
Analysis of High-Dose Ascorbate-Induced Cytotoxicity in Human Glioblastoma Cells and the Role of Dehydroascorbic Acid and Iron

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Cell Viability Assays
2.3. Investigation of Cell Proliferation
2.4. Investigation of Intracellular ROS Levels
2.5. Analysis of Cell Cycle with Propidium Iodide Staining by Flow Cytometry
2.6. Morphological Cell Nucleus Examination
2.7. Immunoblotting
2.8. Statistical Analysis
3. Results
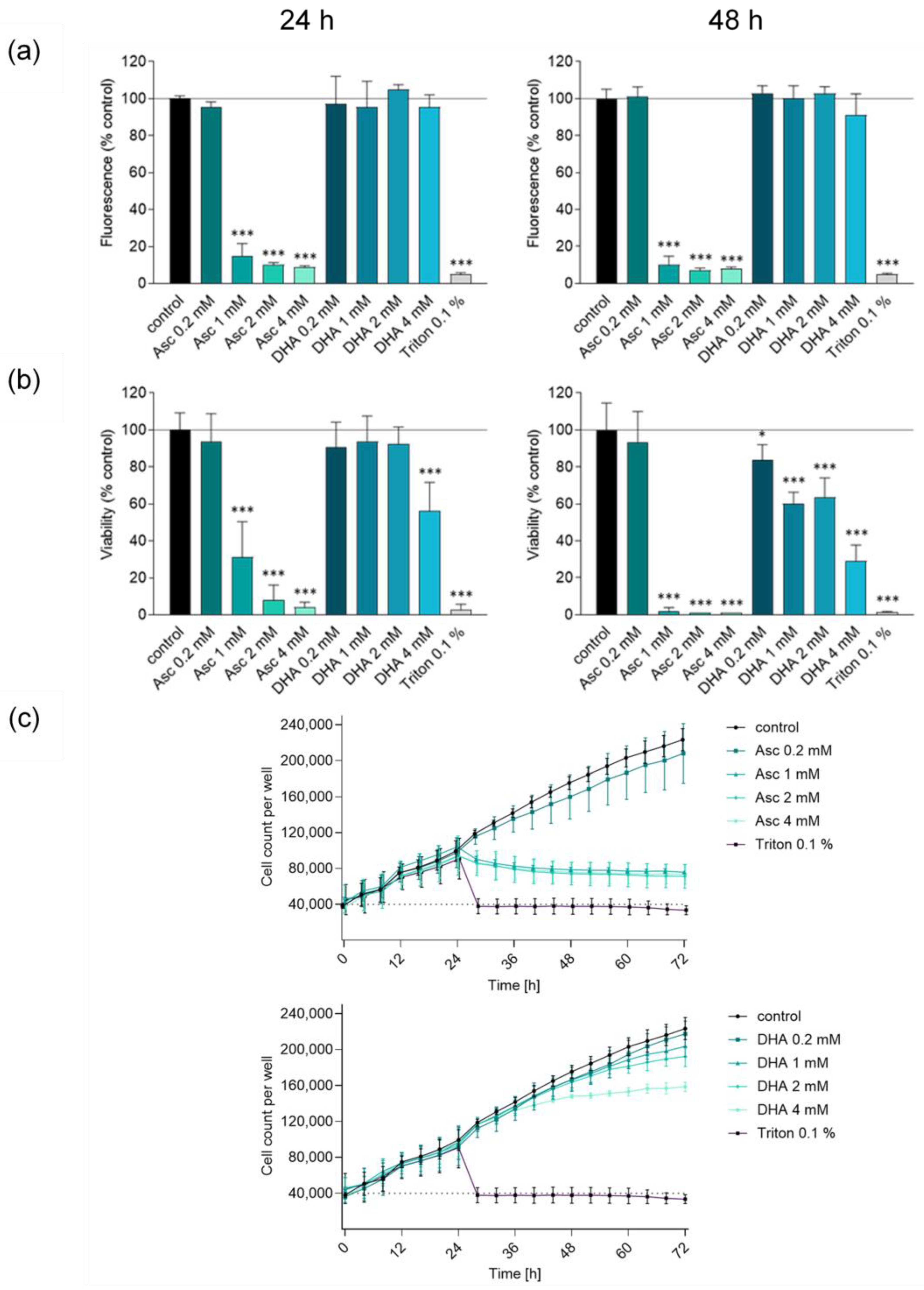
3.1. Ascorbate but Not DHA Induces Cytotoxic Effects in Glioblastoma Cells
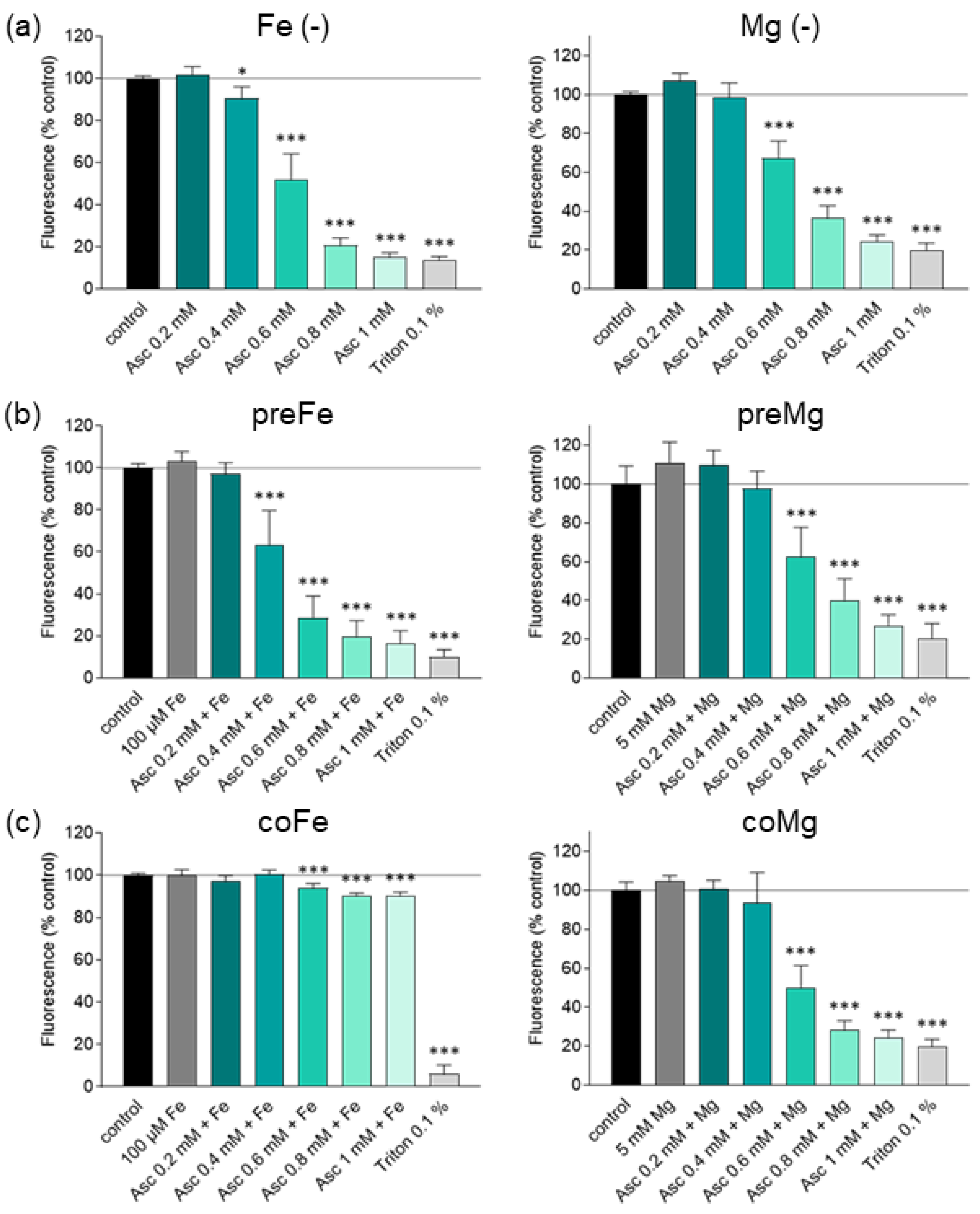
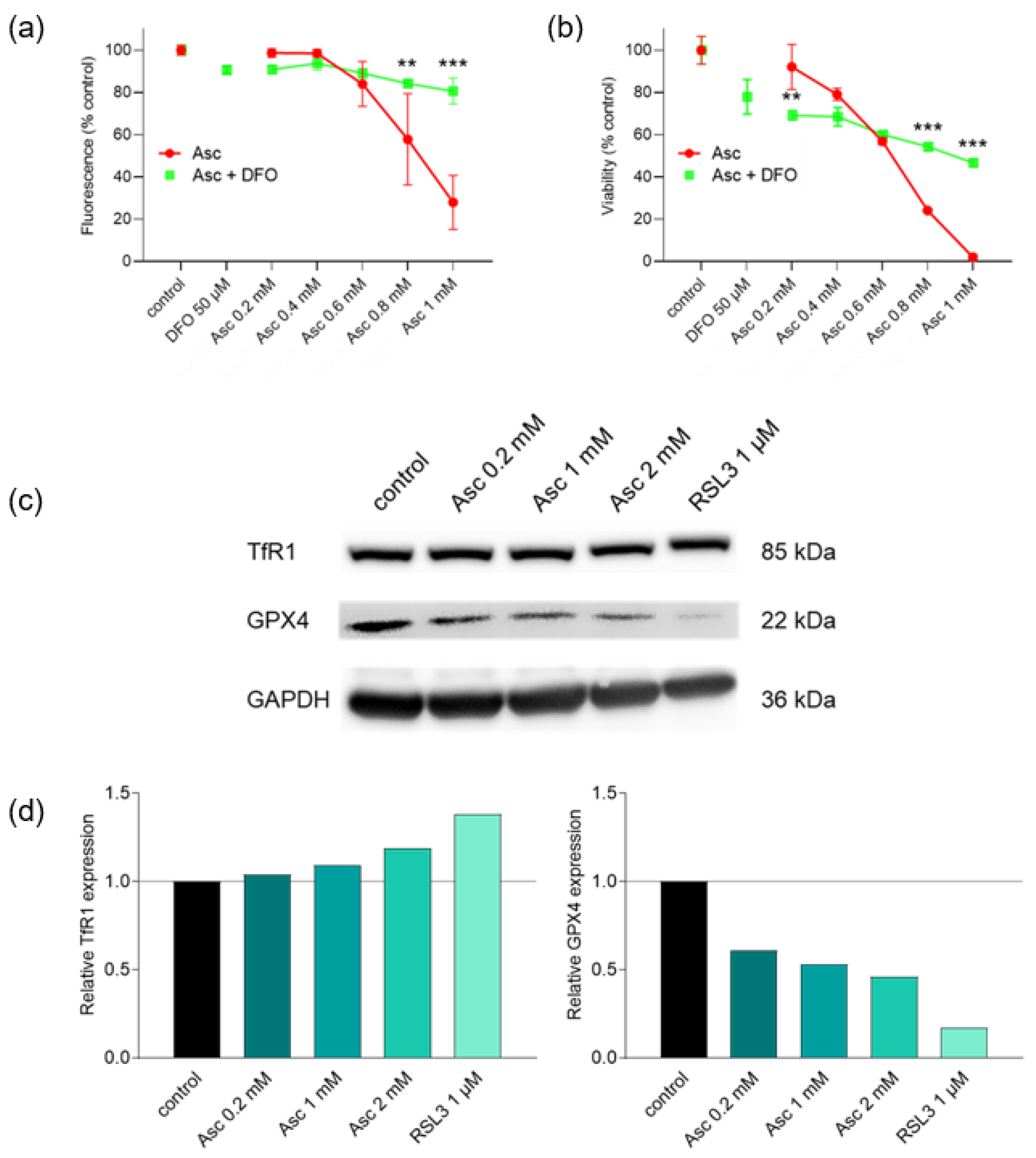
3.2. Effect of Iron and Magnesium on Ascorbate-Induced Cytotoxicity
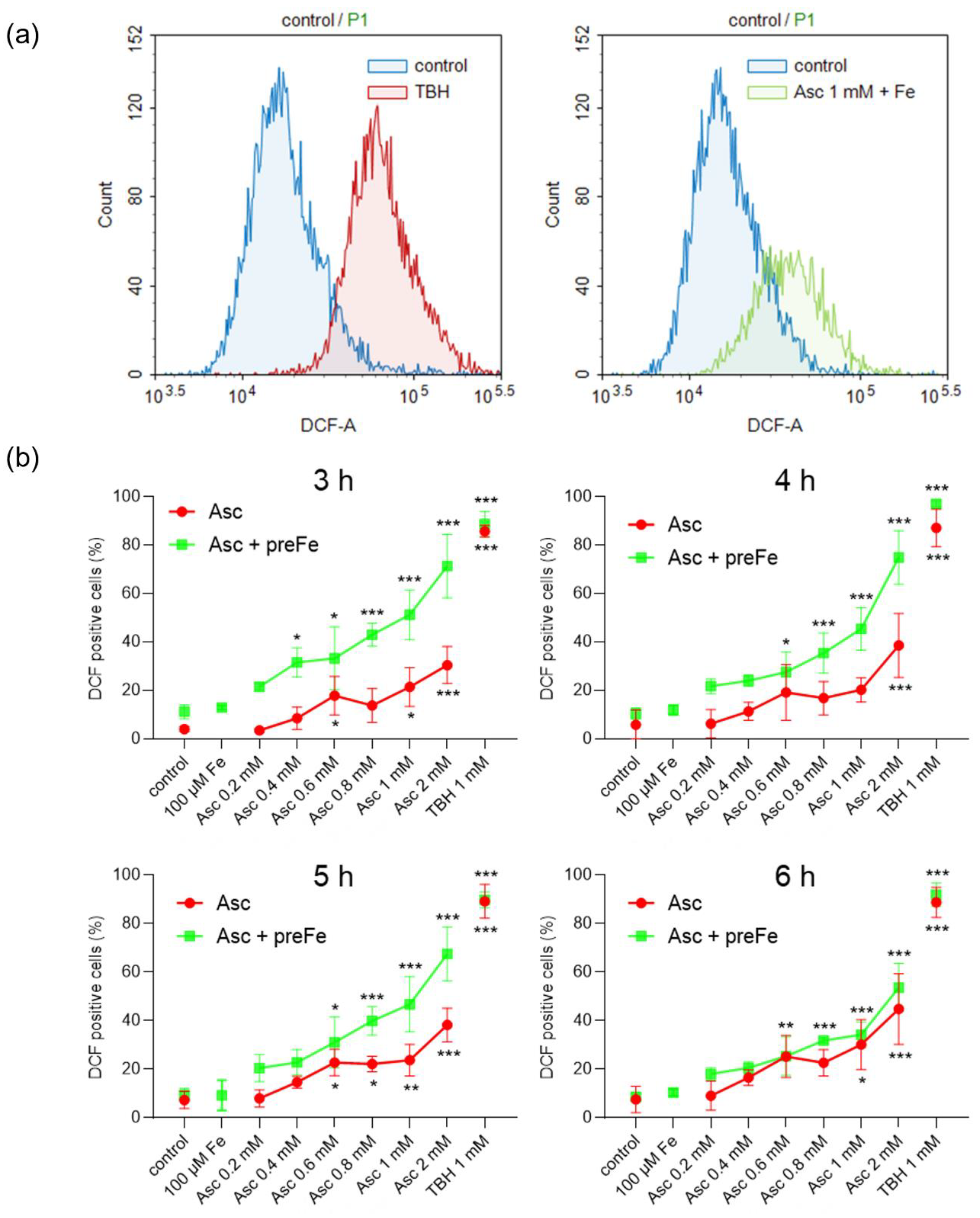
3.3. Potentiation of ROS Formation by Ascorbate and Iron Pre-Treatment
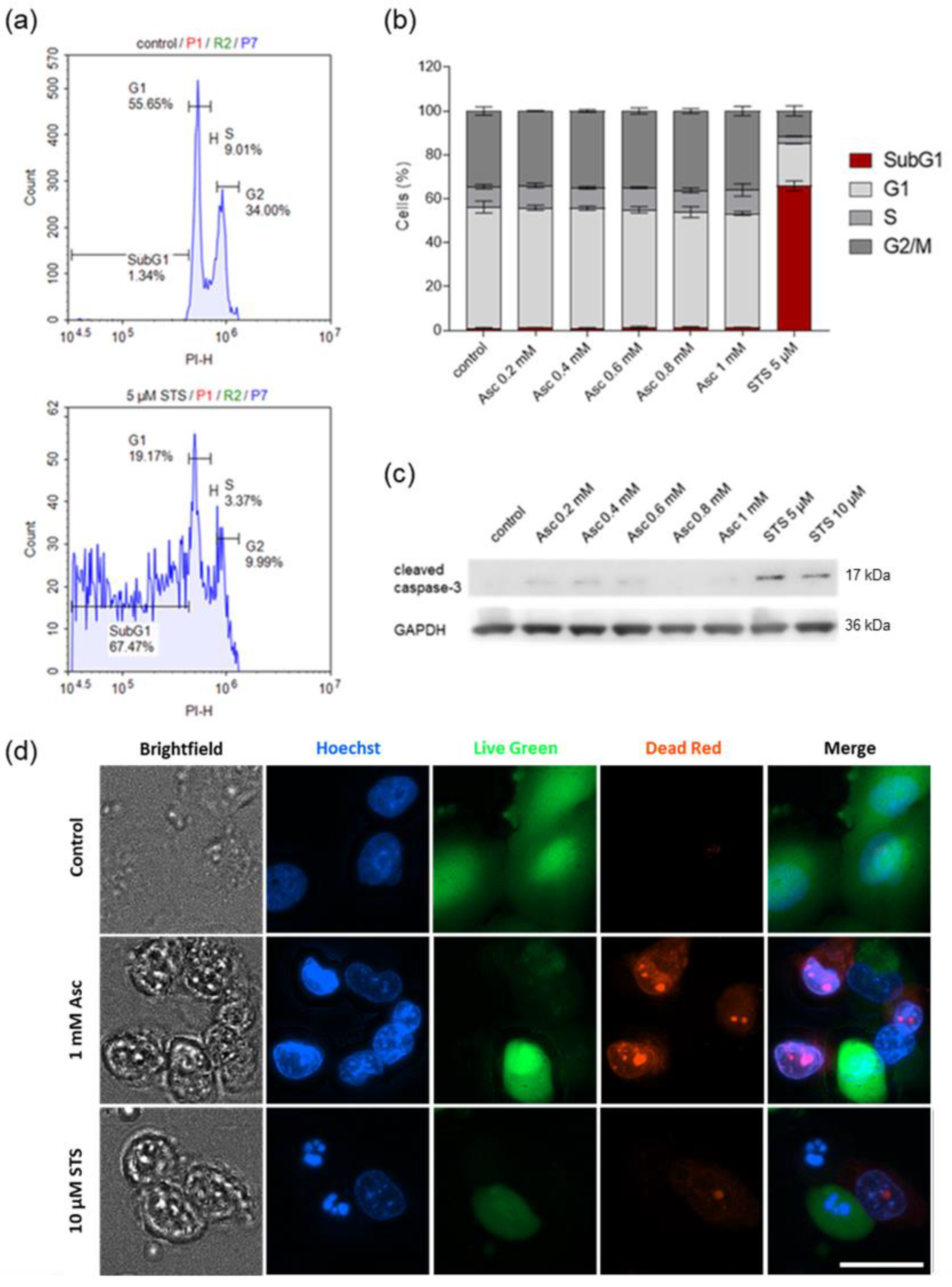
3.4. Ascorbate-Induced Cell Death Shows No Signs of Apoptosis
3.5. Ascorbate-Induced Cell Death Is Iron-Dependent and Shows Signs of Ferroptosis
4. Discussion
5. Conclusions
6. Innovation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Atg4 | autophagy-related protease 4 |
| BBB | blood-brain-barrier |
| BKCa | large-conductance Ca2+-activated K+ channel |
| BSA | bovine serum albumin |
| CQ | chloroquine |
| DHA | dehydroascorbic acid |
| DFO | deferoxamine mesylate |
| DCFH-DA | dichlorodihydrofluorescein diacetate |
| FCS | fetal calf serum |
| Fe | FeCl3 |
| Fe2+ | ferrous iron |
| Fe3+ | ferric iron |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| GPCR | G protein-coupled receptor |
| GPX4 | glutathione peroxidase 4 |
| HIF-1 | hypoxia-inducible factor-1 |
| 5-hmC | 5-hydroxymethylcytosine |
| HRP | horseradish peroxidase |
| LC3B | microtubule-associated proteins 1A/1B light chain 3B |
| LIP | labile iron pool |
| Mg | magnesium |
| MUH | 4-methylumbelliferyl heptanoate |
| NSCLC | non-small cell lung cancer |
| OS | overall survival |
| PI | propidium iodide |
| PMSF | phenylmethanesulfonyl fluoride |
| PVDF | polyvinylidene difluoride |
| RAPA | rapamycin |
| ROS | reactive oxygen species |
| RSL3 | RAS-selective lethal 3 |
| SDS | sodium dodecyl sulfate |
| SRB | sulforhodamine B |
| STS | staurosporine |
| SVCT2 | sodium-dependent vitamin C transporter 2 |
| TBH | tert-butyl hydroperoxide |
| TBST | Tris-buffered saline with Tween-20 |
| TCA | trichloroacetic acid |
| TfR1 | transferrin receptor 1 |
| TMZ | temozolomide |
| TRP | transient receptor potential channel |
References
- Böttger, F.; Vallés-Martí, A.; Cahn, L.; Jimenez, C.R. High-dose intravenous vitamin C, a promising multi-targeting agent in the treatment of cancer. J. Exp. Clin. Cancer Res. 2021, 40, 343. [Google Scholar] [CrossRef] [PubMed]
- Buettner, G.R.; Jurkiewicz, B.A. Catalytic metals, ascorbate and free radicals: Combinations to avoid. Radiat. Res. 1996, 145, 532–541. [Google Scholar] [CrossRef]
- Wang, P.; Shi, Q.; Deng, W.-H.; Yu, J.; Zuo, T.; Mei, F.-C.; Wang, W.-X. Relationship between expression of NADPH oxidase 2 and invasion and prognosis of human gastric cancer. World J. Gastroenterol. 2015, 21, 6271–6279. [Google Scholar] [CrossRef]
- Schoenfeld, J.D.; Sibenaller, Z.A.; Mapuskar, K.A.; Wagner, B.A.; Cramer-Morales, K.L.; Furqan, M.; Sandhu, S.; Carlisle, T.L.; Smith, M.C.; Abu Hejleh, T.; et al. O2- and H2O2-Mediated Disruption of Fe Metabolism Causes the Differential Susceptibility of NSCLC and GBM Cancer Cells to Pharmacological Ascorbate. Cancer Cell 2017, 31, 487–500.e8. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.; Violet, P.-C. Data Triumph at C. Cancer Cell 2017, 31, 467–469. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Pooput, C.; Kirk, K.L.; Krishna, M.C.; Khosh, D.B.; Drisko, J.; Levine, M. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 11105–11109. [Google Scholar] [CrossRef]
- Godoy, A.; Ormazabal, V.; Moraga-Cid, G.; Zúñiga, F.A.; Sotomayor, P.; Barra, V.; Vasquez, O.; Montecinos, V.; Mardones, L.; Guzmán, C.; et al. Mechanistic insights and functional determinants of the transport cycle of the ascorbic acid transporter SVCT2. Activation by sodium and absolute dependence on bivalent cations. J. Biol. Chem. 2007, 282, 615–624. [Google Scholar] [CrossRef]
- Allen, B.G.; Bodeker, K.L.; Smith, M.C.; Monga, V.; Sandhu, S.; Hohl, R.; Carlisle, T.; Brown, H.; Hollenbeck, N.; Vollstedt, S.; et al. First-in-Human Phase I Clinical Trial of Pharmacologic Ascorbate Combined with Radiation and Temozolomide for Newly Diagnosed Glioblastoma. Clin. Cancer Res. 2019, 25, 6590–6597. [Google Scholar] [CrossRef]
- Afonso, M.; Brito, M.A. Therapeutic Options in Neuro-Oncology. Int. J. Mol. Sci. 2022, 23, 5351. [Google Scholar] [CrossRef]
- Martinez-Useros, J.; Martin-Galan, M.; Florez-Cespedes, M.; Garcia-Foncillas, J. Epigenetics of Most Aggressive Solid Tumors: Pathways, Targets and Treatments. Cancers 2021, 13, 3209. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Renner, O.; Burkard, M.; Michels, H.; Vollbracht, C.; Sinnberg, T.; Venturelli, S. Parenteral high-dose ascorbate—A possible approach for the treatment of glioblastoma (Review). Int. J. Oncol. 2021, 58, 35. [Google Scholar] [CrossRef] [PubMed]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef]
- Tan, A.C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef]
- Nualart, F.J.; Rivas, C.I.; Montecinos, V.P.; Godoy, A.S.; Guaiquil, V.H.; Golde, D.W.; Vera, J.C. Recycling of vitamin C by a bystander effect. J. Biol. Chem. 2003, 278, 10128–10133. [Google Scholar] [CrossRef]
- Burkard, M.; Niessner, H.; Leischner, C.; Piotrowsky, A.; Renner, O.; Marongiu, L.; Lauer, U.M.; Busch, C.; Sinnberg, T.; Venturelli, S. High-Dose Ascorbate in Combination with Anti-PD1 Checkpoint Inhibition as Treatment Option for Malignant Melanoma. Cells 2023, 12, 254. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, J.E.; Kahng, J.Y.; Kim, S.H.; Park, J.S.; Yoon, S.J.; Um, J.-Y.; Kim, W.K.; Lee, J.-K.; Park, J.; et al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature 2018, 560, 243–247. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, L.; Wang, S.; Zhao, B.; Lv, H.; Shang, P. Labile iron affects pharmacological ascorbate-induced toxicity in osteosarcoma cell lines. Free Radic. Res. 2020, 54, 385–396. [Google Scholar] [CrossRef]
- Brandt, K.E.; Falls, K.C.; Schoenfeld, J.D.; Rodman, S.N.; Gu, Z.; Zhan, F.; Cullen, J.J.; Wagner, B.A.; Buettner, G.R.; Allen, B.G.; et al. Augmentation of intracellular iron using iron sucrose enhances the toxicity of pharmacological ascorbate in colon cancer cells. Redox Biol. 2018, 14, 82–87. [Google Scholar] [CrossRef]
- Tsuma-Kaneko, M.; Sawanobori, M.; Kawakami, S.; Uno, T.; Nakamura, Y.; Onizuka, M.; Ando, K.; Kawada, H. Iron removal enhances vitamin C-induced apoptosis and growth inhibition of K-562 leukemic cells. Sci. Rep. 2018, 8, 17377. [Google Scholar] [CrossRef] [PubMed]
- Mojić, M.; Bogdanović Pristov, J.; Maksimović-Ivanić, D.; Jones, D.R.; Stanić, M.; Mijatović, S.; Spasojević, I. Extracellular iron diminishes anticancer effects of vitamin C: An in vitro study. Sci. Rep. 2014, 4, 5955. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; Zhao, L.; Yu, J.; Hou, Y.; Ai, N.; Lu, J.-J.; Ge, W.; Chen, X. Exogenous iron impairs the anti-cancer effect of ascorbic acid both in vitro and in vivo. J. Adv. Res. 2023, 46, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Chae, J.S.; Shin, H.; Shin, Y.; Kim, Y.; Kil, E.-J.; Byun, H.-S.; Cho, S.-H.; Park, S.; Lee, S.; et al. Enhanced Anticancer Effect of Adding Magnesium to Vitamin C Therapy: Inhibition of Hormetic Response by SVCT-2 Activation. Transl. Oncol. 2020, 13, 401–409. [Google Scholar] [CrossRef]
- Lőrincz, T.; Holczer, M.; Kapuy, O.; Szarka, A. The Interrelationship of Pharmacologic Ascorbate Induced Cell Death and Ferroptosis. Pathol. Oncol. Res. 2019, 25, 669–679. [Google Scholar] [CrossRef]
- Feng, H.; Schorpp, K.; Jin, J.; Yozwiak, C.E.; Hoffstrom, B.G.; Decker, A.M.; Rajbhandari, P.; Stokes, M.E.; Bender, H.G.; Csuka, J.M.; et al. Transferrin Receptor Is a Specific Ferroptosis Marker. Cell Rep. 2020, 30, 3411–3423.e7. [Google Scholar] [CrossRef]
- Wang, X.; Xu, S.; Zhang, L.; Cheng, X.; Yu, H.; Bao, J.; Lu, R. Vitamin C induces ferroptosis in anaplastic thyroid cancer cells by ferritinophagy activation. Biochem. Biophys. Res. Commun. 2021, 551, 46–53. [Google Scholar] [CrossRef]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.C.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef]
- El Banna, N.; Hatem, E.; Heneman-Masurel, A.; Léger, T.; Baïlle, D.; Vernis, L.; Garcia, C.; Martineau, S.; Dupuy, C.; Vagner, S.; et al. Redox modifications of cysteine-containing proteins, cell cycle arrest and translation inhibition: Involvement in vitamin C-induced breast cancer cell death. Redox Biol. 2019, 26, 101290. [Google Scholar] [CrossRef]
- Castro, M.L.; Carson, G.M.; McConnell, M.J.; Herst, P.M. High Dose Ascorbate Causes Both Genotoxic and Metabolic Stress in Glioma Cells. Antioxidants 2017, 6, 58. [Google Scholar] [CrossRef]
- Gokturk, D.; Kelebek, H.; Ceylan, S.; Yilmaz, D.M. The Effect of Ascorbic Acid over the Etoposide- and Temozolomide-Mediated Cytotoxicity in Glioblastoma Cell Culture: A Molecular Study. Turk. Neurosurg. 2018, 28, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Deubzer, B.; Mayer, F.; Kuçi, Z.; Niewisch, M.; Merkel, G.; Handgretinger, R.; Bruchelt, G. H(2)O(2)-mediated cytotoxicity of pharmacologic ascorbate concentrations to neuroblastoma cells: Potential role of lactate and ferritin. Cell. Physiol. Biochem. 2010, 25, 767–774. [Google Scholar] [CrossRef]
- Ryszawy, D.; Pudełek, M.; Catapano, J.; Ciarach, M.; Setkowicz, Z.; Konduracka, E.; Madeja, Z.; Czyż, J. High doses of sodium ascorbate interfere with the expansion of glioblastoma multiforme cells in vitro and in vivo. Life Sci. 2019, 232, 116657. [Google Scholar] [CrossRef]
- Yanase, F.; Fujii, T.; Naorungroj, T.; Belletti, A.; Luethi, N.; Carr, A.C.; Young, P.J.; Bellomo, R. Harm of IV High-Dose Vitamin C Therapy in Adult Patients: A Scoping Review. Crit. Care Med. 2020, 48, e620–e628. [Google Scholar] [CrossRef] [PubMed]
- Tolkien, Z.; Stecher, L.; Mander, A.P.; Pereira, D.I.A.; Powell, J.J. Ferrous sulfate supplementation causes significant gastrointestinal side-effects in adults: A systematic review and meta-analysis. PLoS ONE 2015, 10, e0117383. [Google Scholar] [CrossRef]
- Cancelo-Hidalgo, M.J.; Castelo-Branco, C.; Palacios, S.; Haya-Palazuelos, J.; Ciria-Recasens, M.; Manasanch, J.; Pérez-Edo, L. Tolerability of different oral iron supplements: A systematic review. Curr. Med. Res. Opin. 2013, 29, 291–303. [Google Scholar] [CrossRef]
- Gröber, U.; Schmidt, J.; Kisters, K. Magnesium in Prevention and Therapy. Nutrients 2015, 7, 8199–8226. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, C.; Molenaar, I.G.M.; Dachs, G.U.; Currie, M.J.; Sykes, P.H.; Vissers, M.C.M. Low ascorbate levels are associated with increased hypoxia-inducible factor-1 activity and an aggressive tumor phenotype in endometrial cancer. Cancer Res. 2010, 70, 5749–5758. [Google Scholar] [CrossRef]
- Campbell, E.J.; Dachs, G.U.; Morrin, H.R.; Davey, V.C.; Robinson, B.A.; Vissers, M.C.M. Activation of the hypoxia pathway in breast cancer tissue and patient survival are inversely associated with tumor ascorbate levels. BMC Cancer 2019, 19, 307. [Google Scholar] [CrossRef]
- Crake, R.L.I.; Burgess, E.R.; Wiggins, G.A.R.; Magon, N.J.; Das, A.B.; Vissers, M.C.M.; Morrin, H.R.; Royds, J.A.; Slatter, T.L.; Robinson, B.A.; et al. Ascorbate content of clinical glioma tissues is related to tumour grade and to global levels of 5-hydroxymethyl cytosine. Sci. Rep. 2022, 12, 14845. [Google Scholar] [CrossRef]
- Sinnberg, T.; Noor, S.; Venturelli, S.; Berger, A.; Schuler, P.; Garbe, C.; Busch, C. The ROS-induced cytotoxicity of ascorbate is attenuated by hypoxia and HIF-1alpha in the NCI60 cancer cell lines. J. Cell. Mol. Med. 2014, 18, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [PubMed]
- Wagner, B.A.; Witmer, J.R.; van’t Erve, T.J.; Buettner, G.R. An Assay for the Rate of Removal of Extracellular Hydrogen Peroxide by Cells. Redox Biol. 2013, 1, 210–217. [Google Scholar] [CrossRef]
- Klingelhoeffer, C.; Kämmerer, U.; Koospal, M.; Mühling, B.; Schneider, M.; Kapp, M.; Kübler, A.; Germer, C.-T.; Otto, C. Natural resistance to ascorbic acid induced oxidative stress is mainly mediated by catalase activity in human cancer cells and catalase-silencing sensitizes to oxidative stress. BMC Complement. Altern. Med. 2012, 12, 61. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Burska, A.N.; Ilyassova, B.; Dildabek, A.; Khamijan, M.; Begimbetova, D.; Molnár, F.; Sarbassov, D.D. Enhancing an Oxidative “Trojan Horse” Action of Vitamin C with Arsenic Trioxide for Effective Suppression of KRAS-Mutant Cancers: A Promising Path at the Bedside. Cells 2022, 11, 3454. [Google Scholar] [CrossRef]
- Tian, W.; Wang, Z.; Tang, N.-N.; Li, J.-T.; Liu, Y.; Chu, W.-F.; Yang, B.-F. Ascorbic Acid Sensitizes Colorectal Carcinoma to the Cytotoxicity of Arsenic Trioxide via Promoting Reactive Oxygen Species-Dependent Apoptosis and Pyroptosis. Front. Pharmacol. 2020, 11, 123. [Google Scholar] [CrossRef]
- Noguera, N.I.; Pelosi, E.; Angelini, D.F.; Piredda, M.L.; Guerrera, G.; Piras, E.; Battistini, L.; Massai, L.; Berardi, A.; Catalano, G.; et al. High-dose ascorbate and arsenic trioxide selectively kill acute myeloid leukemia and acute promyelocytic leukemia blasts in vitro. Oncotarget 2017, 8, 32550–32565. [Google Scholar] [CrossRef]
- Xu, Z.-Y.; Gao, J.-F.; Zhang, L. Association of CFI, IL-8, TF and TFR2 genetic polymorphisms with age-related macular degeneration in a northeastern Chinese population. Curr. Eye Res. 2022, 47, 786–790. [Google Scholar] [CrossRef]
- Tisato, F.; Marzano, C.; Porchia, M.; Pellei, M.; Santini, C. Copper in diseases and treatments, and copper-based anticancer strategies. Med. Res. Rev. 2010, 30, 708–749. [Google Scholar] [CrossRef] [PubMed]
- Freund, E.; Liedtke, K.-R.; Miebach, L.; Wende, K.; Heidecke, A.; Kaushik, N.K.; Choi, E.H.; Partecke, L.-I.; Bekeschus, S. Identification of Two Kinase Inhibitors with Synergistic Toxicity with Low-Dose Hydrogen Peroxide in Colorectal Cancer Cells in vitro. Cancers 2020, 12, 122. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Shen, Z.; Yang, Q.; Sui, F.; Pu, J.; Ma, J.; Ma, S.; Yao, D.; Ji, M.; Hou, P. Vitamin C kills thyroid cancer cells through ROS-dependent inhibition of MAPK/ERK and PI3K/AKT pathways via distinct mechanisms. Theranostics 2019, 9, 4461–4473. [Google Scholar] [CrossRef]
- Graczyk-Jarzynka, A.; Goral, A.; Muchowicz, A.; Zagozdzon, R.; Winiarska, M.; Bajor, M.; Trzeciecka, A.; Fidyt, K.; Krupka, J.A.; Cyran, J.; et al. Inhibition of thioredoxin-dependent H2O2 removal sensitizes malignant B-cells to pharmacological ascorbate. Redox Biol. 2019, 21, 101062. [Google Scholar] [CrossRef]
- Murillo, M.I.; Gaiddon, C.; Le Lagadec, R. Targeting of the intracellular redox balance by metal complexes towards anticancer therapy. Front. Chem. 2022, 10, 967337. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Azad, M.B.; Chen, Y.; Gibson, S.B. Regulation of autophagy by reactive oxygen species (ROS): Implications for cancer progression and treatment. Antioxid. Redox Signal. 2009, 11, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Oronowicz, J.; Reinhard, J.; Reinach, P.S.; Ludwiczak, S.; Luo, H.; Omar Ba Salem, M.H.; Kraemer, M.M.; Biebermann, H.; Kakkassery, V.; Mergler, S. Ascorbate-induced oxidative stress mediates TRP channel activation and cytotoxicity in human etoposide-sensitive and -resistant retinoblastoma cells. Lab. Investig. 2021, 101, 70–88. [Google Scholar] [CrossRef]
- Li, L.; Li, S.; Hu, C.; Zhou, L.; Zhang, Y.; Wang, M.; Qi, Z. BKCa channel is a molecular target of vitamin C to protect against ischemic brain stroke. Mol. Membr. Biol. 2019, 35, 9–20. [Google Scholar] [CrossRef]
- Ren, J.G.; Xia, H.L.; Just, T.; Dai, Y.R. Hydroxyl radical-induced apoptosis in human tumor cells is associated with telomere shortening but not telomerase inhibition and caspase activation. FEBS Lett. 2001, 488, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Tóth, S.Z.; Lőrincz, T.; Szarka, A. Concentration Does Matter: The Beneficial and Potentially Harmful Effects of Ascorbate in Humans and Plants. Antioxid. Redox Signal. 2018, 29, 1516–1533. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Dielschneider, R.F.; Henson, E.S.; Xiao, W.; Choquette, T.R.; Blankstein, A.R.; Chen, Y.; Gibson, S.B. Ferroptosis and autophagy induced cell death occur independently after siramesine and lapatinib treatment in breast cancer cells. PLoS ONE 2017, 12, e0182921. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piotrowsky, A.; Burkard, M.; Hammerschmidt, K.; Ruple, H.K.; Nonnenmacher, P.; Schumacher, M.; Leischner, C.; Berchtold, S.; Marongiu, L.; Kufer, T.A.; et al. Analysis of High-Dose Ascorbate-Induced Cytotoxicity in Human Glioblastoma Cells and the Role of Dehydroascorbic Acid and Iron. Antioxidants 2024, 13, 1095. https://doi.org/10.3390/antiox13091095
Piotrowsky A, Burkard M, Hammerschmidt K, Ruple HK, Nonnenmacher P, Schumacher M, Leischner C, Berchtold S, Marongiu L, Kufer TA, et al. Analysis of High-Dose Ascorbate-Induced Cytotoxicity in Human Glioblastoma Cells and the Role of Dehydroascorbic Acid and Iron. Antioxidants. 2024; 13(9):1095. https://doi.org/10.3390/antiox13091095
Chicago/Turabian StylePiotrowsky, Alban, Markus Burkard, Katharina Hammerschmidt, Hannah K. Ruple, Pia Nonnenmacher, Monika Schumacher, Christian Leischner, Susanne Berchtold, Luigi Marongiu, Thomas A. Kufer, and et al. 2024. "Analysis of High-Dose Ascorbate-Induced Cytotoxicity in Human Glioblastoma Cells and the Role of Dehydroascorbic Acid and Iron" Antioxidants 13, no. 9: 1095. https://doi.org/10.3390/antiox13091095