humphries, K.M.; Szweda, L.I. Reversible Inhibition of α-Ketoglutarate Dehydrogenase by Hydrogen Peroxide: Glutathionylation and Protection of Lipoic Acid. Biochemistry 2008, 47, 473–478. [Google Scholar] [CrossRef]
- Bulteau, A.-L.; Lundberg, K.C.; Ikeda-Saito, M.; Isaya, G.; Szweda, L.I. Reversible redox-dependent modulation of mitochondrial aconitase and proteolytic activity during in vivo cardiac ischemia/reperfusion. Proc. Natl. Acad. Sci. USA 2005, 102, 5987–5991. [Google Scholar] [CrossRef]
- Giangregorio, N.; Palmieri, F.; Indiveri, C. Glutathione controls the redox state of the mitochondrial carnitine/acylcarnitine carrier Cys residues by glutathionylation. Biochim. Biophys. Acta-Gen. Subj. 2013, 1830, 5299–5304. [Google Scholar] [CrossRef] [PubMed]
- Willems, P.H.G.M.; Rossignol, R.; Dieteren, C.E.J.; Murphy, M.P.; Koopman, W.J.H. Redox Homeostasis and Mitochondrial Dynamics. Cell Metab. 2015, 22, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Vrettou, S.; Wirth, B. S-Glutathionylation and S-Nitrosylation in Mitochondria: Focus on Homeostasis and Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 15849. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.J.; Kim, H.; Choi, H.-J.; Lee, S.; Kim, K. Protein Glutathionylation in the Pathogenesis of Neurodegenerative Diseases. Oxid. Med. Cell. Longev. 2017, 2017, 2818565. [Google Scholar] [CrossRef] [PubMed]
- Newman, S.F.; Sultana, R.; Perluigi, M.; Coccia, R.; Cai, J.; Pierce, W.M.; Klein, J.B.; Turner, D.M.; Butterfield, D.A. An increase in S-glutathionylated proteins in the Alzheimer’s disease inferior parietal lobule, a proteomics approach. J. Neurosci. Res. 2007, 85, 1506–1514. [Google Scholar] [CrossRef] [PubMed]
- Dinoto, L.; Deture, M.A.; Purich, D.L. Structural insights into Alzheimer filament assembly pathways based on site-directed mutagenesis and S-glutathionylation of three-repeat neuronal Tau protein. Microsc. Res. Tech. 2005, 67, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Hutson, S.M.; Wallin, R.; Hall, T.R. Identification of mitochondrial branched chain aminotransferase and its isoforms in rat tissues. J. Biol. Chem. 1992, 267, 15681–15686. [Google Scholar] [CrossRef] [PubMed]
- El Kodsi, D.N.; Tokarew, J.M.; Sengupta, R.; Lengacher, N.A.; Chatterji, A.; Nguyen, A.P.; Boston, H.; Jiang, Q.; Palmberg, C.; Pileggi, C.; et al. Parkin coregulates glutathione metabolism in adult mammalian brain. Acta Neuropathol. Commun. 2023, 11, 19. [Google Scholar] [CrossRef]
- Johnson, W.M.; Golczak, M.; Choe, K.; Curran, P.L.; Miller, O.G.; Yao, C.; Wang, W.; Lin, J.; Milkovic, N.M.; Ray, A.; et al. Regulation of DJ-1 by Glutaredoxin 1 in Vivo: Implications for Parkinson’s Disease. Biochemistry 2016, 55, 4519–4532. [Google Scholar] [CrossRef]
- Redler, R.L.; Wilcox, K.C.; Proctor, E.A.; Fee, L.; Caplow, M.; Dokholyan, N.V. Glutathionylation at Cys-111 Induces Dissociation of Wild Type and FALS Mutant SOD1 Dimers. Biochemistry 2011, 50, 7057–7066. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Ryu, H.; Kowall, N.W. Differential regulation of neuronal and inducible nitric oxide synthase (NOS) in the spinal cord of mutant SOD1 (G93A) ALS mice. Biochem. Biophys. Res. Commun. 2009, 387, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Viappiani, S.; Nicolescu, A.C.; Holt, A.; Sawicki, G.; Crawford, B.D.; León, H.; van Mulligen, T.; Schulz, R. Activation and modulation of 72 kDa matrix metalloproteinase-2 by peroxynitrite and glutathione. Biochem. Pharmacol. 2009, 77, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.; Subbaram, S.; Carrico, P.M.; Melendez, J.A. Redox-control of matrix metalloproteinase-1: A critical link between free radicals, matrix remodeling and degenerative disease. Respir. Physiol. Neurobiol. 2010, 174, 299–306. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Giustarini, D.; Colombo, R.; Rossi, R.; Milzani, A. Protein carbonylation in human diseases. Trends Mol. Med. 2003, 9, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Reed, T.T.; Butterfield, D.A. Protein Carbonylation in Brains of Subjects with Selected Neurodegenerative Disorders. In Protein Carbonylation; Wiley: Hoboken, NJ, USA, 2017; pp. 167–205. [Google Scholar]
- Sharma, A.; Weber, D.; Raupbach, J.; Dakal, T.C.; Fließbach, K.; Ramirez, A.; Grune, T.; Wüllner, U. Advanced glycation end products and protein carbonyl levels in plasma reveal sex-specific differences in Parkinson’s and Alzheimer’s disease. Redox Biol. 2020, 34, 101546. [Google Scholar] [CrossRef]
- Sadowska-Bartosz, I.; Adamczyk-Sowa, M.; Galiniak, S.; Mucha, S.; Pierzchala, K.; Bartosz, G. Oxidative modification of serum proteins in multiple sclerosis. Neurochem. Int. 2013, 63, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, L.H.; Shipston, M.J. The Physiology of Protein S-acylation. Physiol. Rev. 2015, 95, 341–376. [Google Scholar] [CrossRef]
- Mesquita, F.S.; Abrami, L.; Linder, M.E.; Bamji, S.X.; Dickinson, B.C.; van der Goot, F.G. Mechanisms and functions of protein S-acylation. Nat. Rev. Mol. Cell Biol. 2024, 25, 488–509. [Google Scholar] [CrossRef]
- Conibear, E.; Davis, N.G. Palmitoylation and depalmitoylation dynamics at a glance. J. Cell Sci. 2010, 123, 4007–4010. [Google Scholar] [CrossRef]
- Hornemann, T. Palmitoylation and depalmitoylation defects. J. Inherit. Metab. Dis. 2015, 38, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Ramzan, F.; Abrar, F.; Mishra, G.G.; Liao, L.M.Q.; Martin, D.D.O. Lost in traffic: Consequences of altered palmitoylation in neurodegeneration. Front. Physiol. 2023, 14, 1166125. [Google Scholar] [CrossRef] [PubMed]
- Fukata, Y.; Fukata, M. Protein palmitoylation in neuronal development and synaptic plasticity. Nat. Rev. Neurosci. 2010, 11, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.S.; Martin, D.D.O.; Butland, S.L.; Lavallée-Adam, M.; Calzolari, D.; Kay, C.; Yates, J.R.; Hayden, M.R. Curation of the Mammalian Palmitoylome Indicates a Pivotal Role for Palmitoylation in Diseases and Disorders of the Nervous System and Cancers. PLoS Comput. Biol. 2015, 11, e1004405. [Google Scholar] [CrossRef] [PubMed]
- Yanai, A.; Huang, K.; Kang, R.; Singaraja, R.R.; Arstikaitis, P.; Gan, L.; Orban, P.C.; Mullard, A.; Cowan, C.M.; Raymond, L.A.; et al. Palmitoylation of huntingtin by HIP14is essential for its trafficking and function. Nat. Neurosci. 2006, 9, 824–831. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, R.; Barren, C.; Kovacs, D.M. Palmitoylation of Amyloid Precursor Protein Regulates Amyloidogenic Processing in Lipid Rafts. J. Neurosci. 2013, 33, 11169–11183. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Vetrivel, K.S.; Drisdel, R.C.; Meckler, X.; Gong, P.; Leem, J.Y.; Li, T.; Carter, M.; Chen, Y.; Nguyen, P.; et al. S-Palmitoylation of γ-Secretase Subunits Nicastrin and APH-1. J. Biol. Chem. 2009, 284, 1373–1384. [Google Scholar] [CrossRef]
- Koegl, M.; Zlatkine, P.; Ley, S.C.; Courtneidge, S.A.; Magee, A.I. Palmitoylation of multiple Src-family kinases at a homologous N-terminal motif. Biochem. J. 1994, 303, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Antinone, S.E.; Ghadge, G.D.; Lam, T.T.; Wang, L.; Roos, R.P.; Green, W.N. Palmitoylation of Superoxide Dismutase 1 (SOD1) Is Increased for Familial Amyotrophic Lateral Sclerosis-linked SOD1 Mutants. J. Biol. Chem. 2013, 288, 21606–21617. [Google Scholar] [CrossRef]
- Jung, D.; Bachmann, H.S. Regulation of protein prenylation. Biomed. Pharmacother. 2023, 164, 114915. [Google Scholar] [CrossRef]
- Zhang, F.L.; Casey, P.J. PROTEIN PRENYLATION: Molecular Mechanisms and Functional Consequences. Annu. Rev. Biochem. 1996, 65, 241–269. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Shen, N.; Wang, X.; Jiang, S.; Xue, B.; Li, C. Protein prenylation and human diseases: A balance of protein farnesylation and geranylgeranylation. Sci. China Life Sci. 2015, 58, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.L.; Vassar, R. Isoprenoids and Alzheimer’s disease: A complex relationship. Neurobiol. Dis. 2006, 22, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Eckert, G.P.; Hooff, G.P.; Strandjord, D.M.; Igbavboa, U.; Volmer, D.A.; Müller, W.E.; Wood, W.G. Regulation of the brain isoprenoids farnesyl- and geranylgeranylpyrophosphate is altered in male Alzheimer patients. Neurobiol. Dis. 2009, 35, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Hooff, G.P.; Peters, I.; Wood, W.G.; Müller, W.E.; Eckert, G.P. Modulation of Cholesterol, Farnesylpyrophosphate, and Geranylgeranylpyrophosphate in Neuroblastoma SH-SY5Y-APP695 Cells: Impact on Amyloid Beta-Protein Production. Mol. Neurobiol. 2010, 41, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Huo, J.; Xin, C.; Yang, J.; Liu, Q.; Dong, H.; Li, R.; Liu, Y. RABGGTB plays a critical role in ALS pathogenesis. Brain Res. Bull. 2024, 206, 110833. [Google Scholar] [CrossRef] [PubMed]
- Jo, A.; Lee, Y.; Kam, T.-I.; Kang, S.-U.; Neifert, S.; Karuppagounder, S.S.; Khang, R.; Kang, H.; Park, H.; Chou, S.-C.; et al. PARIS farnesylation prevents neurodegeneration in models of Parkinson’s disease. Sci. Transl. Med. 2021, 13, eaax8891. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-M.; Tang, X.-Q. Homocysteinylation and Sulfhydration in Diseases. Curr. Neuropharmacol. 2022, 20, 1726–1735. [Google Scholar] [CrossRef]
- Jakubowski, H. Molecular basis of homocysteine toxicity in humans. Cell. Mol. Life Sci. 2004, 61, 470–487. [Google Scholar] [CrossRef]
- Sass, J.O.; Nakanishi, T.; Sato, T.; Sperl, W.; Shimizu, A. S-Homocysteinylation of transthyretin is detected in plasma and serum of humans with different types of hyperhomocysteinemia. Biochem. Biophys. Res. Commun. 2003, 310, 242–246. [Google Scholar] [CrossRef]
- Carey, A.; Fossati, S. Hypertension and hyperhomocysteinemia as modifiable risk factors for Alzheimer’s disease and dementia: New evidence, potential therapeutic strategies, and biomarkers. Alzheimer’s Dement. 2023, 19, 671–695. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Canteli, M.; Paul, J.; Norris, E.H.; Bronstein, R.; Ahn, H.J.; Zamolodchikov, D.; Bhuvanendran, S.; Fenz, K.M.; Strickland, S. Fibrinogen and β-Amyloid Association Alters Thrombosis and Fibrinolysis: A Possible Contributing Factor to Alzheimer’s Disease. Neuron 2010, 66, 695–709. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.C.; Kruyer, A.; Yao, Y.; Feierman, E.; Richards, A.; Strickland, S.; Norris, E.H. Hyperhomocysteinemia exacerbates Alzheimer’s disease pathology by way of the β-amyloid fibrinogen interaction. J. Thromb. Haemost. 2016, 14, 1442–1452. [Google Scholar] [CrossRef] [PubMed]
- Riquier, S.; Breton, J.; Abbas, K.; Cornu, D.; Bouton, C.; Drapier, J.-C. Peroxiredoxin post-translational modifications by redox messengers. Redox Biol. 2014, 2, 777–785. [Google Scholar] [CrossRef]
- McDowell, G.S.; Philpott, A. New Insights into the Role of Ubiquitylation of Proteins. Int. Rev. Cell Mol. Biol. 2016, 325, 35–88. [Google Scholar] [PubMed]
- McDowell, G.S.; Philpott, A. Non-canonical ubiquitylation: Mechanisms and consequences. Int. J. Biochem. Cell Biol. 2013, 45, 1833–1842. [Google Scholar] [CrossRef] [PubMed]
- McClellan, A.J.; Laugesen, S.H.; Ellgaard, L. Cellular functions and molecular mechanisms of non-lysine ubiquitination. Open Biol. 2019, 9, 190147. [Google Scholar] [CrossRef]
- Sabatelli, M.; Marangi, G.; Conte, A.; Tasca, G.; Zollino, M.; Lattante, S. New ALS-Related Genes Expand the Spectrum Paradigm of Amyotrophic Lateral Sclerosis. Brain Pathol. 2016, 26, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Barber, S.C.; Shaw, P.J. Oxidative stress in ALS: Key role in motor neuron injury and therapeutic target. Free Radic. Biol. Med. 2010, 48, 629–641. [Google Scholar] [CrossRef]
- Jankovic, M.; Novakovic, I.; Gamil Anwar Dawod, P.; Gamil Anwar Dawod, A.; Drinic, A.; Abdel Motaleb, F.I.; Ducic, S.; Nikolic, D. Current Concepts on Genetic Aspects of Mitochondrial Dysfunction in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2021, 22, 9832. [Google Scholar] [CrossRef]
- Jagaraj, C.J.; Parakh, S.; Atkin, J.D. Emerging Evidence Highlighting the Importance of Redox Dysregulation in the Pathogenesis of Amyotrophic Lateral Sclerosis (ALS). Front. Cell. Neurosci. 2021, 14, 581950. [Google Scholar] [CrossRef] [PubMed]
- Valle, C.; Carrì, M.T. Cysteine Modifications in the Pathogenesis of ALS. Front. Mol. Neurosci. 2017, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Meister, A. Glutathione metabolism and its selective modification. J. Biol. Chem. 1988, 263, 17205–17208. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J. Redox signaling: An evolution from free radicals to aging. Free Radic. Biol. Med. 2016, 97, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Golko-Perez, S.; Amit, T.; Bar-Am, O.; Youdim, M.B.H.; Weinreb, O. A Novel Iron Chelator-Radical Scavenger Ameliorates Motor Dysfunction and Improves Life Span and Mitochondrial Biogenesis in SOD1G93A ALS Mice. Neurotox. Res. 2017, 31, 230–244. [Google Scholar] [CrossRef] [PubMed]
- Petillon, C.; Hergesheimer, R.; Puy, H.; Corcia, P.; Vourc’h, P.; Andres, C.; Karim, Z.; Blasco, H. The relevancy of data regarding the metabolism of iron to our understanding of deregulated mechanisms in ALS; hypotheses and pitfalls. Front. Neurosci. 2019, 13, 1031. [Google Scholar] [CrossRef] [PubMed]
- Silva-Adaya, D.; Gonsebatt, M.E.; Guevara, J. Thioredoxin System Regulation in the Central Nervous System: Experimental Models and Clinical Evidence. Oxid. Med. Cell. Longev. 2014, 2014, 590808. [Google Scholar] [CrossRef] [PubMed]
- Chae, H.Z.; Kim, H.J.; Kang, S.W.; Rhee, S.G. Characterization of three isoforms of mammalian peroxiredoxin that reduce peroxides in the presence of thioredoxin. Diabetes Res. Clin. Pract. 1999, 45, 101–112. [Google Scholar] [CrossRef]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef]
- Malaspina, A.; Kaushik, N.; De Belleroche, J. Differential expression of 14 genes in amyotrophic lateral sclerosis spinal cord detected using gridded cDNA arrays. J. Neurochem. 2001, 77, 132–145. [Google Scholar] [CrossRef]
- Ogawa, Y.; Kosaka, H.; Nakanishi, T.; Shimizu, A.; Ohoi, N.; Shouji, H.; Yanagihara, T.; Sakoda, S. Stability of Mutant Superoxide Dismutase-1 Associated with Familial Amyotrophic Lateral Sclerosis Determines the Manner of Copper Release and Induction of Thioredoxin in Erythrocytes. Biochem. Biophys. Res. Commun. 1997, 241, 251–257. [Google Scholar] [CrossRef]
- Asensi, M.; Sastre, J.; Pallardo, F.V.; Lloret, A.; Lehner, M.; Garcia-de-la Asuncion, J.; Viña, J. [23] Ratio of reduced to oxidized glutathione as indicator of oxidative stress status and DNA damage. Methods Enzymol. 1999, 299, 267–276. [Google Scholar]
- Ehrhart, J.; Smith, A.J.; Kuzmin-Nichols, N.; Zesiewicz, T.A.; Jahan, I.; Shytle, R.D.; Kim, S.-H.; Sanberg, C.D.; Vu, T.H.; Gooch, C.L.; et al. Humoral factors in ALS patients during disease progression. J. Neuroinflamm. 2015, 12, 127. [Google Scholar] [CrossRef]
- Chi, L.; Ke, Y.; Luo, C.; Gozal, D.; Liu, R. Depletion of reduced glutathione enhances motor neuron degeneration in vitro and in vivo. Neuroscience 2007, 144, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Chae, H.Z.; Kim, K. Peroxiredoxins: A historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic. Biol. Med. 2005, 38, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Wood, Z.A.; Schröder, E.; Robin Harris, J.; Poole, L.B. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 2003, 28, 32–40. [Google Scholar] [CrossRef]
- Rhee, S.G. Overview on Peroxiredoxin. Mol. Cells 2016, 39, 1–5. [Google Scholar] [CrossRef]
- Szeliga, M. Peroxiredoxins in Neurodegenerative Diseases. Antioxidants 2020, 9, 1203. [Google Scholar] [CrossRef] [PubMed]
- Wood-Allum, C.A. Impairment of mitochondrial anti-oxidant defence in SOD1-related motor neuron injury and amelioration by ebselen. Brain 2006, 129, 1693–1709. [Google Scholar] [CrossRef]
- Kriznik, A.; Libiad, M.; Le Cordier, H.; Boukhenouna, S.; Toledano, M.B.; Rahuel-Clermont, S. Dynamics of a Key Conformational Transition in the Mechanism of Peroxiredoxin Sulfinylation. ACS Catal. 2020, 10, 3326–3339. [Google Scholar] [CrossRef]
- Strey, C.W.; Spellman, D.; Stieber, A.; Gonatas, J.O.; Wang, X.; Lambris, J.D.; Gonatas, N.K. Dysregulation of Stathmin, a Microtubule-Destabilizing Protein, and Up-Regulation of Hsp25, Hsp27, and the Antioxidant Peroxiredoxin 6 in a Mouse Model of Familial Amyotrophic Lateral Sclerosis. Am. J. Pathol. 2004, 165, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.; Heath, P.R.; Kirby, J.; Wharton, S.B.; Cookson, M.R.; Menzies, F.M.; Banks, R.E.; Shaw, P.J. Analysis of the Cytosolic Proteome in a Cell Culture Model of Familial Amyotrophic Lateral Sclerosis Reveals Alterations to the Proteasome, Antioxidant Defenses, and Nitric Oxide Synthetic Pathways. J. Biol. Chem. 2003, 278, 6371–6383. [Google Scholar] [CrossRef] [PubMed]
- Devos, D.; Moreau, C.; Kyheng, M.; Garçon, G.; Rolland, A.S.; Blasco, H.; Gelé, P.; Lenglet, T.T.; Veyrat-Durebex, C.; Corcia, P.; et al. Author Correction: A ferroptosis–based panel of prognostic biomarkers for Amyotrophic Lateral Sclerosis. Sci. Rep. 2019, 9, 2918, Erratum in Sci. Rep. 2020, 10, 3312. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, H.; Manfredi, G. Different regulation of wild-type and mutant Cu,Zn superoxide dismutase localization in mammalian mitochondria. Hum. Mol. Genet. 2008, 17, 3303–3317. [Google Scholar] [CrossRef] [PubMed]
- Culotta, V.C.; Klomp, L.W.J.; Strain, J.; Casareno, R.L.B.; Krems, B.; Gitlin, J.D. The Copper Chaperone for Superoxide Dismutase. J. Biol. Chem. 1997, 272, 23469–23472. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, H.; Manfredi, G. Import, maturation, and function of SOD1 and its copper chaperone CCS in the mitochondrial intermembrane space. Antioxid. Redox Signal. 2010, 13, 1375–1384. [Google Scholar] [CrossRef]
- Harraz, M.M.; Marden, J.J.; Zhou, W.; Zhang, Y.; Williams, A.; Sharov, V.S.; Nelson, K.; Luo, M.; Paulson, H.; Schöneich, C.; et al. SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J. Clin. Investig. 2008, 118, 659–670. [Google Scholar] [CrossRef]
- Tafuri, F.; Ronchi, D.; Magri, F.; Comi, G.P.; Corti, S. SOD1 misplacing and mitochondrial dysfunction in amyotrophic lateral sclerosis pathogenesis. Front. Cell. Neurosci. 2015, 9, 336. [Google Scholar] [CrossRef]
- Sasaki, S.; Warita, H.; Murakami, T.; Shibata, N.; Komori, T.; Abe, K.; Kobayashi, M.; Iwata, M. Ultrastructural study of aggregates in the spinal cord of transgenic mice with a G93A mutant SOD1 gene. Acta Neuropathol. 2005, 109, 247–255. [Google Scholar] [CrossRef]
- Higgins, C.M.J.; Jung, C.; Ding, H.; Xu, Z. Mutant Cu, Zn Superoxide Dismutase that Causes Motoneuron Degeneration Is Present in Mitochondria in the CNS. J. Neurosci. 2002, 22, RC215. [Google Scholar] [CrossRef]
- McAlary, L.; Yerbury, J.J.; Aquilina, J.A. Glutathionylation potentiates benign superoxide dismutase 1 variants to the toxic forms associated with amyotrophic lateral sclerosis. Sci. Rep. 2013, 3, 3275. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E. TDP-43 post-translational modifications in health and disease. Expert Opin. Ther. Targets 2018, 22, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, A.; Myers, M.P.; Buratti, E.; Baralle, F.E. Characterizing TDP-43 interaction with its RNA targets. Nucleic Acids Res. 2013, 41, 5062–5074. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 12. [Google Scholar] [CrossRef] [PubMed]
- Igaz, L.M.; Kwong, L.K.; Xu, Y.; Truax, A.C.; Uryu, K.; Neumann, M.; Clark, C.M.; Elman, L.B.; Miller, B.L.; Grossman, M.; et al. Enrichment of C-Terminal Fragments in TAR DNA-Binding Protein-43 Cytoplasmic Inclusions in Brain but not in Spinal Cord of Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Am. J. Pathol. 2008, 173, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Bozzo, F.; Salvatori, I.; Iacovelli, F.; Mirra, A.; Rossi, S.; Cozzolino, M.; Falconi, M.; Valle, C.; Carrì, M.T. Structural insights into the multi-determinant aggregation of TDP-43 in motor neuron-like cells. Neurobiol. Dis. 2016, 94, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Gao, K.; Jankovic, J. The role of FUS gene variants in neurodegenerative diseases. Nat. Rev. Neurol. 2014, 10, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Da Cruz, S.; Cleveland, D.W. Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Curr. Opin. Neurobiol. 2011, 21, 904–919. [Google Scholar] [CrossRef] [PubMed]
- Dormann, D.; Rodde, R.; Edbauer, D.; Bentmann, E.; Fischer, I.; Hruscha, A.; Than, M.E.; Mackenzie, I.R.A.; Capell, A.; Schmid, B.; et al. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 2010, 29, 2841–2857. [Google Scholar] [CrossRef]
- Deng, Q.; Holler, C.J.; Taylor, G.; Hudson, K.F.; Watkins, W.; Gearing, M.; Ito, D.; Murray, M.E.; Dickson, D.W.; Seyfried, N.T.; et al. FUS is Phosphorylated by DNA-PK and Accumulates in the Cytoplasm after DNA Damage. J. Neurosci. 2014, 34, 7802–7813. [Google Scholar] [CrossRef]
- Monahan, Z.; Ryan, V.H.; Janke, A.M.; Burke, K.A.; Rhoads, S.N.; Zerze, G.H.; O’Meally, R.; Dignon, G.L.; Conicella, A.E.; Zheng, W.; et al. Phosphorylation of the FUS low-complexity domain disrupts phase separation, aggregation, and toxicity. EMBO J. 2017, 36, 2951–2967. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.J.; Lee, S.; Choi, H.-J.; Han, Y.J.; Jeon, Y.-M.; Jo, M.; Lee, S.; Nahm, M.; Lim, S.M.; Kim, S.H.; et al. Therapeutic modulation of GSTO activity rescues FUS-associated neurotoxicity via deglutathionylation in ALS disease models. Dev. Cell 2022, 57, 783–798. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.K. Protein Disulfide Isomerase and the Endoplasmic Reticulum in Amyotrophic Lateral Sclerosis. J. Neurosci. 2010, 30, 3865–3867. [Google Scholar] [CrossRef] [PubMed]
- Parakh, S.; Shadfar, S.; Perri, E.R.; Ragagnin, A.M.G.; Piattoni, C.V.; Fogolín, M.B.; Yuan, K.C.; Shahheydari, H.; Don, E.K.; Thomas, C.J.; et al. The Redox Activity of Protein Disulfide Isomerase Inhibits ALS Phenotypes in Cellular and Zebrafish Models. iScience 2020, 23, 101097. [Google Scholar] [CrossRef] [PubMed]
- Rozas, P.; Pinto, C.; Martínez Traub, F.; Díaz, R.; Pérez, V.; Becerra, D.; Ojeda, P.; Ojeda, J.; Wright, M.T.; Mella, J.; et al. Protein disulfide isomerase ERp57 protects early muscle denervation in experimental ALS. Acta Neuropathol. Commun. 2021, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, C.E.; Carroll, K.S. Cysteine-Mediated Redox Signaling: Chemistry, Biology, and Tools for Discovery. Chem. Rev. 2013, 113, 4633–4679. [Google Scholar] [CrossRef] [PubMed]
- Fra, A.; Yoboue, E.D.; Sitia, R. Cysteines as Redox Molecular Switches and Targets of Disease. Front. Mol. Neurosci. 2017, 10, 167. [Google Scholar] [CrossRef] [PubMed]
- Farg, M.A.; Soo, K.Y.; Walker, A.K.; Pham, H.; Orian, J.; Horne, M.K.; Warraich, S.T.; Williams, K.L.; Blair, I.P.; Atkin, J.D. Mutant FUS induces endoplasmic reticulum stress in amyotrophic lateral sclerosis and interacts with protein disulfide-isomerase. Neurobiol. Aging 2012, 33, 2855–2868. [Google Scholar] [CrossRef] [PubMed]
- Jeon, G.S.; Nakamura, T.; Lee, J.-S.; Choi, W.-J.; Ahn, S.-W.; Lee, K.-W.; Sung, J.-J.; Lipton, S.A. Potential Effect of S-Nitrosylated Protein Disulfide Isomerase on Mutant SOD1 Aggregation and Neuronal Cell Death in Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2014, 49, 796–807. [Google Scholar] [CrossRef]
- Schonhoff, C.M.; Matsuoka, M.; Tummala, H.; Johnson, M.A.; Estevéz, A.G.; Wu, R.; Kamaid, A.; Ricart, K.C.; Hashimoto, Y.; Gaston, B.; et al. S-nitrosothiol depletion in amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2006, 103, 2404–2409. [Google Scholar] [CrossRef]
- Liu, Y.-J.; Chern, Y. Contribution of Energy Dysfunction to Impaired Protein Translation in Neurodegenerative Diseases. Front. Cell. Neurosci. 2021, 15, 15. [Google Scholar] [CrossRef]
- Hinchy, E.C.; Gruszczyk, A.V.; Willows, R.; Navaratnam, N.; Hall, A.R.; Bates, G.; Bright, T.P.; Krieg, T.; Carling, D.; Murphy, M.P. Mitochondria-derived ROS activate AMP-activated protein kinase (AMPK) indirectly. J. Biol. Chem. 2018, 293, 17208–17217. [Google Scholar] [CrossRef] [PubMed]
- Zmijewski, J.W.; Banerjee, S.; Bae, H.; Friggeri, A.; Lazarowski, E.R.; Abraham, E. Exposure to Hydrogen Peroxide Induces Oxidation and Activation of AMP-activated Protein Kinase*. J. Biol. Chem. 2010, 285, 33154–33164. [Google Scholar] [CrossRef]
- Shao, D.; Oka, S.; Liu, T.; Zhai, P.; Ago, T.; Sciarretta, S.; Li, H.; Sadoshima, J. A Redox-Dependent Mechanism for Regulation of AMPK Activation by Thioredoxin1 during Energy Starvation. Cell Metab. 2014, 19, 232–245. [Google Scholar] [CrossRef]
- Lim, M.A.; Selak, M.A.; Xiang, Z.; Krainc, D.; Neve, R.L.; Kraemer, B.C.; Watts, J.L.; Kalb, R.G. Reduced Activity of AMP-Activated Protein Kinase Protects against Genetic Models of Motor Neuron Disease. J. Neurosci. 2012, 32, 1123–1141. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-J.; Ju, T.-C.; Chen, H.-M.; Jang, Y.-S.; Lee, L.-M.; Lai, H.-L.; Tai, H.-C.; Fang, J.-M.; Lin, Y.-L.; Tu, P.-H.; et al. Activation of AMP-activated protein kinase α1 mediates mislocalization of TDP-43 in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2015, 24, 787–801. [Google Scholar] [CrossRef]
- Yuan, M.; Yan, R.; Zhang, Y.; Qiu, Y.; Jiang, Z.; Liu, H.; Wang, Y.; Sun, L.; Zhang, H.; Gao, P. CARS senses cysteine deprivation to activate AMPK for cell survival. EMBO J. 2021, 40, e108028. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.; Zhang, Y.; Botchway, B.O.A.; Huang, M.; Lu, Q.; Liu, X. Quercetin activates the Sestrin2/AMPK/SIRT1 axis to improve amyotrophic lateral sclerosis. Biomed. Pharmacother. 2023, 161, 114515. [Google Scholar] [CrossRef]
- Ruderman, N.B.; Julia Xu, X.; Nelson, L.; Cacicedo, J.M.; Saha, A.K.; Lan, F.; Ido, Y. AMPK and SIRT1: A long-standing partnership? Am. J. Physiol. Metab. 2010, 298, E751–E760. [Google Scholar] [CrossRef]
- Yun, Y.C.; Jeong, S.; Kim, S.H.; Cho, G. Reduced sirtuin 1/adenosine monophosphate-activated protein kinase in amyotrophic lateral sclerosis patient-derived mesenchymal stem cells can be restored by resveratrol. J. Tissue Eng. Regen. Med. 2018, 13, 110–115. [Google Scholar] [CrossRef]
- Kefalakes, E.; Böselt, S.; Sarikidi, A.; Ettcheto, M.; Bursch, F.; Naujock, M.; Stanslowsky, N.; Schmuck, M.; Barenys, M.; Wegner, F.; et al. Characterizing the multiple roles of FGF-2 in SOD1 G93A ALS mice in vivo and in vitro. J. Cell. Physiol. 2019, 234, 7395–7410. [Google Scholar] [CrossRef]
- Müller, H.-M.; Steringer, J.P.; Wegehingel, S.; Bleicken, S.; Münster, M.; Dimou, E.; Unger, S.; Weidmann, G.; Andreas, H.; García-Sáez, A.J.; et al. Formation of Disulfide Bridges Drives Oligomerization, Membrane Pore Formation, and Translocation of Fibroblast Growth Factor 2 to Cell Surfaces. J. Biol. Chem. 2015, 290, 8925–8937. [Google Scholar] [CrossRef]
- Thau, N.; Jungnickel, J.; Knippenberg, S.; Ratzka, A.; Dengler, R.; Petri, S.; Grothe, C. Prolonged survival and milder impairment of motor function in the SOD1 ALS mouse model devoid of fibroblast growth factor 2. Neurobiol. Dis. 2012, 47, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Wang, Y.; Cheng, H.; Zhang, Q.; Ge, W.; Guo, D. RedoxDB—A curated database for experimentally verified protein oxidative modification. Bioinformatics 2012, 28, 2551–2552. [Google Scholar] [CrossRef]
- Li, Z.; Li, S.; Luo, M.; Jhong, J.-H.; Li, W.; Yao, L.; Pang, Y.; Wang, Z.; Wang, R.; Ma, R.; et al. dbPTM in 2022: An updated database for exploring regulatory networks and functional associations of protein post-translational modifications. Nucleic Acids Res. 2022, 50, D471–D479. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhang, Q.; Li, S.; Cheng, B.; Xue, H.; Wei, Z.; Shao, T.; Liu, Z.-X.; Cheng, H.; Wang, Z. iCysMod: An integrative database for protein cysteine modifications in eukaryotes. Brief. Bioinform. 2021, 22, bbaa400. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Perluigi, M. Redox Proteomics: A Key Tool for New Insights into Protein Modification with Relevance to Disease. Antioxid. Redox Signal. 2017, 26, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Cadenas-Garrido, P.; Schonvandt-Alarcos, A.; Herrera-Quintana, L.; Vázquez-Lorente, H.; Santamaría-Quiles, A.; Ruiz de Francisco, J.; Moya-Escudero, M.; Martín-Oliva, D.; Martín-Guerrero, S.M.; Rodríguez-Santana, C.; et al. Using Redox Proteomics to Gain New Insights into Neurodegenerative Disease and Protein Modification. Antioxidants 2024, 13, 127. [Google Scholar] [CrossRef]
- Gu, L.; Robinson, R.A.S. Proteomic approaches to quantify cysteine reversible modifications in aging and neurodegenerative diseases. PROTEOMICS–Clin. Appl. 2016, 10, 1159–1177. [Google Scholar] [CrossRef]
- Zhong, Q.; Xiao, X.; Qiu, Y.; Xu, Z.; Chen, C.; Chong, B.; Zhao, X.; Hai, S.; Li, S.; An, Z.; et al. Protein posttranslational modifications in health and diseases: Functions, regulatory mechanisms, and therapeutic implications. Med. Commun. 2023, 4, e261. [Google Scholar] [CrossRef]
- Rani, N.; Sahu, M.; Ambasta, R.K.; Kumar, P. Triaging between post-translational modification of cell cycle regulators and their therapeutics in neurodegenerative diseases. Ageing Res. Rev. 2024, 94, 102174. [Google Scholar] [CrossRef] [PubMed]
- Paterniti, I.; Cordaro, M.; Campolo, M.; Siracusa, R.; Cornelius, C.; Navarra, M.; Cuzzocrea, S.; Esposito, E. Neuroprotection by Association of Palmitoylethanolamide with Luteolin in Experimental Alzheimer’s Disease Models: The Control of Neuroinflammation. CNS Neurol. Disord.-Drug Targets 2014, 13, 1530–1541. [Google Scholar] [CrossRef] [PubMed]
- Assogna, M.; Casula, E.P.; Borghi, I.; Bonnì, S.; Samà, D.; Motta, C.; Di Lorenzo, F.; D’Acunto, A.; Porrazzini, F.; Minei, M.; et al. Effects of Palmitoylethanolamide Combined with Luteoline on Frontal Lobe Functions, High Frequency Oscillations, and GABAergic Transmission in Patients with Frontotemporal Dementia. J. Alzheimer’s Dis. 2020, 76, 1297–1308. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| PTM | Protein | NDD |
|---|---|---|
| ROS-mediated | ||
| Disulfide bond | SOD1 | ALS [16] |
| Tau | AD [17] | |
| TDP43 | ALS, FTD [18] | |
| Sulfinylation and sulfonation | DJ-1 (PARK7) | PD [19] |
| Parkin | PD [20] | |
| SOD1 | ALS [21] | |
| TTR | MS [22] | |
| RNS-mediated | ||
| S-nitrosylation | CDK5 | AD [23] |
| DJ-1 | PD [24,25] | |
| DRP1 | AD [26], HD [27] | |
| GAPDH | HD [28], PD [29] | |
| GOSPEL | PD [30] | |
| Parkin | PD [31] | |
| PDI | ALS [32] | |
| PRX2 | PD [33] | |
| UCHL1 | PD [34] | |
| XIAP | PD [35] | |
| RSS-mediated | ||
| S-sulfhydration (persulfidation) | Akt | AD [36] |
| GSK3β | AD [37] | |
| Parkin | PD [38] | |
| TTR | MS [22,39] | |
| Sirt1 | PD [40] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Percio, A.; Cicchinelli, M.; Masci, D.; Summo, M.; Urbani, A.; Greco, V. Oxidative Cysteine Post Translational Modifications Drive the Redox Code Underlying Neurodegeneration and Amyotrophic Lateral Sclerosis. Antioxidants 2024, 13, 883. https://doi.org/10.3390/antiox13080883
Percio A, Cicchinelli M, Masci D, Summo M, Urbani A, Greco V. Oxidative Cysteine Post Translational Modifications Drive the Redox Code Underlying Neurodegeneration and Amyotrophic Lateral Sclerosis. Antioxidants. 2024; 13(8):883. https://doi.org/10.3390/antiox13080883
Chicago/Turabian StylePercio, Anna, Michela Cicchinelli, Domiziana Masci, Mariagrazia Summo, Andrea Urbani, and Viviana Greco. 2024. "Oxidative Cysteine Post Translational Modifications Drive the Redox Code Underlying Neurodegeneration and Amyotrophic Lateral Sclerosis" Antioxidants 13, no. 8: 883. https://doi.org/10.3390/antiox13080883