

Downregulation of Iron–Sulfur Cluster Biogenesis May Contribute to Hyperglycemia-Mediated Diabetic Peripheral Neuropathy in Murine Models
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Glucose and Insulin Tolerance Tests
2.3. Hot Plate Test
2.4. Rotarod Test
2.5. Beam-Walk Test
2.6. Electron Microscopy
2.7. Histological Assays
2.8. Ferrozine Iron Assays
2.9. Labile Iron Pool (LIP) Measurements
2.10. The Enzymatic Activities of Aconitase, Complexes I and II, and Mitochondrial Membrane Potential (MMP) Levels
2.11. ROS Detection Assays
2.12. Western Blot Assays
2.13. Quantitative Real-Time PCR
2.14. Statistics Analysis
3. Results
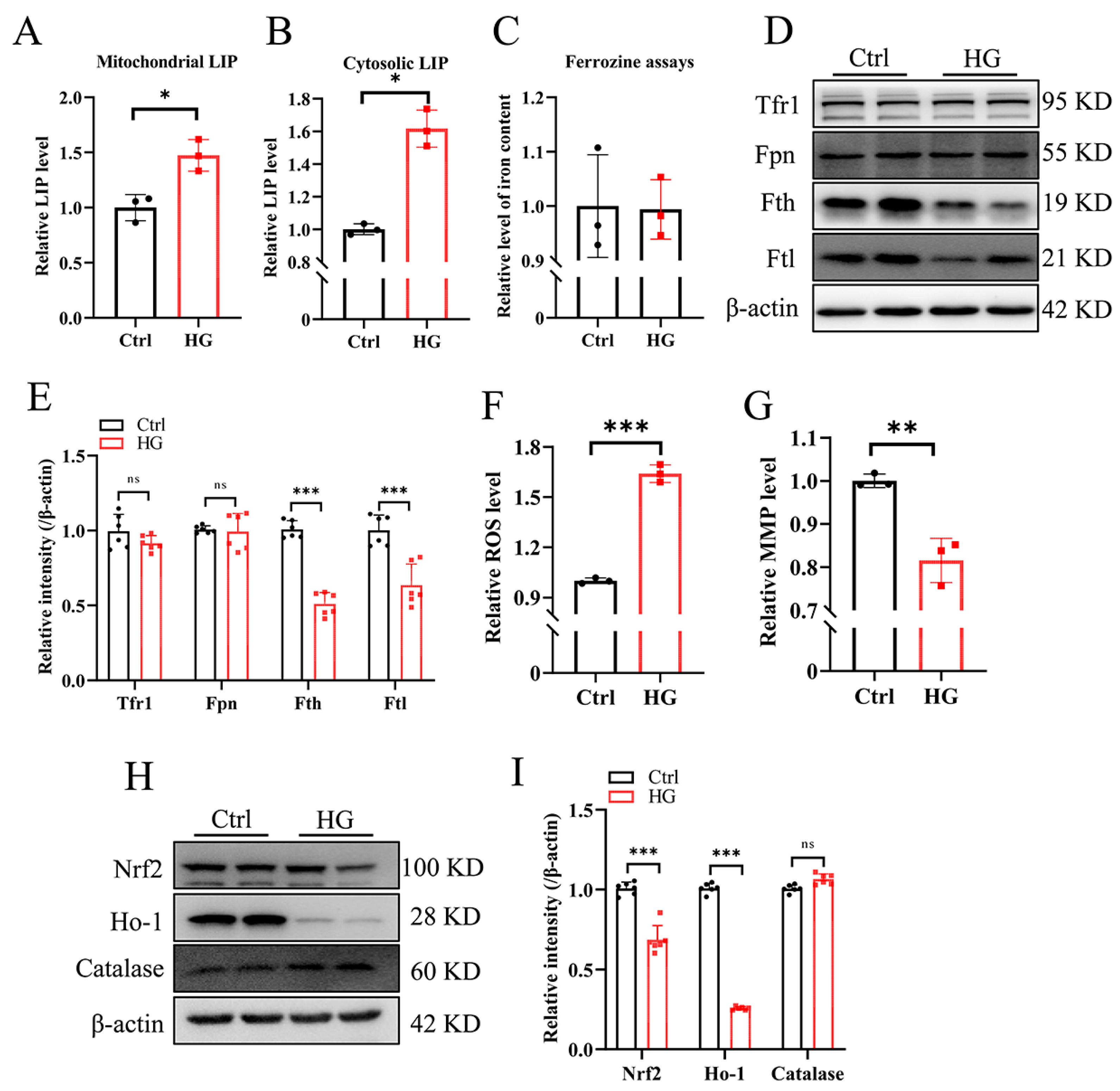
3.1. High Glucose Downregulates Fe-S-Cluster Biogenesis and Fe-S-Containing Enzymatic Activities in RSC96 Cells
3.2. High-Glucose Treatment Increases the Cellular Labile Iron Pool and Promotes ROS Production in RSC96 Cells
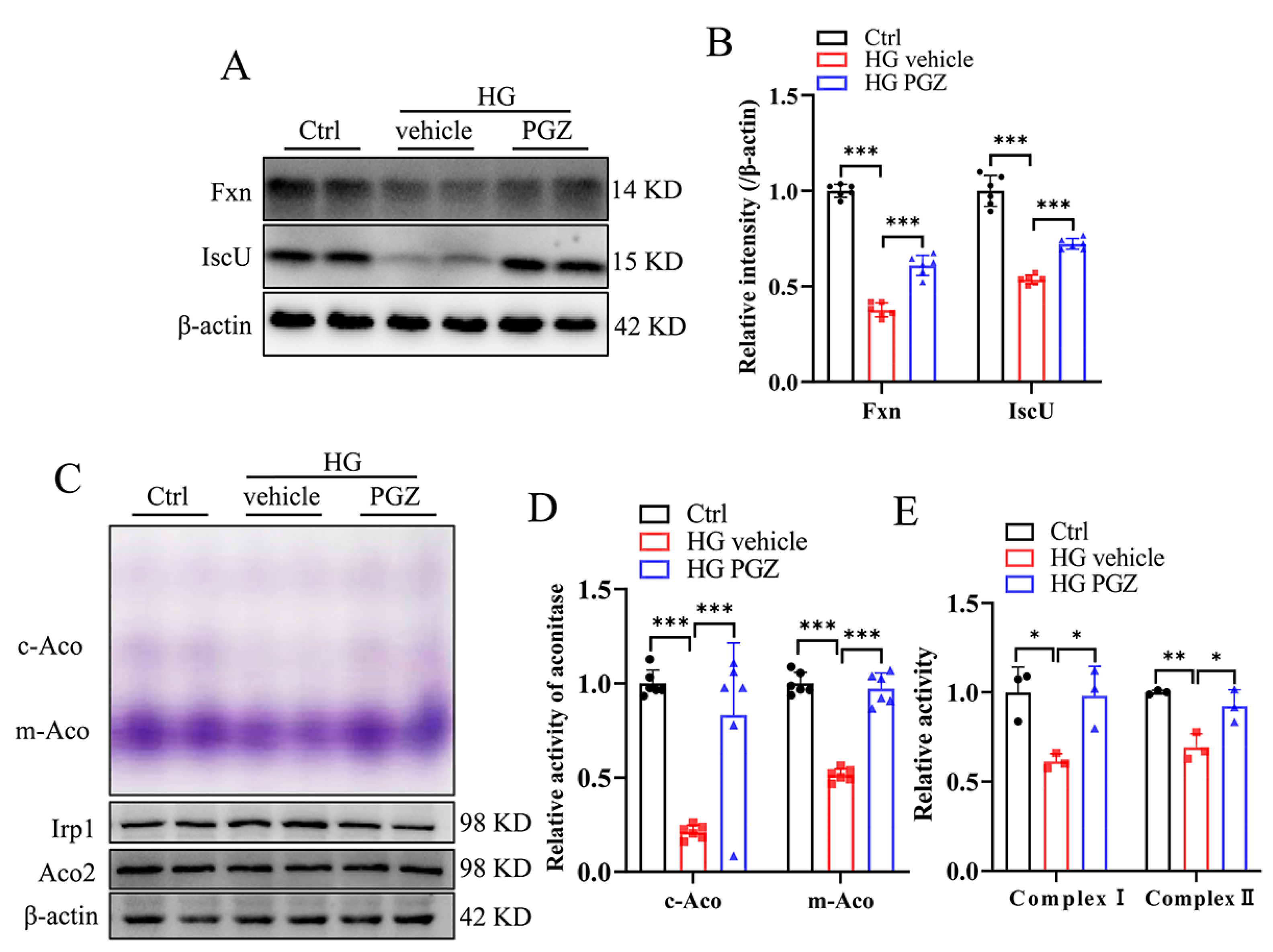
3.3. Pioglitazone Treatment Increases the Expression of Genes Involved in the Biosynthesis of Fe-S Clusters and Recovers Iron Homeostasis and Redox Balance in RSC96 Cells
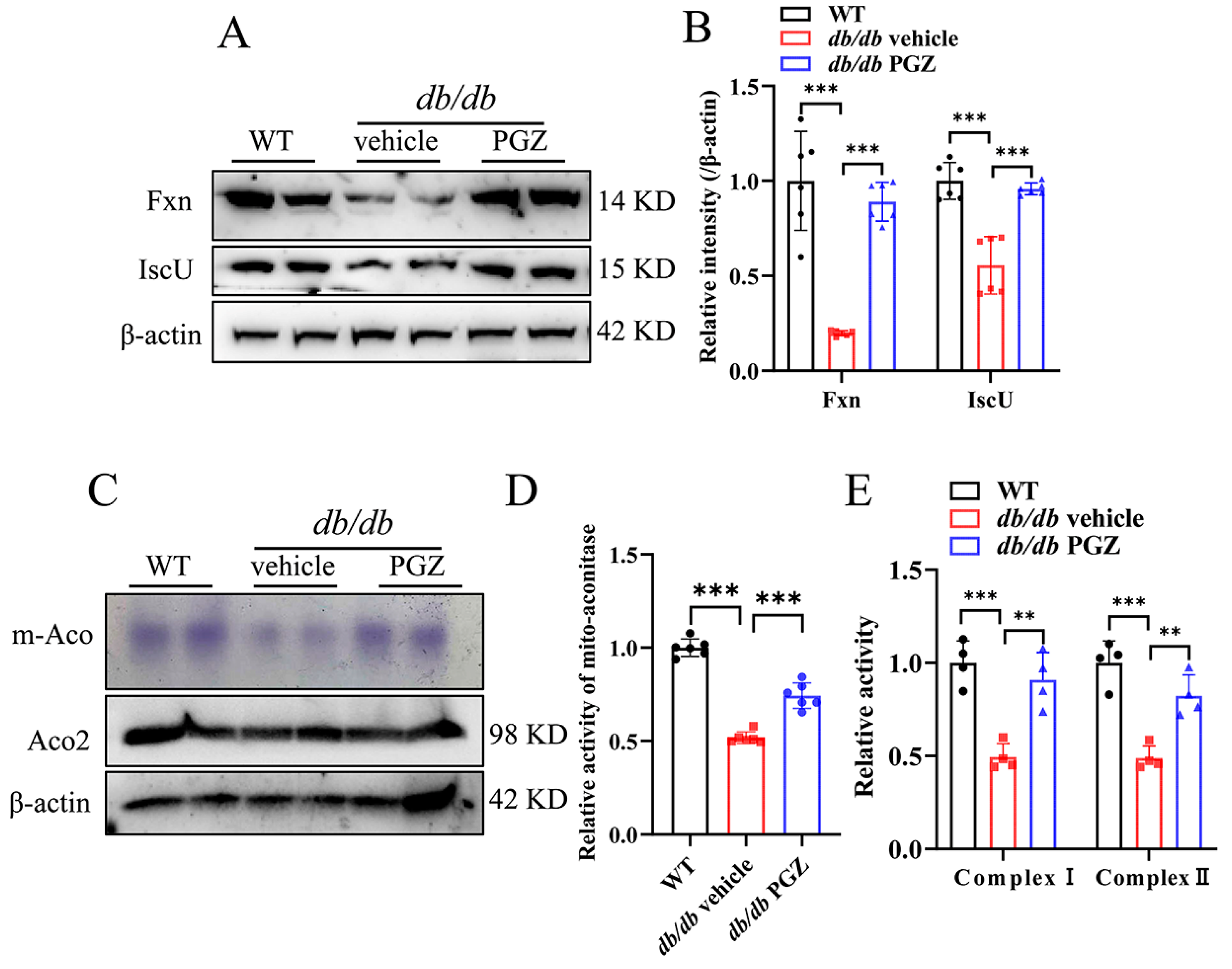
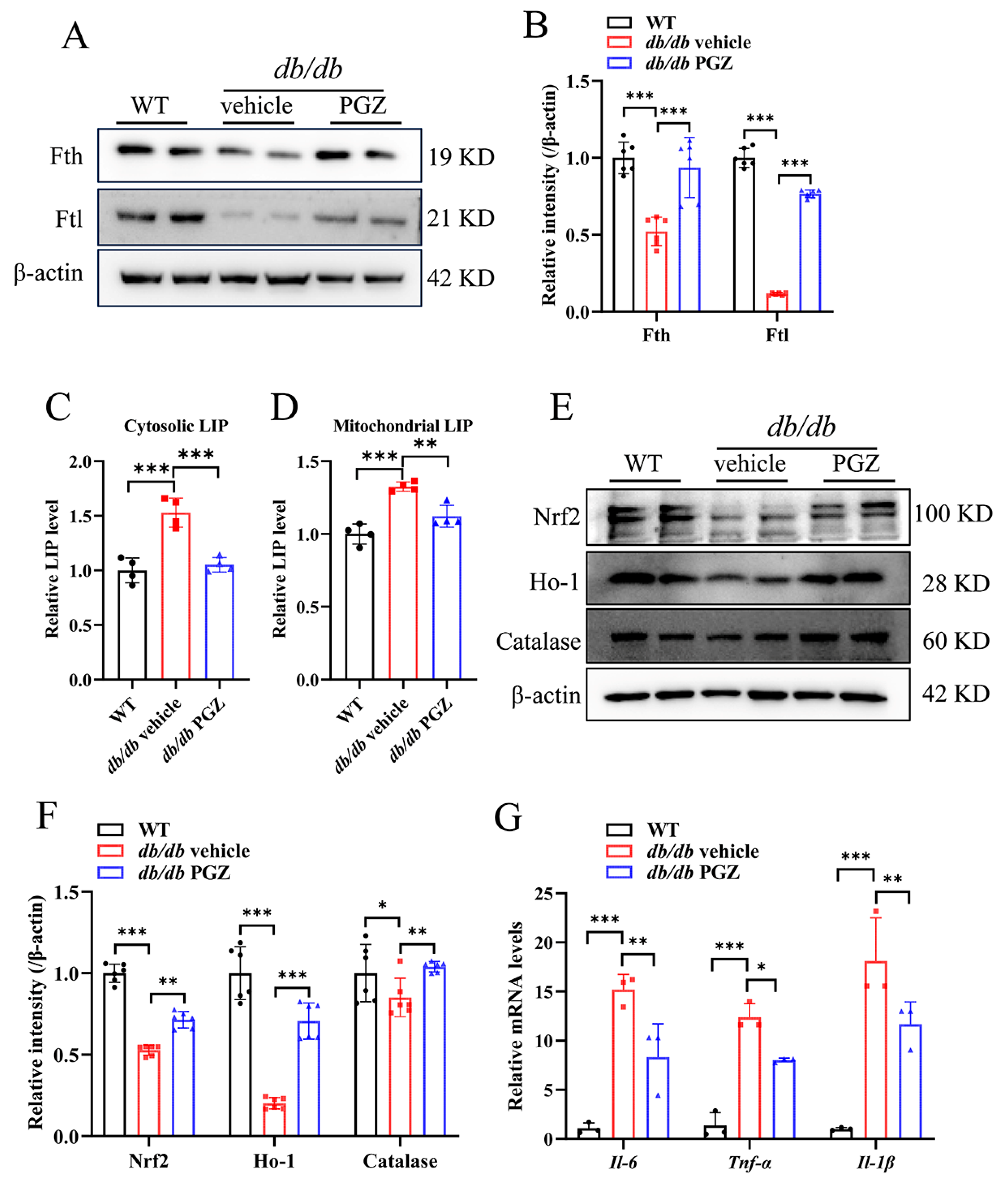
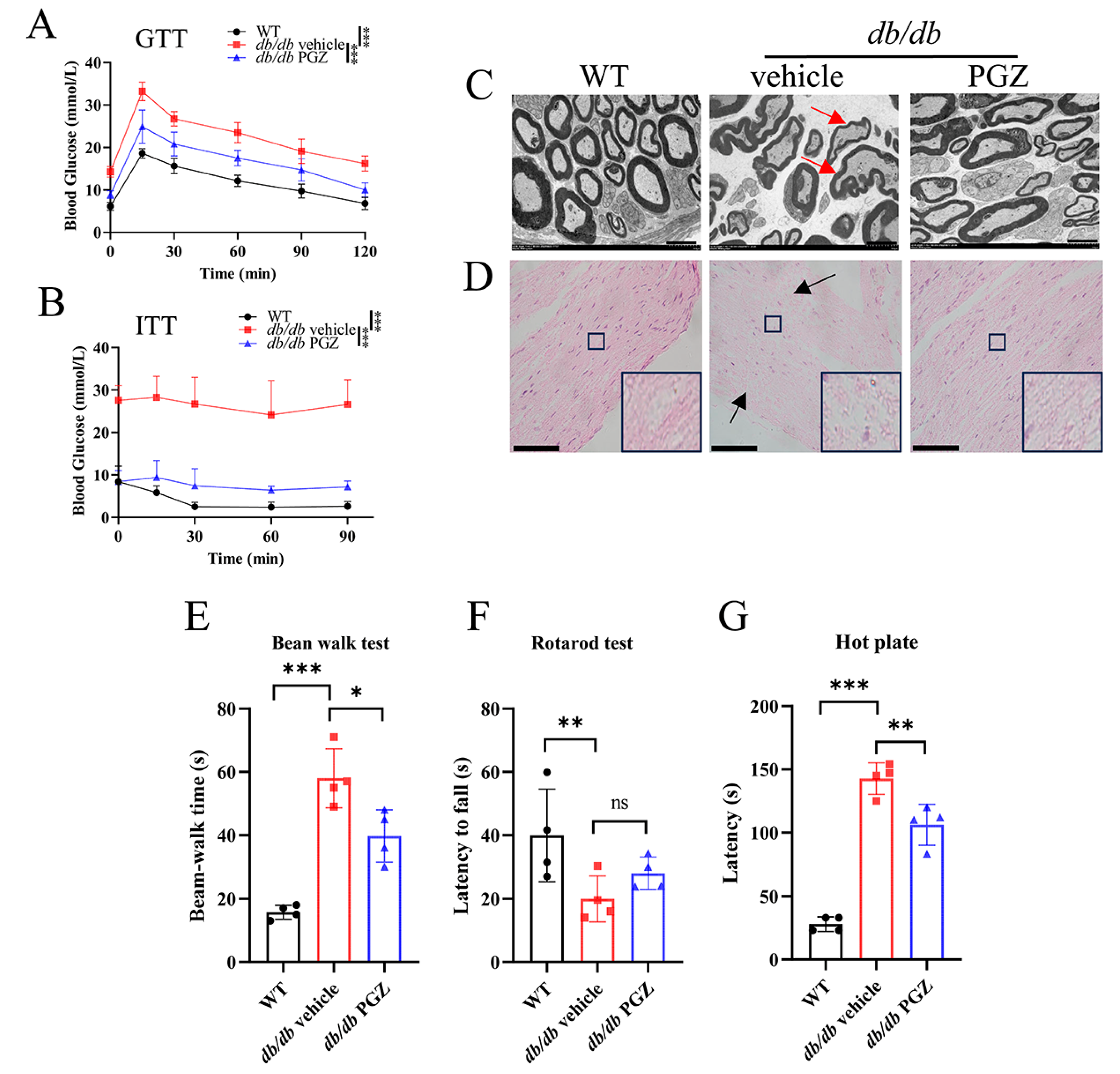
3.4. Rescue of Fe-S Cluster Biosynthesis by Pioglitazone Treatment Correlates Well with the Increase in the Enzymatic Activities of Fe-S Protein and with the Improvement of Iron Homeostasis and Inflammation in Sciatic Nerve
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Elafros, M.A.; Andersen, H.; Bennett, D.L.; Savelieff, M.G.; Viswanathan, V.; Callaghan, B.C.; Feldman, E.L. Towards prevention of diabetic peripheral neuropathy: Clinical presentation, pathogenesis, and new treatments. Lancet Neurol. 2022, 21, 922–936. [Google Scholar] [CrossRef] [PubMed]
- Selvarajah, D.; Kar, D.; Khunti, K.; Davies, M.J.; Scott, A.R.; Walker, J.; Tesfaye, S. Diabetic peripheral neuropathy: Advances in diagnosis and strategies for screening and early intervention. Lancet Diabetes Endocrinol. 2019, 7, 938–948. [Google Scholar] [CrossRef] [PubMed]
- Majd, H.; Amin, S.; Ghazizadeh, Z.; Cesiulis, A.; Arroyo, E.; Lankford, K.; Majd, A.; Farahvashi, S.; Chemel, A.K.; Okoye, M.; et al. Deriving Schwann cells from hPSCs enables disease modeling and drug discovery for diabetic peripheral neuropathy. Cell Stem Cell 2023, 30, 632–647.e10. [Google Scholar] [CrossRef]
- Li, K. Iron Pathophysiology in Friedreich’s Ataxia. Adv. Exp. Med. Biol. 2019, 1173, 125–143. [Google Scholar] [CrossRef] [PubMed]
- Lill, R.; Freibert, S.A. Mechanisms of Mitochondrial Iron-Sulfur Protein Biogenesis. Annu. Rev. Biochem. 2020, 89, 471–499. [Google Scholar] [CrossRef]
- Mochel, F.; Knight, M.A.; Tong, W.H.; Hernandez, D.; Ayyad, K.; Taivassalo, T.; Andersen, P.M.; Singleton, A.; Rouault, T.A.; Fischbeck, K.H.; et al. Splice mutation in the iron-sulfur cluster scaffold protein ISCU causes myopathy with exercise intolerance. Am. J. Hum. Genet. 2008, 82, 652–660. [Google Scholar] [CrossRef]
- Obis, È.; Irazusta, V.; Sanchís, D.; Ros, J.; Tamarit, J. Frataxin deficiency in neonatal rat ventricular myocytes targets mitochondria and lipid metabolism. Free Radic. Biol. Med. 2014, 73, 21–33. [Google Scholar] [CrossRef]
- Wong, A.; Yang, J.; Cavadini, P.; Gellera, C.; Lonnerdal, B.; Taroni, F.; Cortopassi, G. The Friedreich’s ataxia mutation confers cellular sensitivity to oxidant stress which is rescued by chelators of iron and calcium and inhibitors of apoptosis. Hum. Mol. Genet. 1999, 8, 425–430. [Google Scholar] [CrossRef]
- Chantrel-Groussard, K.; Geromel, V.; Puccio, H.; Koenig, M.; Munnich, A.; Rötig, A.; Rustin, P. Disabled early recruitment of antioxidant defenses in Friedreich’s ataxia. Hum. Mol. Genet. 2001, 10, 2061–2067. [Google Scholar] [CrossRef]
- Koeppen, A.H.; Mazurkiewicz, J.E. Friedreich ataxia: Neuropathology revised. J. Neuropathol. Exp. Neurol. 2013, 72, 78–90. [Google Scholar] [CrossRef]
- Koeppen, A.H.; Morral, J.A.; Davis, A.N.; Qian, J.; Petrocine, S.V.; Knutson, M.D.; Gibson, W.M.; Cusack, M.J.; Li, D. The dorsal root ganglion in Friedreich’s ataxia. Acta Neuropathol. 2009, 118, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Rosenblatt, S.; Miskin, B.; Glazer, N.B.; Prince, M.J.; Robertson, K.E. The impact of pioglitazone on glycemic control and atherogenic dyslipidemia in patients with type 2 diabetes mellitus. Coron. Artery Dis. 2001, 12, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Poupon-Bejuit, L.; Rocha-Ferreira, E.; Thornton, C.; Hagberg, H.; Rahim, A.A. Neuroprotective Effects of Diabetes Drugs for the Treatment of Neonatal Hypoxia-Ischemia Encephalopathy. Front. Cell. Neurosci. 2020, 14, 112. [Google Scholar] [CrossRef]
- Kuate Defo, A.; Bakula, V.; Pisaturo, A.; Labos, C.; Wing, S.S.; Daskalopoulou, S.S. Diabetes, antidiabetic medications and risk of dementia: A systematic umbrella review and meta-analysis. Diabetes Obes. Metab. 2024, 26, 441–462. [Google Scholar] [CrossRef] [PubMed]
- Fadini, G.P.; Longato, E.; Morieri, M.L.; Del Prato, S.; Avogaro, A.; Solini, A. Long-term benefits of dapagliflozin on renal outcomes of type 2 diabetes under routine care: A comparative effectiveness study on propensity score matched cohorts at low renal risk. Lancet Reg. Health Eur. 2024, 38, 100847. [Google Scholar] [CrossRef]
- Sun, Y.G.; Zhao, Z.Q.; Meng, X.L.; Yin, J.; Liu, X.Y.; Chen, Z.F. Cellular basis of itch sensation. Science 2009, 325, 1531–1534. [Google Scholar] [CrossRef]
- Liu, Y.; Cai, J.; Shen, J.; Dong, W.; Xu, L.; Fang, M.; Lin, Y.; Liu, J.; Ding, Y.; Qiao, T.; et al. SS-31 efficacy in a mouse model of Friedreich ataxia by upregulation of frataxin expression. Hum. Mol. Genet. 2021, 31, 176–188. [Google Scholar] [CrossRef]
- Xu, L.; Sun, Z.; Xing, Z.; Liu, Y.; Zhao, H.; Tang, Z.; Luo, Y.; Hao, S.; Li, K. Cur@SF NPs alleviate Friedreich’s ataxia in a mouse model through synergistic iron chelation and antioxidation. J. Nanobiotechnol. 2022, 20, 118. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhao, H.; Hao, S.; Shang, L.; Wu, J.; Song, C.; Meyron-Holtz, E.G.; Qiao, T.; Li, K. Iron regulatory protein deficiency compromises mitochondrial function in murine embryonic fibroblasts. Sci. Rep. 2018, 8, 5118. [Google Scholar] [CrossRef]
- Li, H.; Liu, Y.; Shang, L.; Cai, J.; Wu, J.; Zhang, W.; Pu, X.; Dong, W.; Qiao, T.; Li, K. Iron regulatory protein 2 modulates the switch from aerobic glycolysis to oxidative phosphorylation in mouse embryonic fibroblasts. Proc. Natl. Acad. Sci. USA 2019, 116, 9871–9876. [Google Scholar] [CrossRef]
- Yin, Y.; Qu, H.; Yang, Q.; Fang, Z.; Gao, R. Astragaloside IV alleviates Schwann cell injury in diabetic peripheral neuropathy by regulating microRNA-155-mediated autophagy. Phytomedicine 2021, 92, 153749. [Google Scholar] [CrossRef]
- Lin, Q.; Li, K.; Chen, Y.; Xie, J.; Wu, C.; Cui, C.; Deng, B. Oxidative Stress in Diabetic Peripheral Neuropathy: Pathway and Mechanism-Based Treatment. Mol. Neurobiol. 2023, 60, 4574–4594. [Google Scholar] [CrossRef]
- Rodríguez-Pascau, L.; Britti, E.; Calap-Quintana, P.; Dong, Y.N.; Vergara, C.; Delaspre, F.; Medina-Carbonero, M.; Tamarit, J.; Pallardó, F.V.; Gonzalez-Cabo, P.; et al. PPAR gamma agonist leriglitazone improves frataxin-loss impairments in cellular and animal models of Friedreich Ataxia. Neurobiol. Dis. 2021, 148, 105162. [Google Scholar] [CrossRef]
- Marmolino, D.; Acquaviva, F.; Pinelli, M.; Monticelli, A.; Castaldo, I.; Filla, A.; Cocozza, S. PPAR-gamma agonist Azelaoyl PAF increases frataxin protein and mRNA expression: New implications for the Friedreich’s ataxia therapy. Cerebellum 2009, 8, 98–103. [Google Scholar] [CrossRef]
- Kawasaki, F.; Matsuda, M.; Kanda, Y.; Inoue, H.; Kaku, K. Structural and functional analysis of pancreatic islets preserved by pioglitazone in db/db mice. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E510–E518. [Google Scholar] [CrossRef]
- Hinder, L.M.; Park, M.; Rumora, A.E.; Hur, J.; Eichinger, F.; Pennathur, S.; Kretzler, M.; Brosius, F.C., 3rd; Feldman, E.L. Comparative RNA-Seq transcriptome analyses reveal distinct metabolic pathways in diabetic nerve and kidney disease. J. Cell. Mol. Med. 2017, 21, 2140–2152. [Google Scholar] [CrossRef]
- Mert, T.; Sahin, E.; Yaman, S.; Sahin, M. Effects of immune cell-targeted treatments result from the suppression of neuronal oxidative stress and inflammation in experimental diabetic rats. Naunyn Schmiedebergs Arch. Pharmacol. 2020, 393, 1293–1302. [Google Scholar] [CrossRef]
- Cashman, C.R.; Höke, A. Mechanisms of distal axonal degeneration in peripheral neuropathies. Neurosci. Lett. 2015, 596, 33–50. [Google Scholar] [CrossRef]
- Lennertz, R.C.; Medler, K.A.; Bain, J.L.; Wright, D.E.; Stucky, C.L. Impaired sensory nerve function and axon morphology in mice with diabetic neuropathy. J. Neurophysiol. 2011, 106, 905–914. [Google Scholar] [CrossRef]
- Xu, X.F.; Zhang, D.D.; Liao, J.C.; Xiao, L.; Wang, Q.; Qiu, W. Galanin and its receptor system promote the repair of injured sciatic nerves in diabetic rats. Neural Regen. Res. 2016, 11, 1517–1526. [Google Scholar] [CrossRef]
- Guo, D.; Lu, X.; Xu, X.; Gou, H.; Wang, Z.; Cao, Y.; Luo, X. Therapeutic Effect of Vinorine on Sciatic Nerve Injured Rat. Neurochem. Res. 2018, 43, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Rolim, L.C.; da Silva, E.M.; Flumignan, R.L.; Abreu, M.M.; Dib, S.A. Acetyl-L-carnitine for the treatment of diabetic peripheral neuropathy. Cochrane Database Syst. Rev. 2019, 6, Cd011265. [Google Scholar] [CrossRef]
- Cnop, M.; Mulder, H.; Igoillo-Esteve, M. Diabetes in Friedreich ataxia. J. Neurochem. 2013, 126 (Suppl. S1), 94–102. [Google Scholar] [CrossRef]
- Turchi, R.; Tortolici, F.; Guidobaldi, G.; Iacovelli, F.; Falconi, M.; Rufini, S.; Faraonio, R.; Casagrande, V.; Federici, M.; De Angelis, L.; et al. Frataxin deficiency induces lipid accumulation and affects thermogenesis in brown adipose tissue. Cell Death Dis. 2020, 11, 51. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Inzucchi, S.; Abdul-Ghani, M.; Nissen, S.E. Pioglitazone: The forgotten, cost-effective cardioprotective drug for type 2 diabetes. Diabetes Vasc. Dis. Res. 2019, 16, 133–143. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, L.; Huang, F.; Sun, Z.; Zhang, J.; Xia, S.; Zhao, H.; Liu, Y.; Yang, L.; Ding, Y.; Bian, D.; et al. Downregulation of Iron–Sulfur Cluster Biogenesis May Contribute to Hyperglycemia-Mediated Diabetic Peripheral Neuropathy in Murine Models. Antioxidants 2024, 13, 1036. https://doi.org/10.3390/antiox13091036
Wu L, Huang F, Sun Z, Zhang J, Xia S, Zhao H, Liu Y, Yang L, Ding Y, Bian D, et al. Downregulation of Iron–Sulfur Cluster Biogenesis May Contribute to Hyperglycemia-Mediated Diabetic Peripheral Neuropathy in Murine Models. Antioxidants. 2024; 13(9):1036. https://doi.org/10.3390/antiox13091036
Chicago/Turabian StyleWu, Lin, Fei Huang, Zichen Sun, Jinghua Zhang, Siyu Xia, Hongting Zhao, Yutong Liu, Lu Yang, Yibing Ding, Dezhi Bian, and et al. 2024. "Downregulation of Iron–Sulfur Cluster Biogenesis May Contribute to Hyperglycemia-Mediated Diabetic Peripheral Neuropathy in Murine Models" Antioxidants 13, no. 9: 1036. https://doi.org/10.3390/antiox13091036