

Unraveling the Protective Role of Oleocanthal and Its Oxidation Product, Oleocanthalic Acid, against Neuroinflammation

 , , ,
, , ,  , , , ,
, , , ,  ,
,  , and
, and 
Abstract

1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Culture and Treatments
2.3. Cell Viability Assay
2.4. NO Production
2.5. DCFH-DA Assay
2.6. MCB Assay
2.7. Real-Time Polymerase Chain Reaction (PCR)
2.8. Preparation of Nuclear and Cytosolic Fractions
2.9. Western Immunoblotting
2.10. TLR4 Surface Expression
2.11. CD14 Surface Expression
2.12. BV-2 Phagocytic Activity
2.13. Molecular Docking Studies
2.14. Proteomic Analysis
2.15. Spot Digestion and Protein Identification
2.16. Bioinformatic Analysis
2.17. Statistical Analysis
3. Results
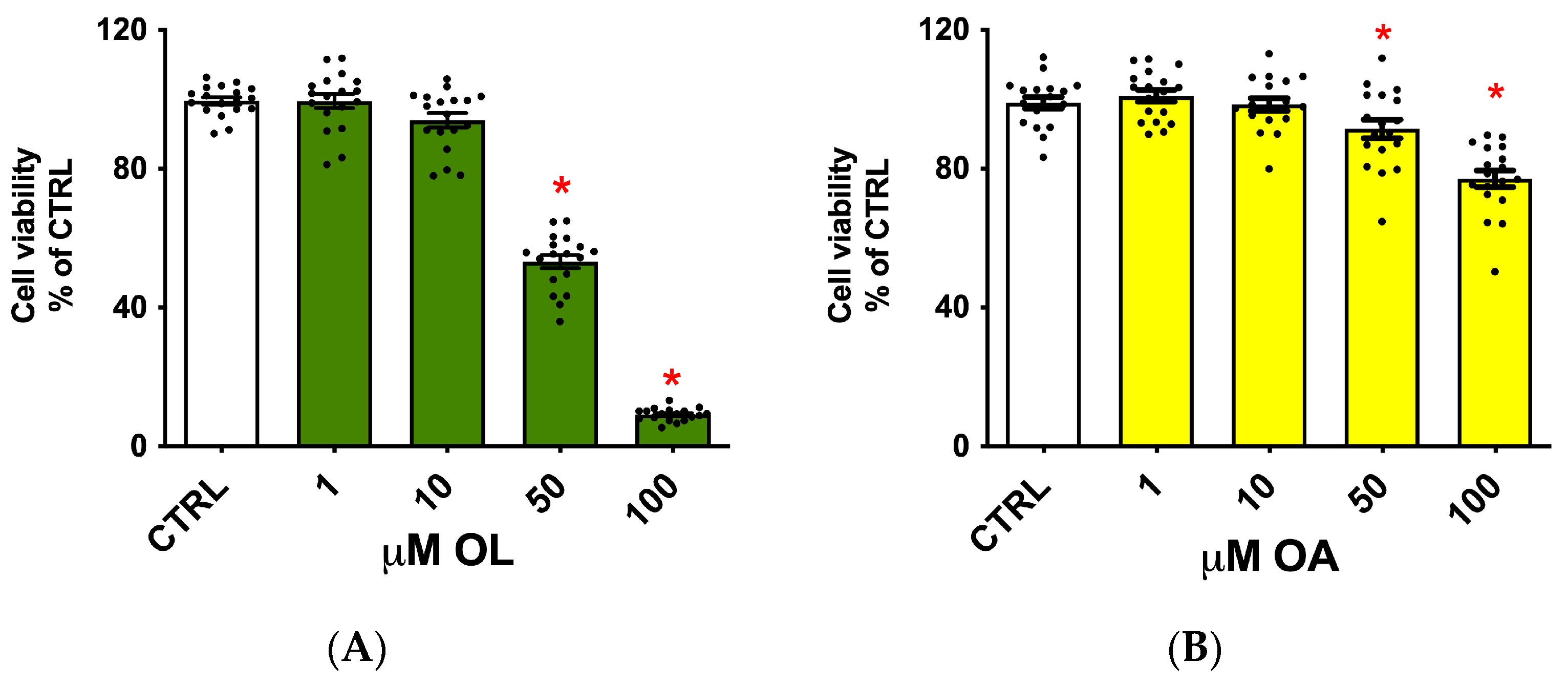
3.1. Evaluation of the Oleocanthal and Oleocanthalic Acid Potential Cytotoxicity
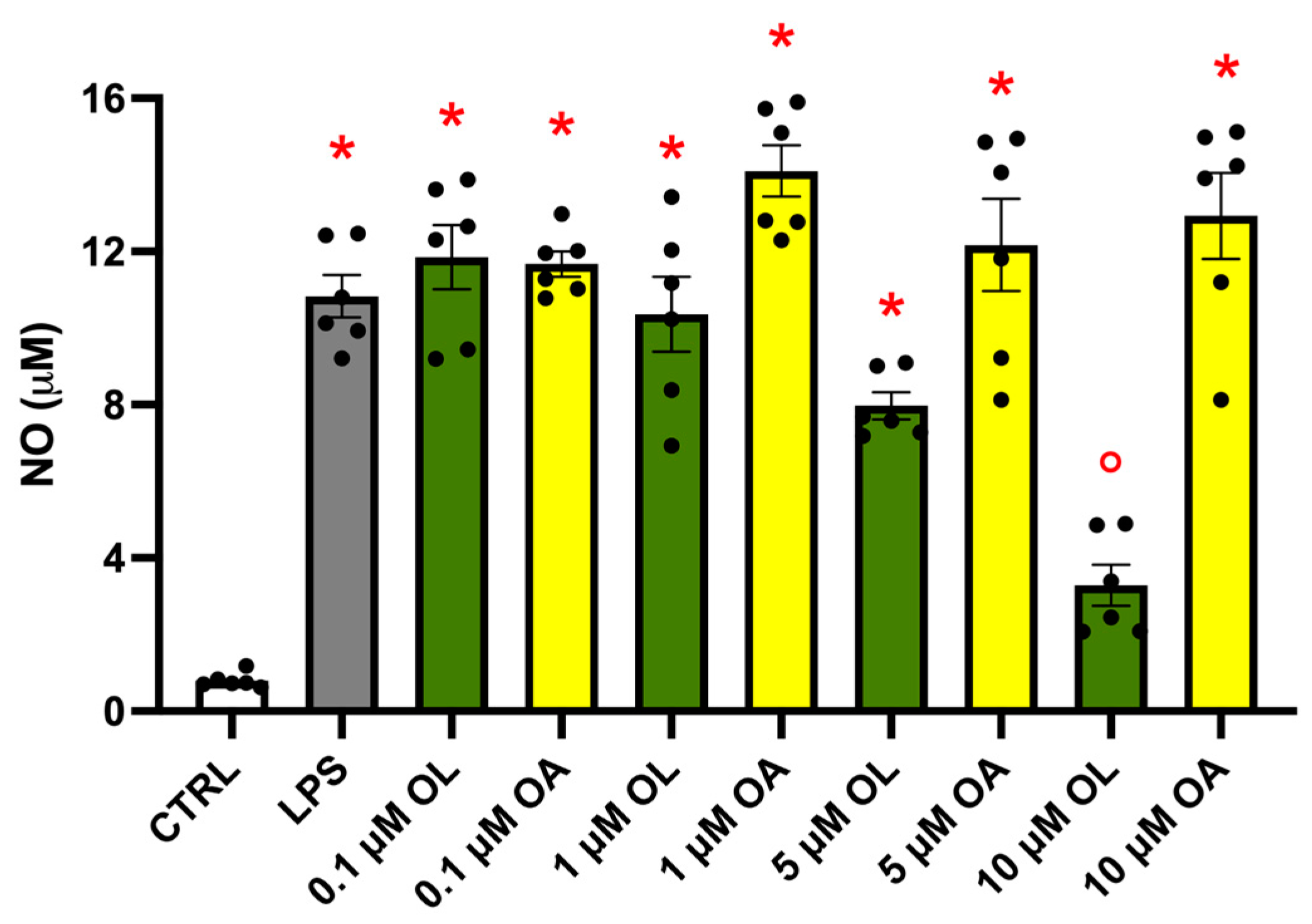
3.2. Effect of OL and OA Treatment on NO Production in LPS-Activated BV-2 Cells
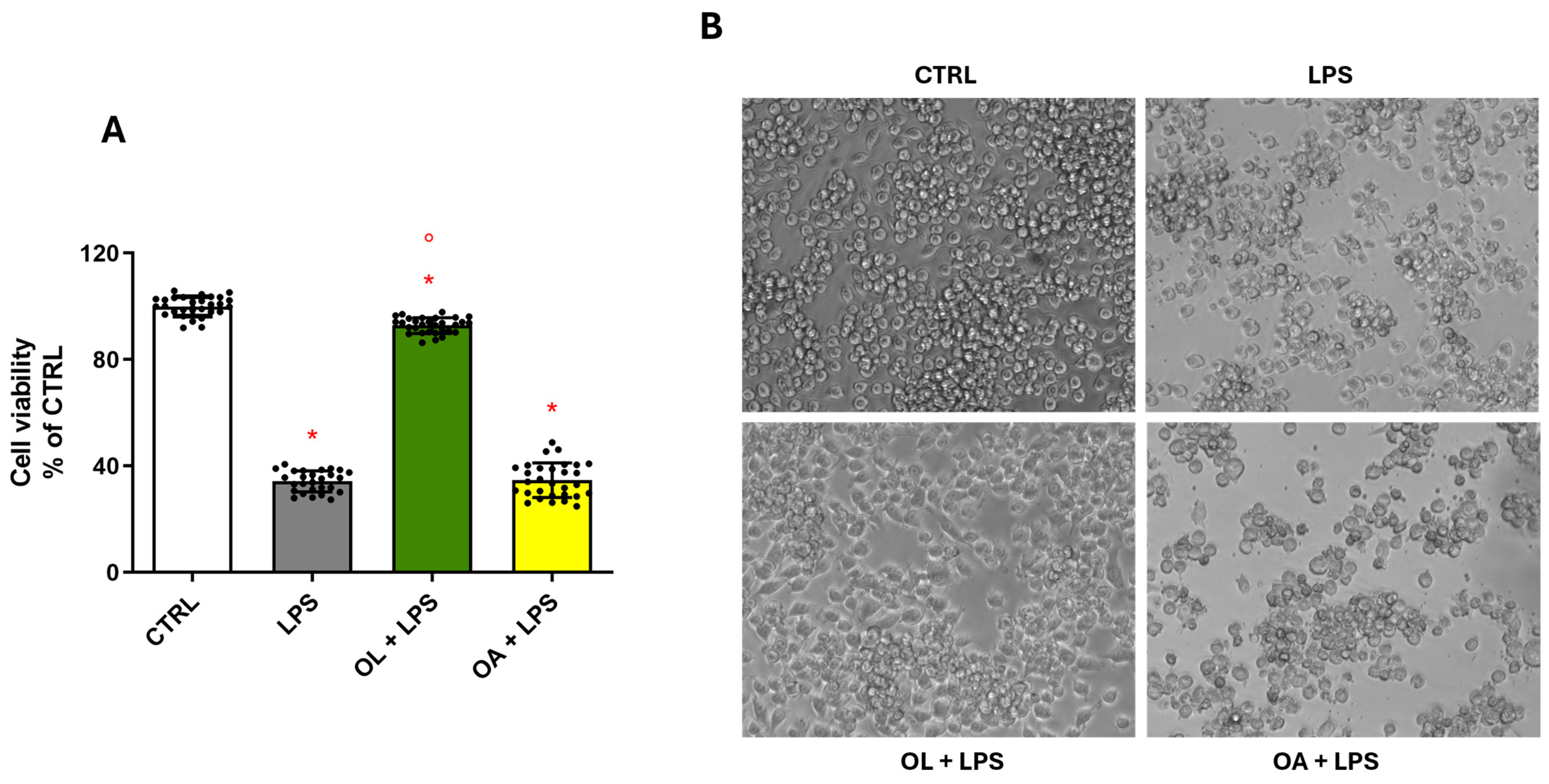
3.3. OL Counteracts LPS-Induced Damage in BV-2 Cells
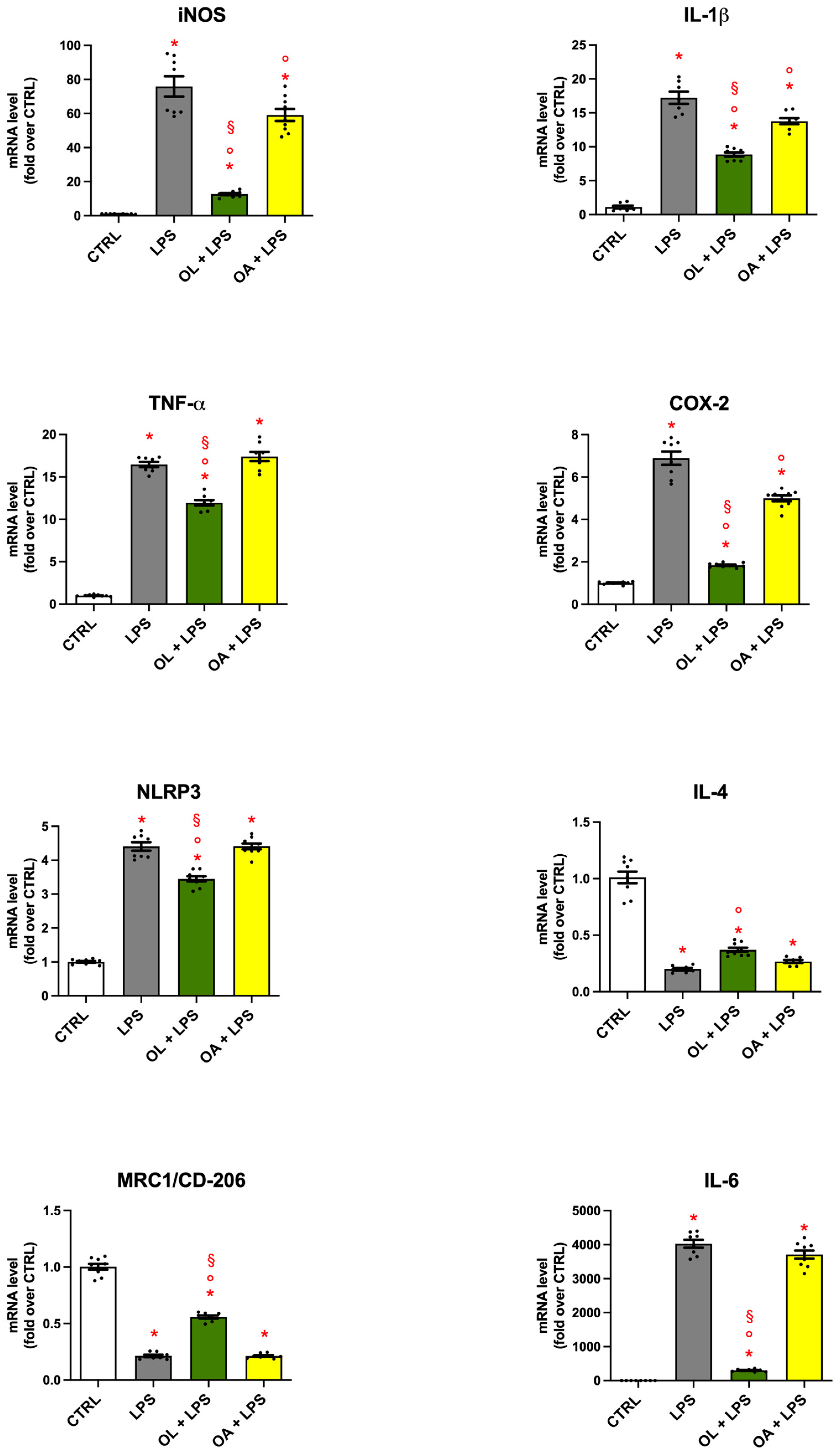
3.4. Gene Expression of Pro- and Anti-Inflammatory Mediators in BV-2 Cells Treated with OL and OA
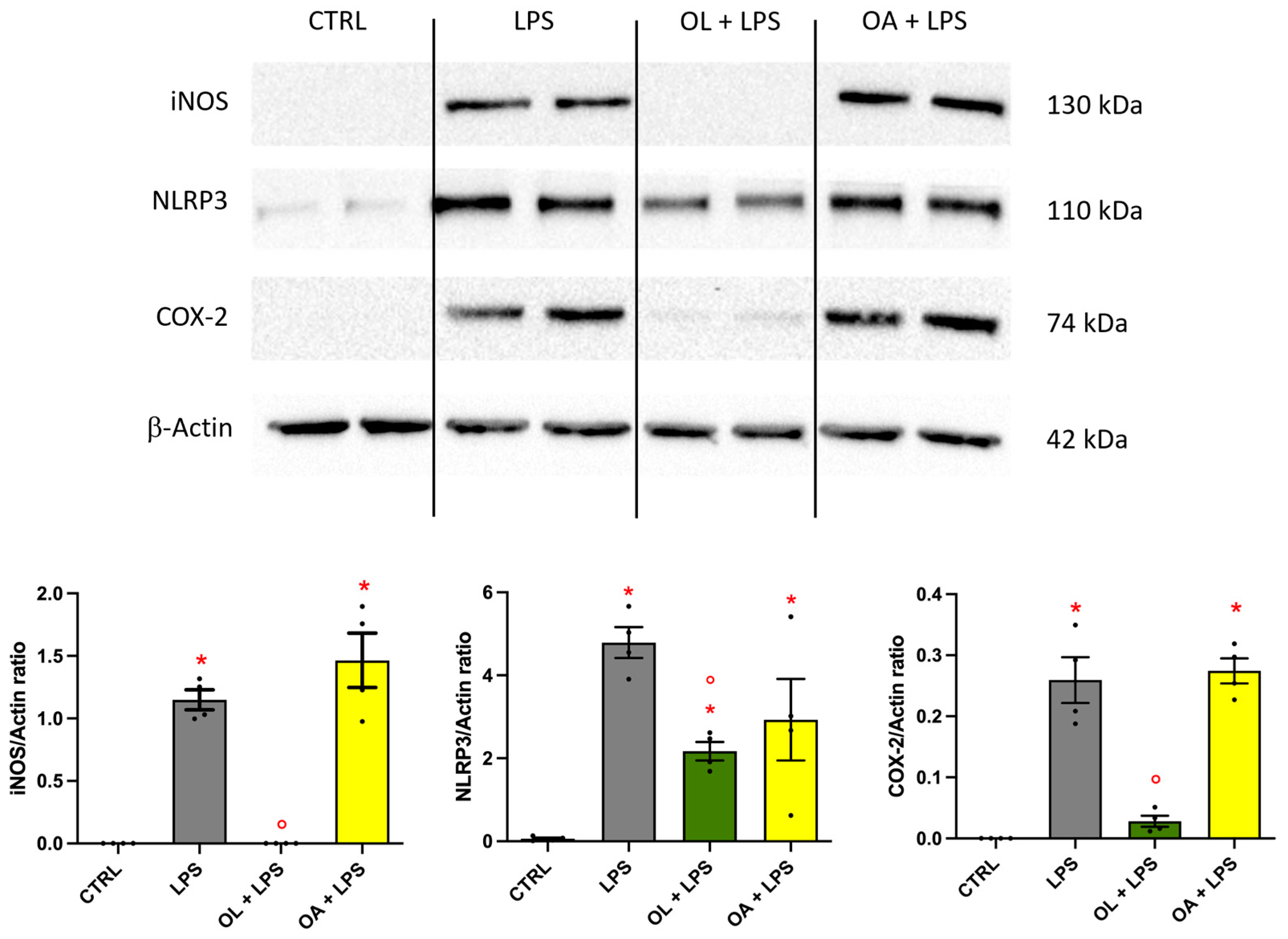
3.5. Oleocanthal Effect on iNOS, NLRP3, and COX-2 Protein Expression
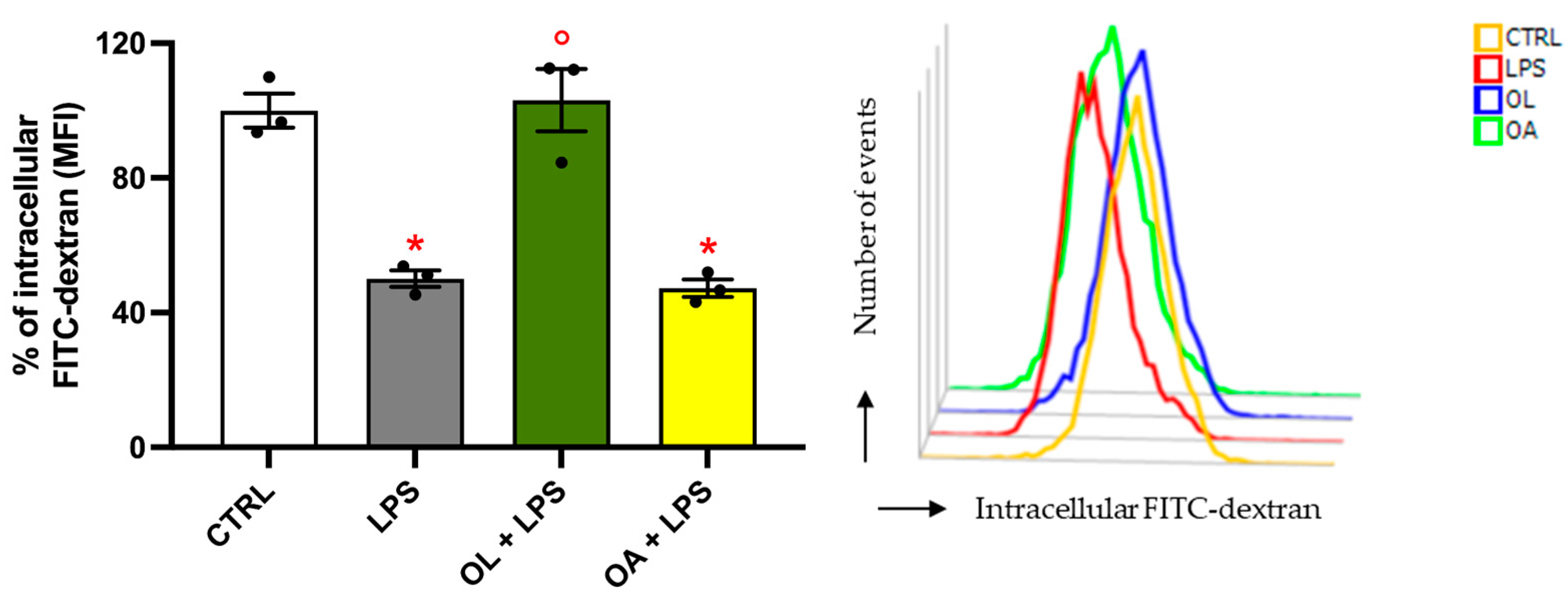
3.6. Oleocanthal Increases Phagocytic Activity in LPS-Stimulated BV-2 Cells
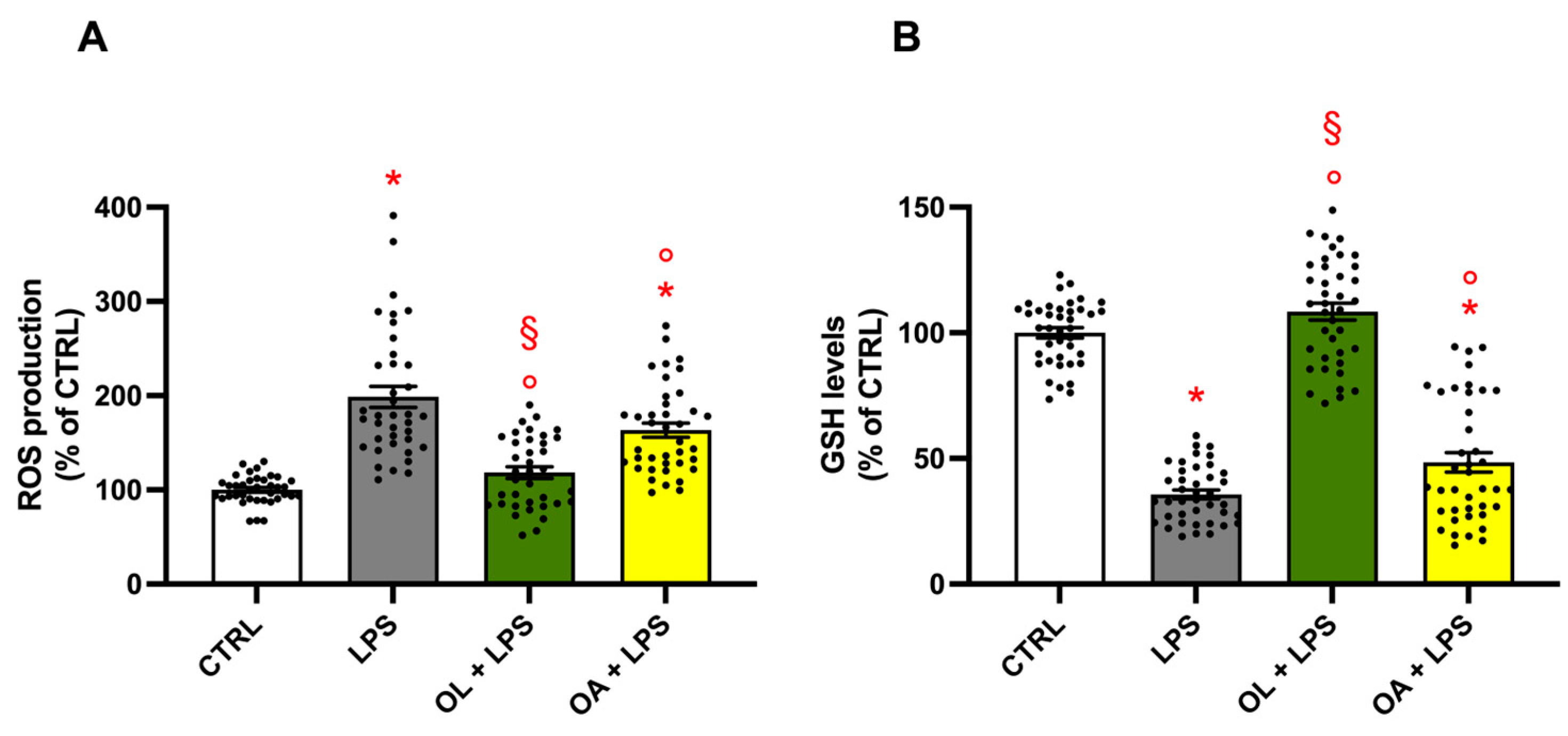
3.7. Antioxidant Activity of Oleocanthal and Oleocanthalic Acid in LPS-Stimulated BV-2 Cells
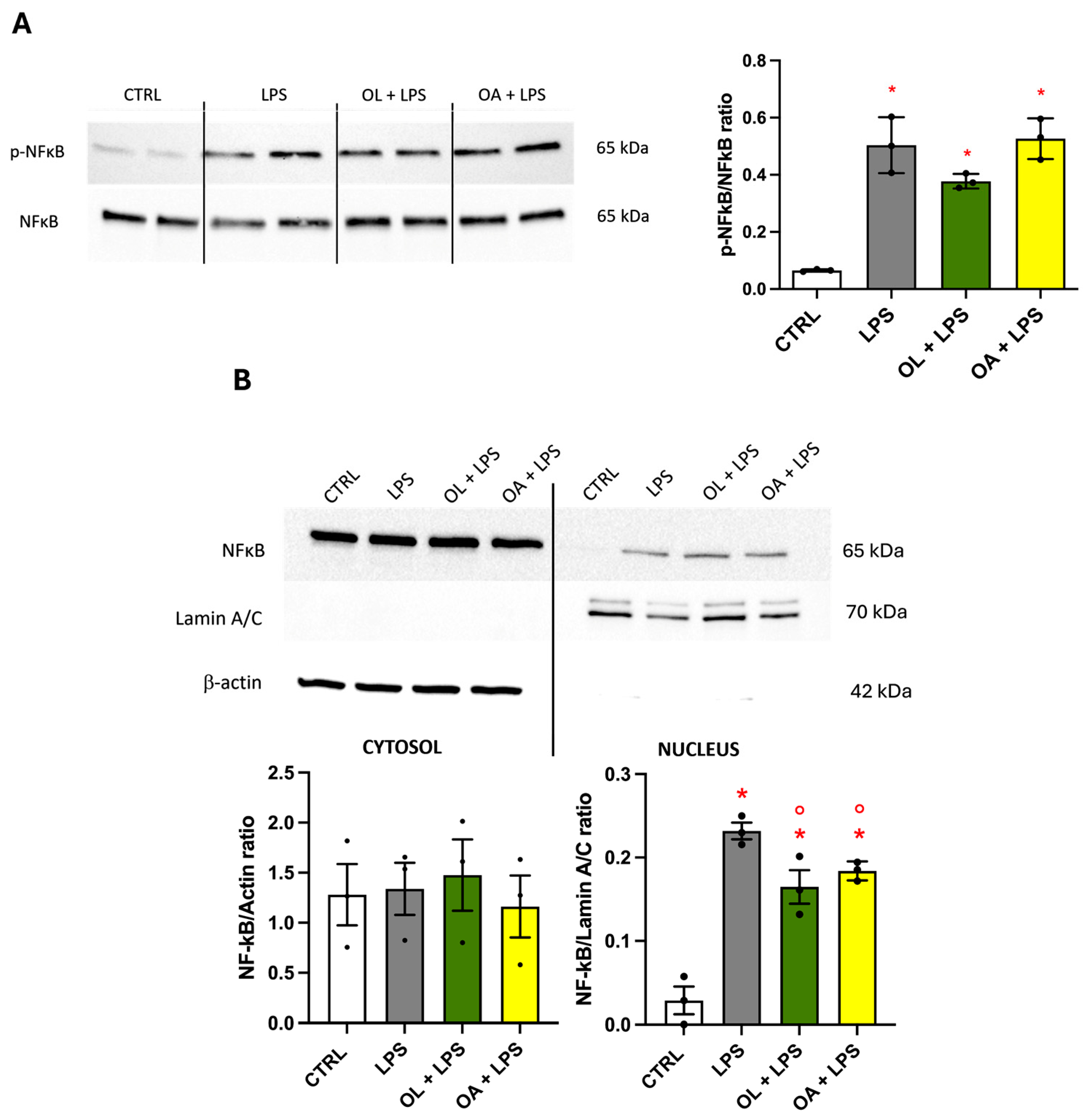
3.8. NF-κB Transcription Factor, Effect on Its Phosphorylation, and Migration to the Nucleus
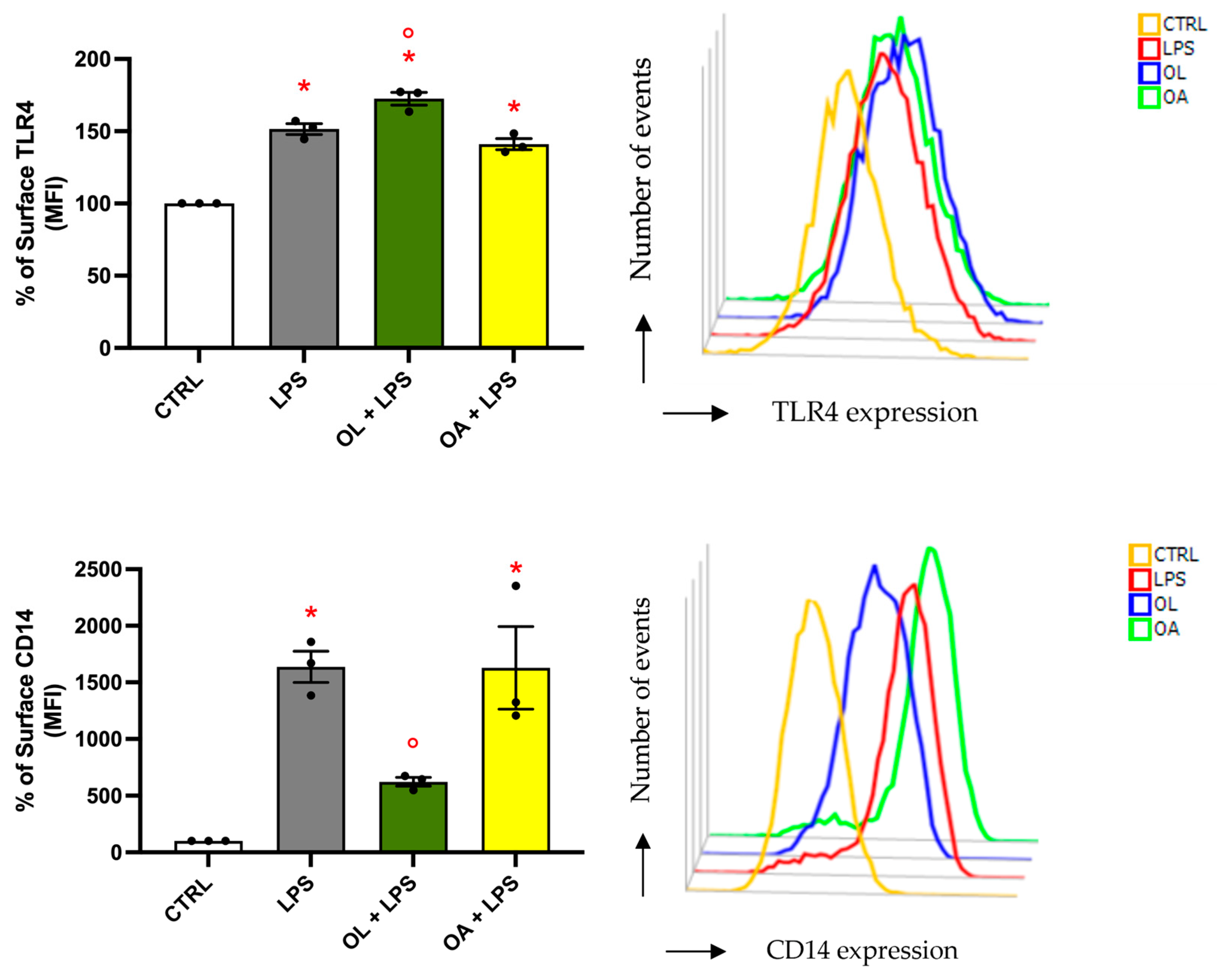
3.9. Analysis of TLR4 and CD14 Surface Expression
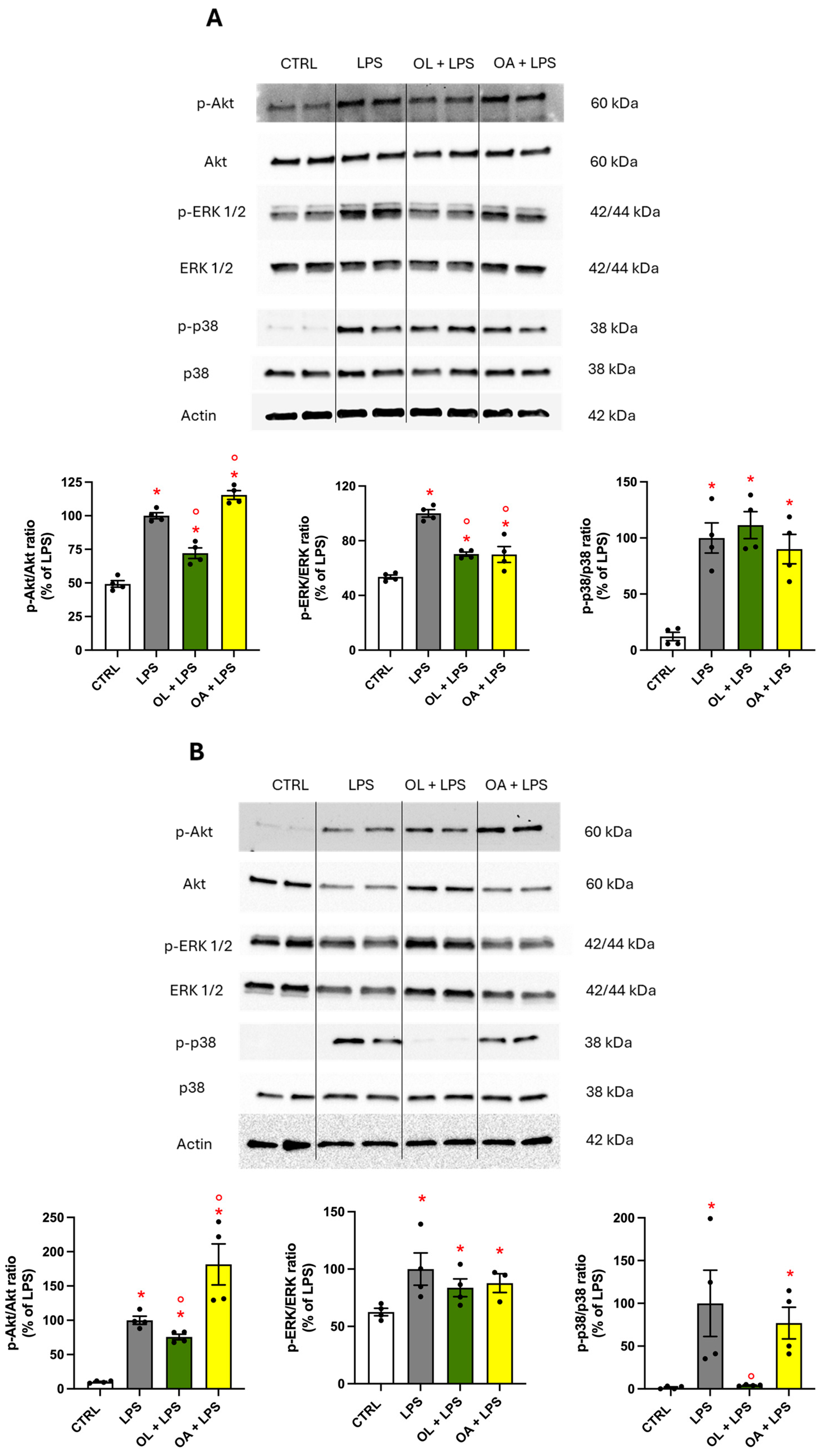
3.10. Modulation of MAPKs and Protein Kinase Akt Activation
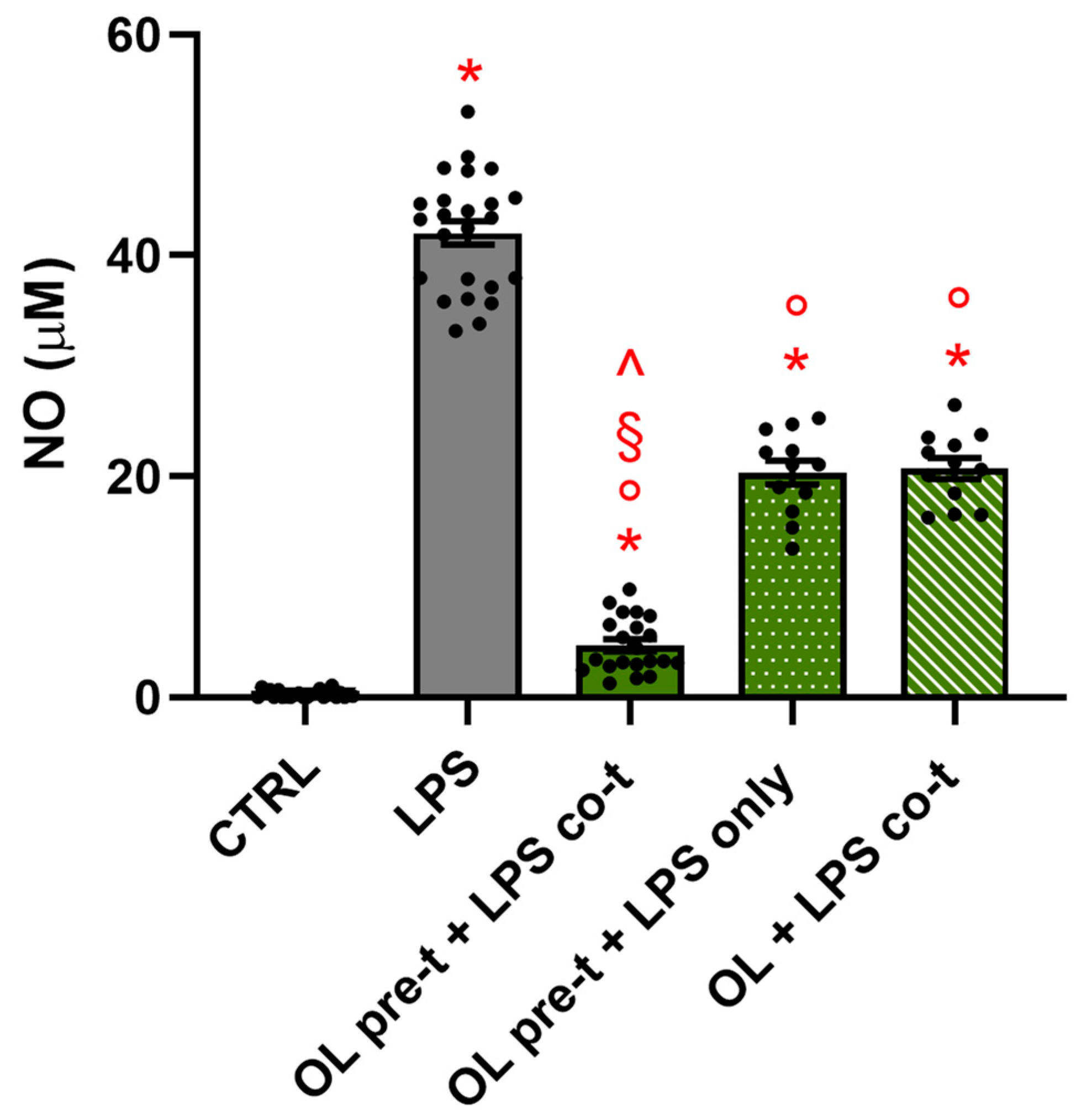
3.11. The Timing of the Treatment Is Crucial for OL’s Anti-Inflammatory Activity
3.12. Proteomic Analysis
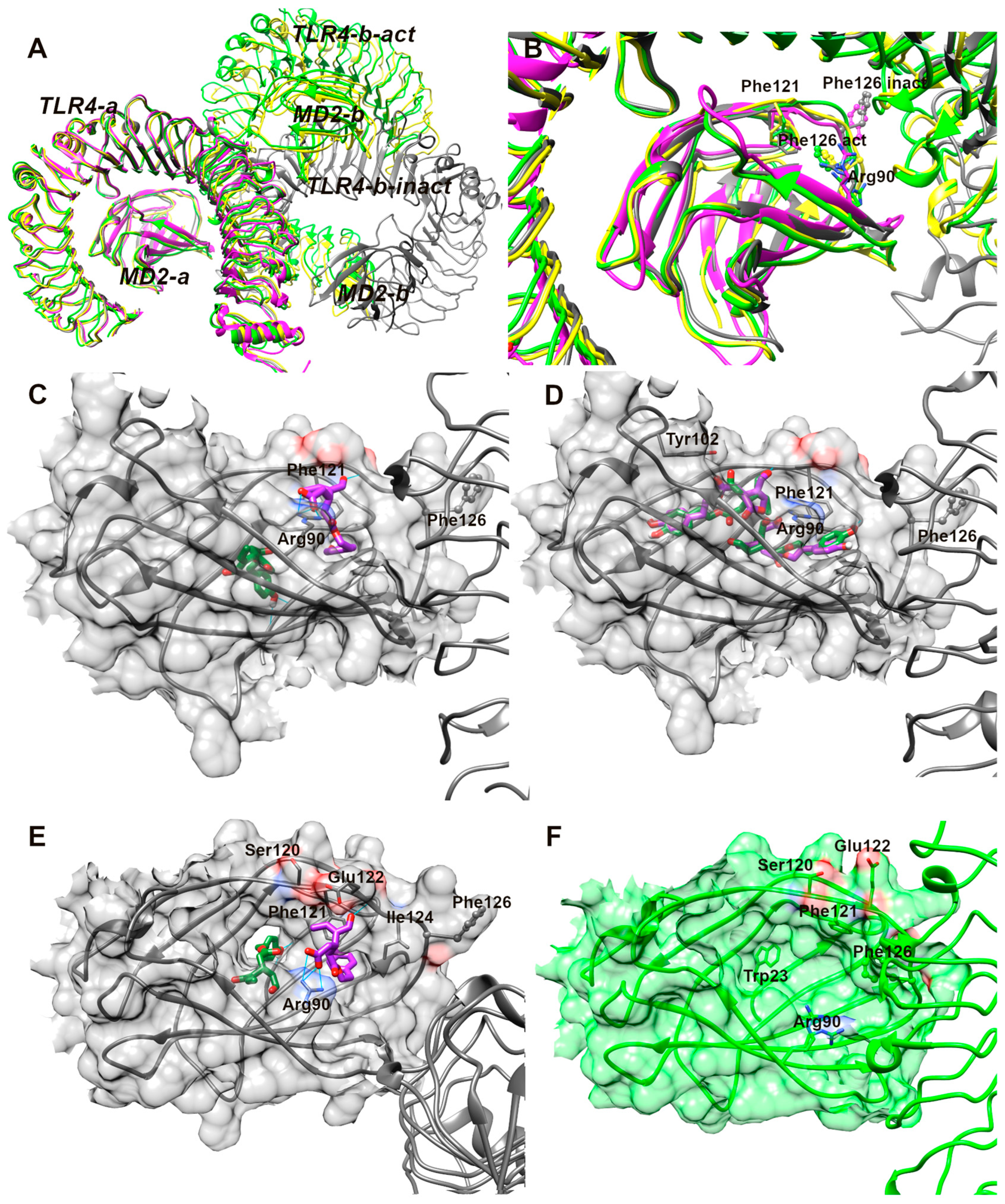
3.13. Analysis of Molecular Docking Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pasteuning-Vuhman, S.; de Jongh, R.; Timmers, A.; Pasterkamp, R.J. Towards Advanced iPSC-Based Drug Development for Neurodegenerative Disease. Trends Mol. Med. 2021, 27, 263–279. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Goh, E.L.K.; Chan, J.K.Y. Neural Cells for Neurodegenerative Diseases in Clinical Trials. Stem Cells Transl. Med. 2023, 12, 510–526. [Google Scholar] [CrossRef]
- Angeloni, C.; Malaguti, M.; Prata, C.; Freschi, M.; Barbalace, M.C.; Hrelia, S. Mechanisms Underlying Neurodegenerative Disorders and Potential Neuroprotective Activity of Agrifood By-Products. Antioxidants 2022, 12, 94. [Google Scholar] [CrossRef] [PubMed]
- Schain, M.; Kreisl, W.C. Neuroinflammation in Neurodegenerative Disorders-a Review. Curr. Neurol. Neurosci. Rep. 2017, 17, 25. [Google Scholar] [CrossRef]
- Mayne, K.; White, J.A.; McMurran, C.E.; Rivera, F.J.; de la Fuente, A.G. Aging and Neurodegenerative Disease: Is the Adaptive Immune System a Friend or Foe? Front. Aging Neurosci. 2020, 12, 572090. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.-Q.; Zhou, J.-W. Neuroinflammation in the Central Nervous System: Symphony of Glial Cells. Glia 2019, 67, 1017–1035. [Google Scholar] [CrossRef]
- Spencer, J.P.E.; Vafeiadou, K.; Williams, R.J.; Vauzour, D. Neuroinflammation: Modulation by Flavonoids and Mechanisms of Action. Mol. Aspects Med. 2012, 33, 83–97. [Google Scholar] [CrossRef]
- Wang, Q.; Yao, H.; Liu, W.; Ya, B.; Cheng, H.; Xing, Z.; Wu, Y. Microglia Polarization in Alzheimer’s Disease: Mechanisms and a Potential Therapeutic Target. Front. Aging Neurosci. 2021, 13, 772717. [Google Scholar] [CrossRef]
- Holtman, I.R.; Skola, D.; Glass, C.K. Transcriptional Control of Microglia Phenotypes in Health and Disease. J. Clin. Investig. 2017, 127, 3220–3229. [Google Scholar] [CrossRef]
- Guo, S.; Wang, H.; Yin, Y. Microglia Polarization from M1 to M2 in Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 815347. [Google Scholar] [CrossRef]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 Polarization and Metabolic States. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Darwish, S.F.; Elbadry, A.M.M.; Elbokhomy, A.S.; Salama, G.A.; Salama, R.M. The Dual Face of Microglia (M1/M2) as a Potential Target in the Protective Effect of Nutraceuticals against Neurodegenerative Diseases. Front. Aging 2023, 4, 1231706. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Lively, S.; Schlichter, L.C. Microglia Responses to Pro-Inflammatory Stimuli (LPS, IFNγ+TNFα) and Reprogramming by Resolving Cytokines (IL-4, IL-10). Front. Cell. Neurosci. 2018, 12, 215. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wang, Y.; Sun, Y.; Peng, G. Neuroinflammation as a Potential Therapeutic Target in Alzheimer’s Disease. Clin. Interv. Aging 2022, 17, 665–674. [Google Scholar] [CrossRef]
- Tansey, M.G.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and Immune Dysfunction in Parkinson Disease. Nat. Rev. Immunol. 2022, 22, 657–673. [Google Scholar] [CrossRef]
- Villegas-Aguilar, M.d.C.; Fernández-Ochoa, Á.; Cádiz-Gurrea, M.d.l.L.; Pimentel-Moral, S.; Lozano-Sánchez, J.; Arráez-Román, D.; Segura-Carretero, A. Pleiotropic Biological Effects of Dietary Phenolic Compounds and Their Metabolites on Energy Metabolism, Inflammation and Aging. Molecules 2020, 25, 596. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as Regulators of Metabolism and Healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef]
- Singh, B.; Parsaik, A.K.; Mielke, M.M.; Erwin, P.J.; Knopman, D.S.; Petersen, R.C.; Roberts, R.O. Association of Mediterranean Diet with Mild Cognitive Impairment and Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Alzheimers Dis. JAD 2014, 39, 271–282. [Google Scholar] [CrossRef]
- Psaltopoulou, T.; Sergentanis, T.N.; Panagiotakos, D.B.; Sergentanis, I.N.; Kosti, R.; Scarmeas, N. Mediterranean Diet, Stroke, Cognitive Impairment, and Depression: A Meta-Analysis. Ann. Neurol. 2013, 74, 580–591. [Google Scholar] [CrossRef]
- van den Brink, A.C.; Brouwer-Brolsma, E.M.; Berendsen, A.A.M.; van de Rest, O. The Mediterranean, Dietary Approaches to Stop Hypertension (DASH), and Mediterranean-DASH Intervention for Neurodegenerative Delay (MIND) Diets Are Associated with Less Cognitive Decline and a Lower Risk of Alzheimer’s Disease-A Review. Adv. Nutr. Bethesda Md 2019, 10, 1040–1065. [Google Scholar] [CrossRef] [PubMed]
- Guasch-Ferré, M.; Willett, W.C. The Mediterranean Diet and Health: A Comprehensive Overview. J. Intern. Med. 2021, 290, 549–566. [Google Scholar] [CrossRef] [PubMed]
- Barbalace, M.C.; Zallocco, L.; Beghelli, D.; Ronci, M.; Scortichini, S.; Digiacomo, M.; Macchia, M.; Mazzoni, M.R.; Fiorini, D.; Lucacchini, A.; et al. Antioxidant and Neuroprotective Activity of Extra Virgin Olive Oil Extracts Obtained from Quercetano Cultivar Trees Grown in Different Areas of the Tuscany Region (Italy). Antioxidants 2021, 10, 421. [Google Scholar] [CrossRef]
- Corona, G.; Spencer, J.P.E.; Dessì, M.A. Extra Virgin Olive Oil Phenolics: Absorption, Metabolism, and Biological Activities in the GI Tract. Toxicol. Ind. Health 2009, 25, 285–293. [Google Scholar] [CrossRef]
- Angeloni, C.; Malaguti, M.; Barbalace, M.C.; Hrelia, S. Bioactivity of Olive Oil Phenols in Neuroprotection. Int. J. Mol. Sci. 2017, 18, 2230. [Google Scholar] [CrossRef]
- Esposito Salsano, J.; Digiacomo, M.; Cuffaro, D.; Bertini, S.; Macchia, M. Content Variations in Oleocanthalic Acid and Other Phenolic Compounds in Extra-Virgin Olive Oil during Storage. Foods 2022, 11, 1354. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Luna, A.; Criado-Navarro, I.; Ledesma-Escobar, C.A.; López-Bascón, M.A.; Priego-Capote, F. The Decrease in the Health Benefits of Extra Virgin Olive Oil during Storage Is Conditioned by the Initial Phenolic Profile. Food Chem. 2021, 336, 127730. [Google Scholar] [CrossRef]
- Antoniadi, L.; Angelis, A.; Stathopoulos, P.; Bata, E.-M.; Papoutsaki, Z.; Halabalaki, M.; Skaltsounis, L.A. Oxidized Forms of Olive Oil Secoiridoids: Semisynthesis, Identification and Correlation with Quality Parameters. Planta Med. 2022, 88, 805–813. [Google Scholar] [CrossRef]
- Nikou, T.; Sakavitsi, M.E.; Kalampokis, E.; Halabalaki, M. Metabolism and Bioavailability of Olive Bioactive Constituents Based on In Vitro, In Vivo and Human Studies. Nutrients 2022, 14, 3773. [Google Scholar] [CrossRef]
- Cuffaro, D.; Pinto, D.; Silva, A.M.; Bertolini, A.; Bertini, S.; Saba, A.; Macchia, M.; Rodrigues, F.; Digiacomo, M. Insights into the Antioxidant/Antiradical Effects and In Vitro Intestinal Permeation of Oleocanthal and Its Metabolites Tyrosol and Oleocanthalic Acid. Molecules 2023, 28, 5150. [Google Scholar] [CrossRef]
- Boskou, D. Olive Oil: Chemistry and Technology, 2nd ed.; AOCS Publishing: New York, NY, USA, 2006; ISBN 978-1-893997-88-2. [Google Scholar]
- Visioli, F.; Davalos, A.; López de Las Hazas, M.-C.; Crespo, M.C.; Tomé-Carneiro, J. An Overview of the Pharmacology of Olive Oil and Its Active Ingredients. Br. J. Pharmacol. 2020, 177, 1316–1330. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-López, P.; Lozano-Sanchez, J.; Borrás-Linares, I.; Emanuelli, T.; Menéndez, J.A.; Segura-Carretero, A. Structure-Biological Activity Relationships of Extra-Virgin Olive Oil Phenolic Compounds: Health Properties and Bioavailability. Antioxidants 2020, 9, 685. [Google Scholar] [CrossRef] [PubMed]
- Nikou, T.; Karampetsou, K.V.; Koutsoni, O.S.; Skaltsounis, A.-L.; Dotsika, E.; Halabalaki, M. Pharmacokinetics and Metabolism Investigation of Oleocanthal. J. Nat. Prod. 2024, 87, 530–543. [Google Scholar] [CrossRef]
- Rivero-Pino, F. Oleocanthal-Characterization, Production, Safety, Functionality and In Vivo Evidences. Food Chem. 2023, 425, 136504. [Google Scholar] [CrossRef] [PubMed]
- Scotece, M.; Conde, J.; Abella, V.; López, V.; Francisco, V.; Ruiz, C.; Campos, V.; Lago, F.; Gomez, R.; Pino, J.; et al. Oleocanthal Inhibits Catabolic and Inflammatory Mediators in LPS-Activated Human Primary Osteoarthritis (OA) Chondrocytes through MAPKs/NF-κB Pathways. Cell. Physiol. Biochem. 2018, 49, 2414–2426. [Google Scholar] [CrossRef]
- Iacono, A.; Gómez, R.; Sperry, J.; Conde, J.; Bianco, G.; Meli, R.; Gómez-Reino, J.J.; Smith, A.B.; Gualillo, O. Effect of Oleocanthal and Its Derivatives on Inflammatory Response Induced by Lipopolysaccharide in a Murine Chondrocyte Cell Line. Arthritis Rheum. 2010, 62, 1675–1682. [Google Scholar] [CrossRef]
- Angeloni, C.; Giusti, L.; Hrelia, S. New Neuroprotective Perspectives in Fighting Oxidative Stress and Improving Cellular Energy Metabolism by Oleocanthal. Neural Regen. Res. 2019, 14, 1217–1218. [Google Scholar] [CrossRef]
- Barbalace, M.C.; Freschi, M.; Rinaldi, I.; Mazzara, E.; Maraldi, T.; Malaguti, M.; Prata, C.; Maggi, F.; Petrelli, R.; Hrelia, S.; et al. Identification of Anti-Neuroinflammatory Bioactive Compounds in Essential Oils and Aqueous Distillation Residues Obtained from Commercial Varieties of Cannabis sativa L. Int. J. Mol. Sci. 2023, 24, 16601. [Google Scholar] [CrossRef]
- Qosa, H.; Batarseh, Y.S.; Mohyeldin, M.M.; El Sayed, K.A.; Keller, J.N.; Kaddoumi, A. Oleocanthal Enhances Amyloid-β Clearance from the Brains of TgSwDI Mice and in Vitro across a Human Blood-Brain Barrier Model. ACS Chem. Neurosci. 2015, 6, 1849–1859. [Google Scholar] [CrossRef]
- Monti, M.C.; Margarucci, L.; Riccio, R.; Casapullo, A. Modulation of Tau Protein Fibrillization by Oleocanthal. J. Nat. Prod. 2012, 75, 1584–1588. [Google Scholar] [CrossRef]
- Montoya, T.; Castejón, M.L.; Sánchez-Hidalgo, M.; González-Benjumea, A.; Fernández-Bolaños, J.G.; Alarcón de-la-Lastra, C. Oleocanthal Modulates LPS-Induced Murine Peritoneal Macrophages Activation via Regulation of Inflammasome, Nrf-2/HO-1, and MAPKs Signaling Pathways. J. Agric. Food Chem. 2019, 67, 5552–5559. [Google Scholar] [CrossRef] [PubMed]
- Tsolakou, A.; Diamantakos, P.; Kalaboki, I.; Mena-Bravo, A.; Priego-Capote, F.; Abdallah, I.M.; Kaddoumi, A.; Melliou, E.; Magiatis, P. Oleocanthalic Acid, a Chemical Marker of Olive Oil Aging and Exposure to a High Storage Temperature with Potential Neuroprotective Activity. J. Agric. Food Chem. 2018, 66, 7337–7346. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Alonso, S.; Mancebo-Campos, V.; Salvador, M.D.; Fregapane, G. Evolution of Major and Minor Components and Oxidation Indices of Virgin Olive Oil during 21 Months Storage at Room Temperature. Food Chem. 2007, 100, 36–42. [Google Scholar] [CrossRef]
- Esposito Salsano, J.; Pinto, D.; Rodrigues, F.; Saba, A.; Manera, C.; Digiacomo, M.; Macchia, M. Oleocanthalic Acid from Extra-Virgin Olive Oil: Analysis, Preparative Isolation and Radical Scavenging Activity. J. Food Compos. Anal. 2022, 105, 104160. [Google Scholar] [CrossRef]
- Maestro, version 9.0. Macromodel. Schrödinger Inc.: Portland, OR, USA, 2009.
- Wang, Y.; Su, L.; Morin, M.D.; Jones, B.T.; Whitby, L.R.; Surakattula, M.M.R.P.; Huang, H.; Shi, H.; Choi, J.H.; Wang, K.; et al. TLR4/MD-2 Activation by a Synthetic Agonist with No Similarity to LPS. Proc. Natl. Acad. Sci. USA 2016, 113, E884–E893. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-I.; Lee, C.J.; Jin, M.S.; Lee, C.-H.; Paik, S.-G.; Lee, H.; Lee, J.-O. Crystal Structure of CD14 and Its Implications for Lipopolysaccharide Signaling. J. Biol. Chem. 2005, 280, 11347–11351. [Google Scholar] [CrossRef]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved Protein-Ligand Docking Using GOLD. Proteins 2003, 52, 609–623. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Giusti, L.; Angeloni, C.; Barbalace, M.C.; Lacerenza, S.; Ciregia, F.; Ronci, M.; Urbani, A.; Manera, C.; Digiacomo, M.; Macchia, M.; et al. A Proteomic Approach to Uncover Neuroprotective Mechanisms of Oleocanthal against Oxidative Stress. Int. J. Mol. Sci. 2018, 19, 2329. [Google Scholar] [CrossRef]
- Polini, B.; Zallocco, L.; Gado, F.; Ferrisi, R.; Ricardi, C.; Zuccarini, M.; Carnicelli, V.; Manera, C.; Ronci, M.; Lucacchini, A.; et al. A Proteomic Approach Identified TFEB as a Key Player in the Protective Action of Novel CB2R Bitopic Ligand FD22a against the Deleterious Effects Induced by β-Amyloid in Glial Cells. Cells 2024, 13, 875. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape Provides a Biologist-Oriented Resource for the Analysis of Systems-Level Datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chen, M.; Huang, X.; Zhang, G.; Zeng, L.; Zhang, G.; Wu, S.; Wang, Y. SRplot: A Free Online Platform for Data Visualization and Graphing. PLoS ONE 2023, 18, e0294236. [Google Scholar] [CrossRef] [PubMed]
- Kuzmich, N.N.; Sivak, K.V.; Chubarev, V.N.; Porozov, Y.B.; Savateeva-Lyubimova, T.N.; Peri, F. TLR4 Signaling Pathway Modulators as Potential Therapeutics in Inflammation and Sepsis. Vaccines 2017, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The Role of Pattern-Recognition Receptors in Innate Immunity: Update on Toll-like Receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Ryu, J.-K.; Kim, S.J.; Rah, S.-H.; Kang, J.I.; Jung, H.E.; Lee, D.; Lee, H.K.; Lee, J.-O.; Park, B.S.; Yoon, T.-Y.; et al. Reconstruction of LPS Transfer Cascade Reveals Structural Determinants within LBP, CD14, and TLR4-MD2 for Efficient LPS Recognition and Transfer. Immunity 2017, 46, 38–50. [Google Scholar] [CrossRef]
- Kim, H.M.; Park, B.S.; Kim, J.-I.; Kim, S.E.; Lee, J.; Oh, S.C.; Enkhbayar, P.; Matsushima, N.; Lee, H.; Yoo, O.J.; et al. Crystal Structure of the TLR4-MD-2 Complex with Bound Endotoxin Antagonist Eritoran. Cell 2007, 130, 906–917. [Google Scholar] [CrossRef]
- Park, B.S.; Song, D.H.; Kim, H.M.; Choi, B.-S.; Lee, H.; Lee, J.-O. The Structural Basis of Lipopolysaccharide Recognition by the TLR4-MD-2 Complex. Nature 2009, 458, 1191–1195. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, G.; Chen, L.; Liu, X.; Fu, W.; Zhang, Y.; Li, C.; Liang, G.; Cai, Y. Insights into the Binding Mode of Curcumin to MD-2: Studies from Molecular Docking, Molecular Dynamics Simulations and Experimental Assessments. Mol. Biosyst. 2015, 11, 1933–1938. [Google Scholar] [CrossRef]
- Belcher, J.D.; Zhang, P.; Nguyen, J.; Kiser, Z.M.; Nath, K.A.; Hu, J.; Trent, J.O.; Vercellotti, G.M. Identification of a Heme Activation Site on the MD-2/TLR4 Complex. Front. Immunol. 2020, 11, 1370. [Google Scholar] [CrossRef]
- Boyd, R.J.; Avramopoulos, D.; Jantzie, L.L.; McCallion, A.S. Neuroinflammation Represents a Common Theme amongst Genetic and Environmental Risk Factors for Alzheimer and Parkinson Diseases. J. Neuroinflammation 2022, 19, 223. [Google Scholar] [CrossRef]
- Cervellati, C.; Trentini, A.; Pecorelli, A.; Valacchi, G. Inflammation in Neurological Disorders: The Thin Boundary Between Brain and Periphery. Antioxid. Redox Signal. 2020, 33, 191–210. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in Neurodegenerative Disorders: The Roles of Microglia and Astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Angeloni, C.; Vauzour, D. Natural Products and Neuroprotection. Int. J. Mol. Sci. 2019, 20, 5570. [Google Scholar] [CrossRef] [PubMed]
- Catorce, M.N.; Gevorkian, G. Evaluation of Anti-Inflammatory Nutraceuticals in LPS-Induced Mouse Neuroinflammation Model: An Update. Curr. Neuropharmacol. 2020, 18, 636–654. [Google Scholar] [CrossRef]
- Rai, R.; Kalar, P.L.; Jat, D.; Mishra, S.K. Naringenin Mitigates Nanoparticulate-Aluminium Induced Neuronal Degeneration in Brain Cortex and Hippocampus through Downregulation of Oxidative Stress and Neuroinflammation. Neurochem. Int. 2024, 178, 105799. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, G.K.; Keast, R.S.J.; Morel, D.; Lin, J.; Pika, J.; Han, Q.; Lee, C.-H.; Smith, A.B.; Breslin, P.A.S. Phytochemistry: Ibuprofen-like Activity in Extra-Virgin Olive Oil. Nature 2005, 437, 45–46. [Google Scholar] [CrossRef]
- Batista, C.R.A.; Gomes, G.F.; Candelario-Jalil, E.; Fiebich, B.L.; de Oliveira, A.C.P. Lipopolysaccharide-Induced Neuroinflammation as a Bridge to Understand Neurodegeneration. Int. J. Mol. Sci. 2019, 20, 2293. [Google Scholar] [CrossRef]
- Brown, G.C.; Camacho, M.; Williams-Gray, C.H. The Endotoxin Hypothesis of Parkinson’s Disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2023, 38, 1143–1155. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Ali, T.; Rehman, S.U.; Kim, M.O. Phytomedicine-Based Potent Antioxidant, Fisetin Protects CNS-Insult LPS-Induced Oxidative Stress-Mediated Neurodegeneration and Memory Impairment. J. Clin. Med. 2019, 8, 850. [Google Scholar] [CrossRef]
- Loffredo, L.; Ettorre, E.; Zicari, A.M.; Inghilleri, M.; Nocella, C.; Perri, L.; Spalice, A.; Fossati, C.; De Lucia, M.C.; Pigozzi, F.; et al. Oxidative Stress and Gut-Derived Lipopolysaccharides in Neurodegenerative Disease: Role of NOX2. Oxid. Med. Cell. Longev. 2020, 2020, 8630275. [Google Scholar] [CrossRef]
- Andreadou, E.G.; Katsipis, G.; Tsolaki, M.; Pantazaki, A.A. Involvement and Relationship of Bacterial Lipopolysaccharides and Cyclooxygenases Levels in Alzheimer’s Disease and Mild Cognitive Impairment Patients. J. Neuroimmunol. 2021, 357, 577561. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Tapia, M.; Mimenza-Alvarado, A.; Granados-Domínguez, L.; Flores-López, A.; López-Barradas, A.; Ortiz, V.; Pérez-Cruz, C.; Sánchez-Vidal, H.; Hernández-Acosta, J.; Ávila-Funes, J.A.; et al. The Gut Microbiota-Brain Axis during Aging, Mild Cognitive Impairment and Dementia: Role of Tau Protein, β-Amyloid and LPS in Serum and Curli Protein in Stool. Nutrients 2023, 15, 932. [Google Scholar] [CrossRef] [PubMed]
- Sandiego, C.M.; Gallezot, J.-D.; Pittman, B.; Nabulsi, N.; Lim, K.; Lin, S.-F.; Matuskey, D.; Lee, J.-Y.; O’Connor, K.C.; Huang, Y.; et al. Imaging Robust Microglial Activation after Lipopolysaccharide Administration in Humans with PET. Proc. Natl. Acad. Sci. USA 2015, 112, 12468–12473. [Google Scholar] [CrossRef] [PubMed]
- Bahador, M.; Cross, A.S. From Therapy to Experimental Model: A Hundred Years of Endotoxin Administration to Human Subjects. J. Endotoxin Res. 2007, 13, 251–279. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Condello, C.; Keene, C.D.; Wang, Y.; Bird, T.D.; Paul, S.M.; Luo, W.; Colonna, M.; Baddeley, D.; Grutzendler, J. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron 2016, 92, 252–264. [Google Scholar] [CrossRef]
- Grubman, A.; Choo, X.Y.; Chew, G.; Ouyang, J.F.; Sun, G.; Croft, N.P.; Rossello, F.J.; Simmons, R.; Buckberry, S.; Landin, D.V.; et al. Transcriptional Signature in Microglia Associated with Aβ Plaque Phagocytosis. Nat. Commun. 2021, 12, 3015. [Google Scholar] [CrossRef]
- Fan, Z.; Okello, A.A.; Brooks, D.J.; Edison, P. Longitudinal Influence of Microglial Activation and Amyloid on Neuronal Function in Alzheimer’s Disease. Brain J. Neurol. 2015, 138, 3685–3698. [Google Scholar] [CrossRef]
- Ismail, E.N.; Jantan, I.; Vidyadaran, S.; Jamal, J.A.; Azmi, N. Phyllanthus Amarus Prevents LPS-Mediated BV2 Microglial Activation via MyD88 and NF-κB Signaling Pathways. BMC Complement. Med. Ther. 2020, 20, 202. [Google Scholar] [CrossRef]
- Wen, J.; Ribeiro, R.; Zhang, Y. Specific PKC Isoforms Regulate LPS-Stimulated iNOS Induction in Murine Microglial Cells. J. Neuroinflammation 2011, 8, 38. [Google Scholar] [CrossRef]
- Duan, Z.; Chen, H.; Miao, W.; He, J.; Xu, D.; Qi, Z.; Yang, L.; Jia, W.; Wu, C. Scutellarin Alleviates Microglia-mediated Neuroinflammation and Apoptosis after Ischemic Stroke through the PI3K/AKT/GSK3β Signaling Pathway. J. Cell Commun. Signal. 2024, 18, e12023. [Google Scholar] [CrossRef]
- Li, J.; Wang, L.; Zeng, Q.; He, J.; Tang, Q.; Wang, K.; He, G. MKP-1 Regulates the Inflammatory Activation of Microglia against Alzheimer’s Disease. CNS Neurosci. Ther. 2023, 30, e14409. [Google Scholar] [CrossRef] [PubMed]
- Leitner, G.R.; Wenzel, T.J.; Marshall, N.; Gates, E.J.; Klegeris, A. Targeting Toll-like Receptor 4 to Modulate Neuroinflammation in Central Nervous System Disorders. Expert Opin. Ther. Targets 2019, 23, 865–882. [Google Scholar] [CrossRef]
- Ciesielska, A.; Matyjek, M.; Kwiatkowska, K. TLR4 and CD14 Trafficking and Its Influence on LPS-Induced pro-Inflammatory Signaling. Cell. Mol. Life Sci. 2021, 78, 1233–1261. [Google Scholar] [CrossRef]
- Zanoni, I.; Ostuni, R.; Marek, L.R.; Barresi, S.; Barbalat, R.; Barton, G.M.; Granucci, F.; Kagan, J.C. CD14 Controls the LPS-Induced Endocytosis of Toll-like Receptor 4. Cell 2011, 147, 868–880. [Google Scholar] [CrossRef] [PubMed]
- Cighetti, R.; Ciaramelli, C.; Sestito, S.E.; Zanoni, I.; Kubik, Ł.; Ardá-Freire, A.; Calabrese, V.; Granucci, F.; Jerala, R.; Martín-Santamaría, S.; et al. Modulation of CD14 and TLR4·MD-2 Activities by a Synthetic Lipid A Mimetic. ChemBioChem Eur. J. Chem. Biol. 2014, 15, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, N.-Y.; Park, J.-Y.; Park, H.-M.; Lim, C.-M.; Kim, J.; Lee, H.P.; Hong, J.T.; Yoon, D.-Y. MMPP Exerts Anti-Inflammatory Effects by Suppressing MD2-Dependent NF-κB and JNK/AP-1 Pathways in THP-1 Monocytes. Pharmaceuticals 2023, 16, 480. [Google Scholar] [CrossRef]
- Gradisar, H.; Keber, M.M.; Pristovsek, P.; Jerala, R. MD-2 as the Target of Curcumin in the Inhibition of Response to LPS. J. Leukoc. Biol. 2007, 82, 968–974. [Google Scholar] [CrossRef]
- Koo, J.E.; Park, Z.-Y.; Kim, N.D.; Lee, J.Y. Sulforaphane Inhibits the Engagement of LPS with TLR4/MD2 Complex by Preferential Binding to Cys133 in MD2. Biochem. Biophys. Res. Commun. 2013, 434, 600–605. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, J.; Ying, S.; Chen, G.; Wu, B.; Xu, T.; Liu, Z.; Liu, X.; Huang, L.; Shan, X.; et al. Discovery of New MD2 Inhibitor from Chalcone Derivatives with Anti-Inflammatory Effects in LPS-Induced Acute Lung Injury. Sci. Rep. 2016, 6, 25130. [Google Scholar] [CrossRef]
- Higginbotham, L.; Ping, L.; Dammer, E.B.; Duong, D.M.; Zhou, M.; Gearing, M.; Hurst, C.; Glass, J.D.; Factor, S.A.; Johnson, E.C.B.; et al. Integrated Proteomics Reveals Brain-Based Cerebrospinal Fluid Biomarkers in Asymptomatic and Symptomatic Alzheimer’s Disease. Sci. Adv. 2020, 6, eaaz9360. [Google Scholar] [CrossRef]
- Zhou, M.; Haque, R.U.; Dammer, E.B.; Duong, D.M.; Ping, L.; Johnson, E.C.B.; Lah, J.J.; Levey, A.I.; Seyfried, N.T. Targeted Mass Spectrometry to Quantify Brain-Derived Cerebrospinal Fluid Biomarkers in Alzheimer’s Disease. Clin. Proteomics 2020, 17, 19. [Google Scholar] [CrossRef] [PubMed]
- Foote, M.; Zhou, Y. 14-3-3 Proteins in Neurological Disorders. Int. J. Biochem. Mol. Biol. 2012, 3, 152–164. [Google Scholar] [PubMed]
- Sathe, G.; Na, C.H.; Renuse, S.; Madugundu, A.K.; Albert, M.; Moghekar, A.; Pandey, A. Quantitative Proteomic Profiling of Cerebrospinal Fluid to Identify Candidate Biomarkers for Alzheimer’s Disease. Proteom. Clin. Appl. 2019, 13, e1800105. [Google Scholar] [CrossRef] [PubMed]
- Bader, J.M.; Geyer, P.E.; Müller, J.B.; Strauss, M.T.; Koch, M.; Leypoldt, F.; Koertvelyessy, P.; Bittner, D.; Schipke, C.G.; Incesoy, E.I.; et al. Proteome Profiling in Cerebrospinal Fluid Reveals Novel Biomarkers of Alzheimer’s Disease. Mol. Syst. Biol. 2020, 16, e9356. [Google Scholar] [CrossRef] [PubMed]
- Fountoulakis, M.; Cairns, N.; Lubec, G. Increased Levels of 14-3-3 Gamma and Epsilon Proteins in Brain of Patients with Alzheimer’s Disease and Down Syndrome. In The Molecular Biology of Down Syndrome; Lubec, G., Ed.; Springer: Vienna, Austria, 1999; pp. 323–335. ISBN 978-3-211-83377-3. [Google Scholar]
- Peng, Y.; Zhu, L.; Bai, Q.; Wang, L.; Li, Q. Serum Level of YWHAG as a Diagnostic Marker of Cognitive Impairment in Parkinson’s Disease Patients. Acta Neurol. Belg. 2024, 124, 879–885. [Google Scholar] [CrossRef]
- Das, J.; Tiwari, M.; Subramanyam, D. Clathrin Light Chains: Not to Be Taken so Lightly. Front. Cell Dev. Biol. 2021, 9, 774587. [Google Scholar] [CrossRef]
- Heerssen, H.; Fetter, R.D.; Davis, G.W. Clathrin Dependence of Synaptic-Vesicle Formation at the Drosophila Neuromuscular Junction. Curr. Biol. CB 2008, 18, 401–409. [Google Scholar] [CrossRef]
- Redlingshöfer, L.; McLeod, F.; Chen, Y.; Camus, M.D.; Burden, J.J.; Palomer, E.; Briant, K.; Dannhauser, P.N.; Salinas, P.C.; Brodsky, F.M. Clathrin Light Chain Diversity Regulates Membrane Deformation In Vitro and Synaptic Vesicle Formation In Vivo. Proc. Natl. Acad. Sci. USA 2020, 117, 23527–23538. [Google Scholar] [CrossRef]
- Fujikura, M.; Iwahara, N.; Hisahara, S.; Kawamata, J.; Matsumura, A.; Yokokawa, K.; Saito, T.; Manabe, T.; Matsushita, T.; Suzuki, S.; et al. CD14 and Toll-Like Receptor 4 Promote Fibrillar Aβ42 Uptake by Microglia through A Clathrin-Mediated Pathway. J. Alzheimers Dis. JAD 2019, 68, 323–337. [Google Scholar] [CrossRef]
- Duarte, A.C.; Hrynchak, M.V.; Gonçalves, I.; Quintela, T.; Santos, C.R.A. Sex Hormone Decline and Amyloid β Synthesis, Transport and Clearance in the Brain. J. Neuroendocrinol. 2016, 28. [Google Scholar] [CrossRef]
- Cheng, Y.; Hu, X.; Liu, C.; Chen, M.; Wang, J.; Wang, M.; Gao, F.; Han, J.; Zhang, C.; Sun, D.; et al. Gelsolin Inhibits the Inflammatory Process Induced by LPS. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 41, 205–212. [Google Scholar] [CrossRef]
- Ji, L.; Zhao, X.; Hua, Z. Potential Therapeutic Implications of Gelsolin in Alzheimer’s Disease. J. Alzheimers Dis. JAD 2015, 44, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Eberhart, T.; Stanley, F.U.; Ricci, L.; Chirico, T.; Ferrarese, R.; Sisti, S.; Scagliola, A.; Baj, A.; Badurek, S.; Sommer, A.; et al. ACOD1 Deficiency Offers Protection in a Mouse Model of Diet-Induced Obesity by Maintaining a Healthy Gut Microbiota. Cell Death Dis. 2024, 15, 105. [Google Scholar] [CrossRef]
- Li, Z.; Zheng, W.; Kong, W.; Zeng, T. Itaconate: A Potent Macrophage Immunomodulator. Inflammation 2023, 46, 1177–1191. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Chen, F.; Wang, N.; Tang, D.; Kang, R. ACOD1 in Immunometabolism and Disease. Cell. Mol. Immunol. 2020, 17, 822–833. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Liu, J.; Tang, D.; Kang, R. The Dual Role of ACOD1 in Inflammation. J. Immunol. 2023, 211, 518–526. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse | RefSeq Accession No |
|---|---|---|---|
| iNOS | CCTCCTCCACCCTACCAAGT | CACCCAAAGTGCTTCAGTCA | NM_010927 |
| COX-2 | TGGGGTGATGAGCAACTATT | AAGGAGCTCTGGGTCAAACT | NM_011198 |
| NLRP3 | GATGCTGGAATTAGACAACTG | GTACATTTCACCCAACTGTAG | NM_145827 |
| TNF-α | CCCCAAAGGGATGAGAAGTTC | CCTCCACTTGGTGGTTTGCT | NM_013693 |
| IL-1β | GTTCCCATTAGACAACTGCACTACAG | GTCGTTGCTTGGTTCTCCTTGTA | NM_008361 |
| IL-6 | GTCTATACCACTTCACAAGTC | TGCATCATCGTTGTTCATAC | NM_031168 |
| IL-4 | CTGGATTCATCGATAAGCTG | TTTGCATGATGCTCTTTAGG | NM_021283 |
| MRC1 | GTTATGAAAGGCAAGGATGG | ATCAGTGAAGGTGGATAGAG | NM_008625 |
| GAPDH | ACCACAGTCCATGCCATCAC | TCCACCACCCTGTTGCTGTA | NM_001289726 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbalace, M.C.; Freschi, M.; Rinaldi, I.; Zallocco, L.; Malaguti, M.; Manera, C.; Ortore, G.; Zuccarini, M.; Ronci, M.; Cuffaro, D.; et al. Unraveling the Protective Role of Oleocanthal and Its Oxidation Product, Oleocanthalic Acid, against Neuroinflammation. Antioxidants 2024, 13, 1074. https://doi.org/10.3390/antiox13091074
Barbalace MC, Freschi M, Rinaldi I, Zallocco L, Malaguti M, Manera C, Ortore G, Zuccarini M, Ronci M, Cuffaro D, et al. Unraveling the Protective Role of Oleocanthal and Its Oxidation Product, Oleocanthalic Acid, against Neuroinflammation. Antioxidants. 2024; 13(9):1074. https://doi.org/10.3390/antiox13091074
Chicago/Turabian StyleBarbalace, Maria Cristina, Michela Freschi, Irene Rinaldi, Lorenzo Zallocco, Marco Malaguti, Clementina Manera, Gabriella Ortore, Mariachiara Zuccarini, Maurizio Ronci, Doretta Cuffaro, and et al. 2024. "Unraveling the Protective Role of Oleocanthal and Its Oxidation Product, Oleocanthalic Acid, against Neuroinflammation" Antioxidants 13, no. 9: 1074. https://doi.org/10.3390/antiox13091074
APA StyleBarbalace, M. C., Freschi, M., Rinaldi, I., Zallocco, L., Malaguti, M., Manera, C., Ortore, G., Zuccarini, M., Ronci, M., Cuffaro, D., Macchia, M., Hrelia, S., Giusti, L., Digiacomo, M., & Angeloni, C. (2024). Unraveling the Protective Role of Oleocanthal and Its Oxidation Product, Oleocanthalic Acid, against Neuroinflammation. Antioxidants, 13(9), 1074. https://doi.org/10.3390/antiox13091074