
Indole-3-Carboxaldehyde Alleviates LPS-Induced Intestinal Inflammation by Inhibiting ROS Production and NLRP3 Inflammasome Activation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Intestinal Epithelial Cell Culture and LPS Treatment
2.3. Animals and Treatments
2.4. Immunofluorescence (IF)
2.5. DCFH-DA, MitoTracker Red, and JC-1 Staining
2.6. Intestinal Epithelial Cell Permeability Analysis
2.7. Western Blotting (WB)
2.8. Real-Time Quantitative PCR (qPCR)
2.9. Enzyme-Linked Immuno-Sorbent Assay (ELISA)
2.10. Statistical Analysis
3. Results
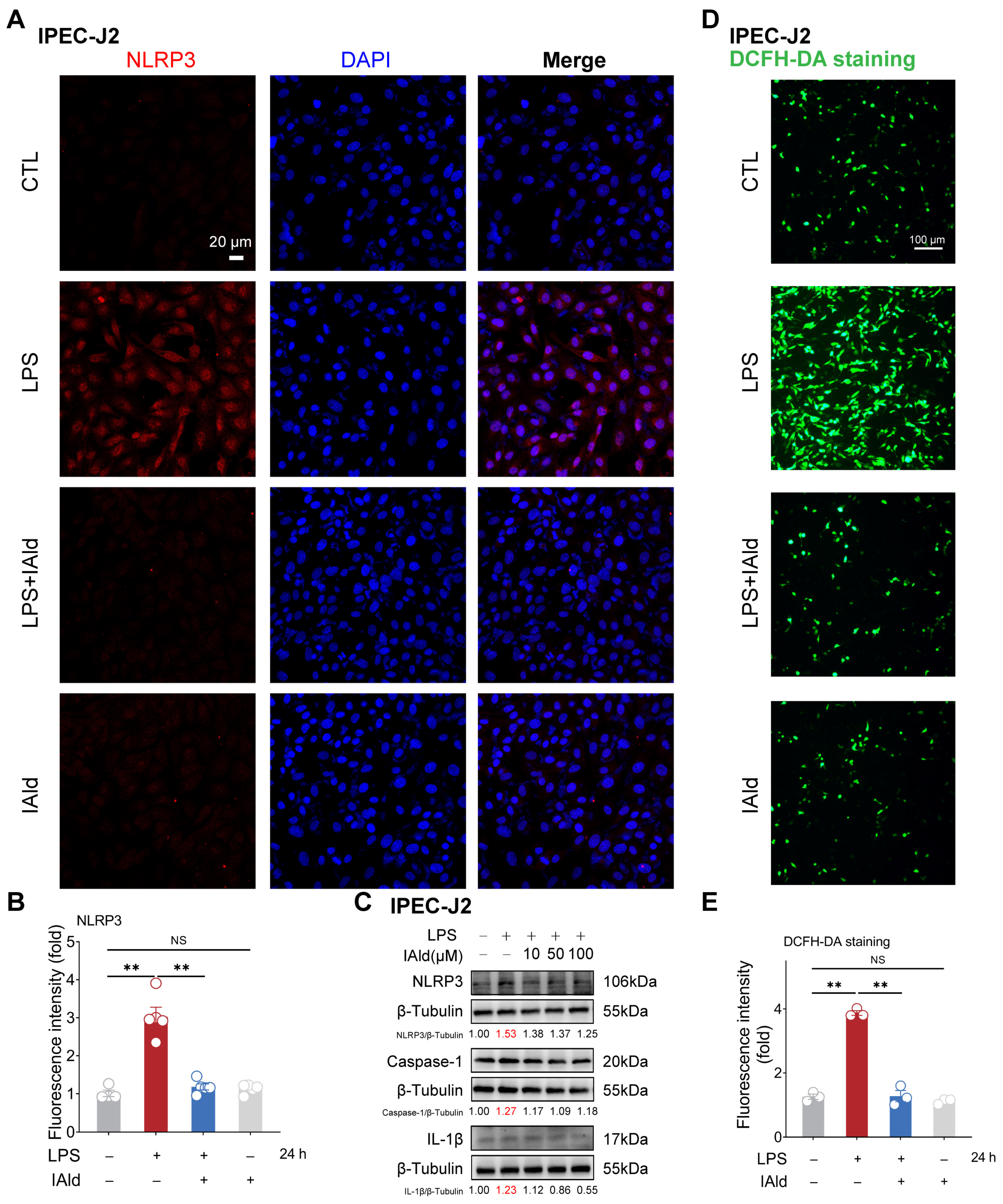
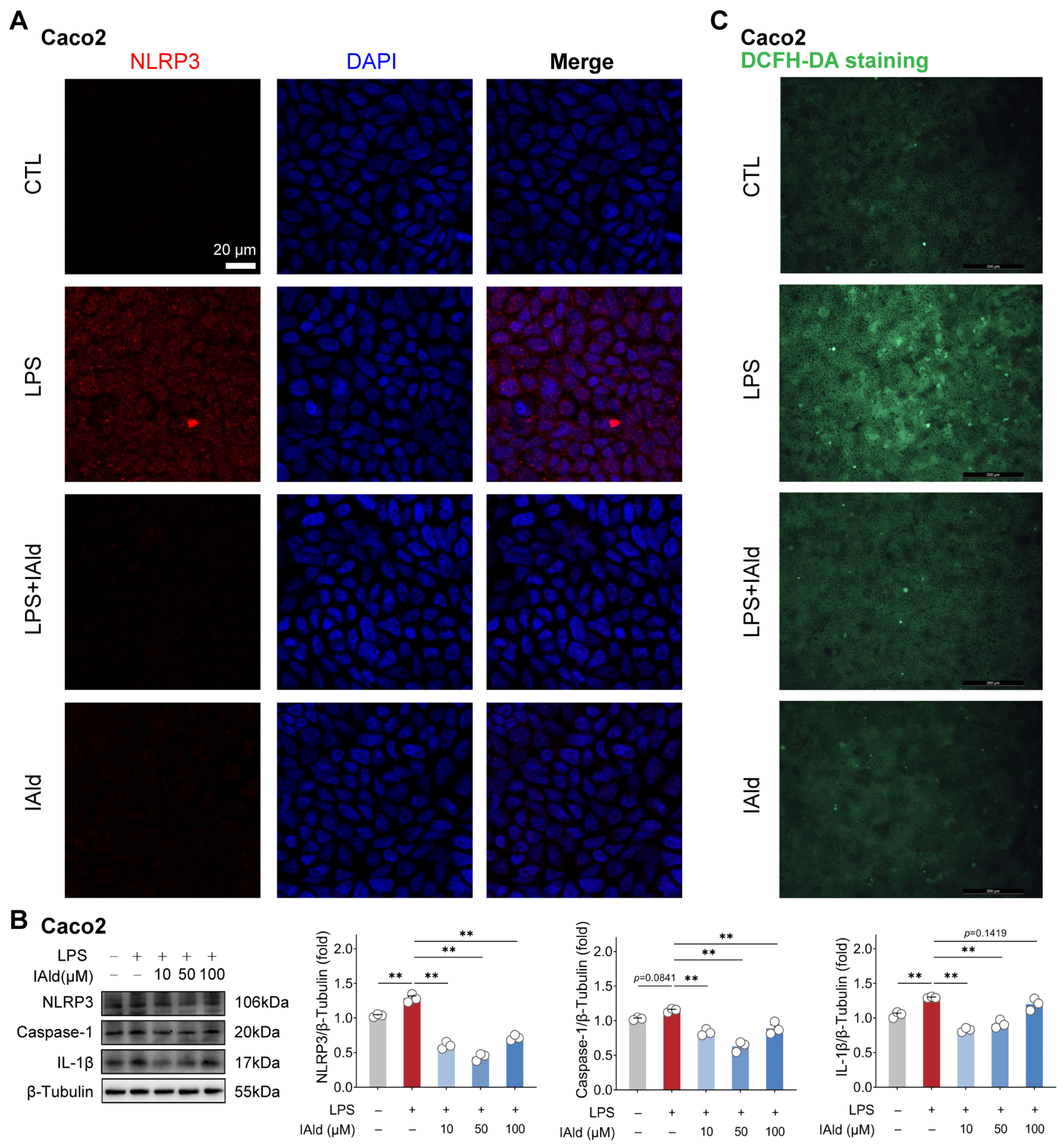
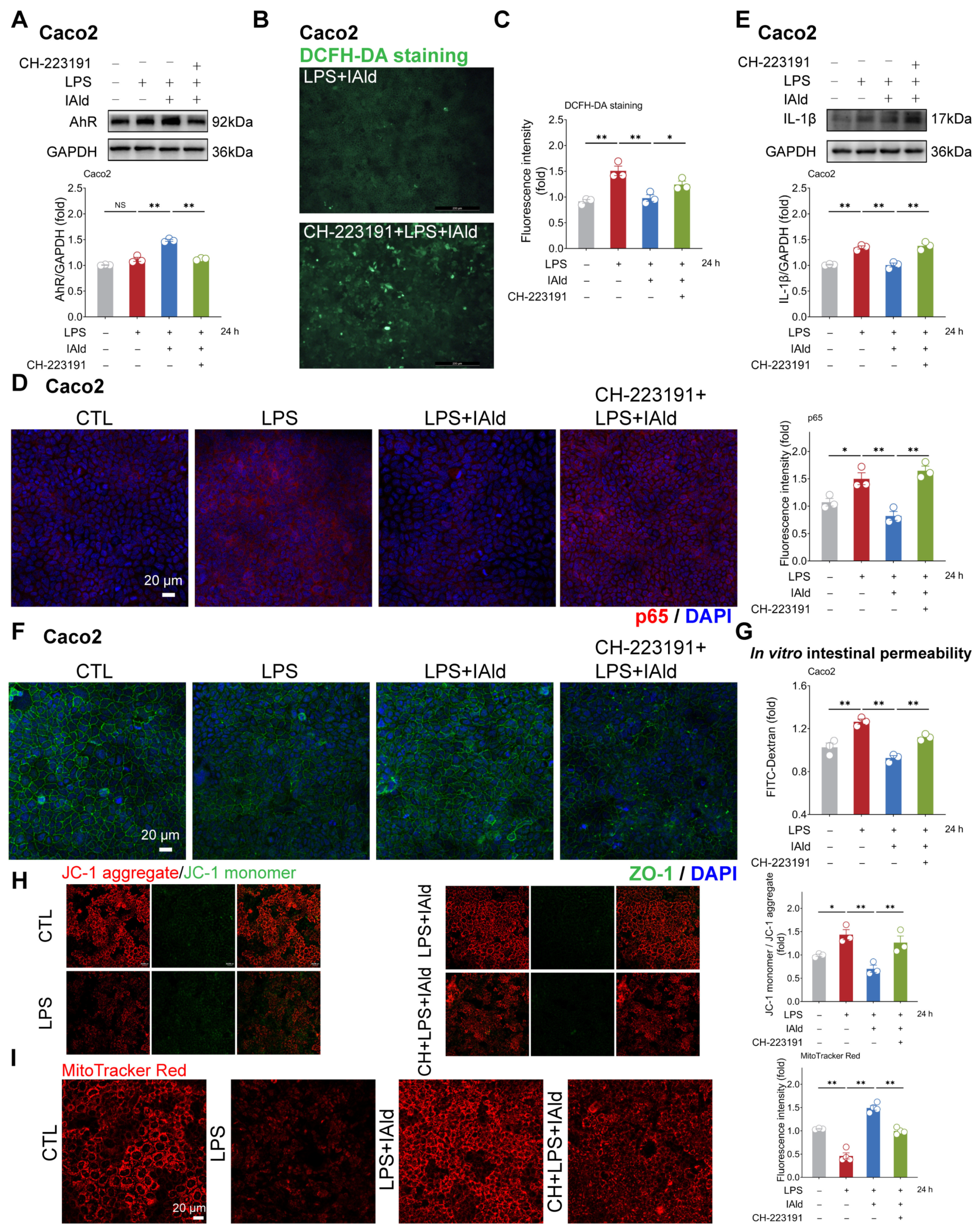
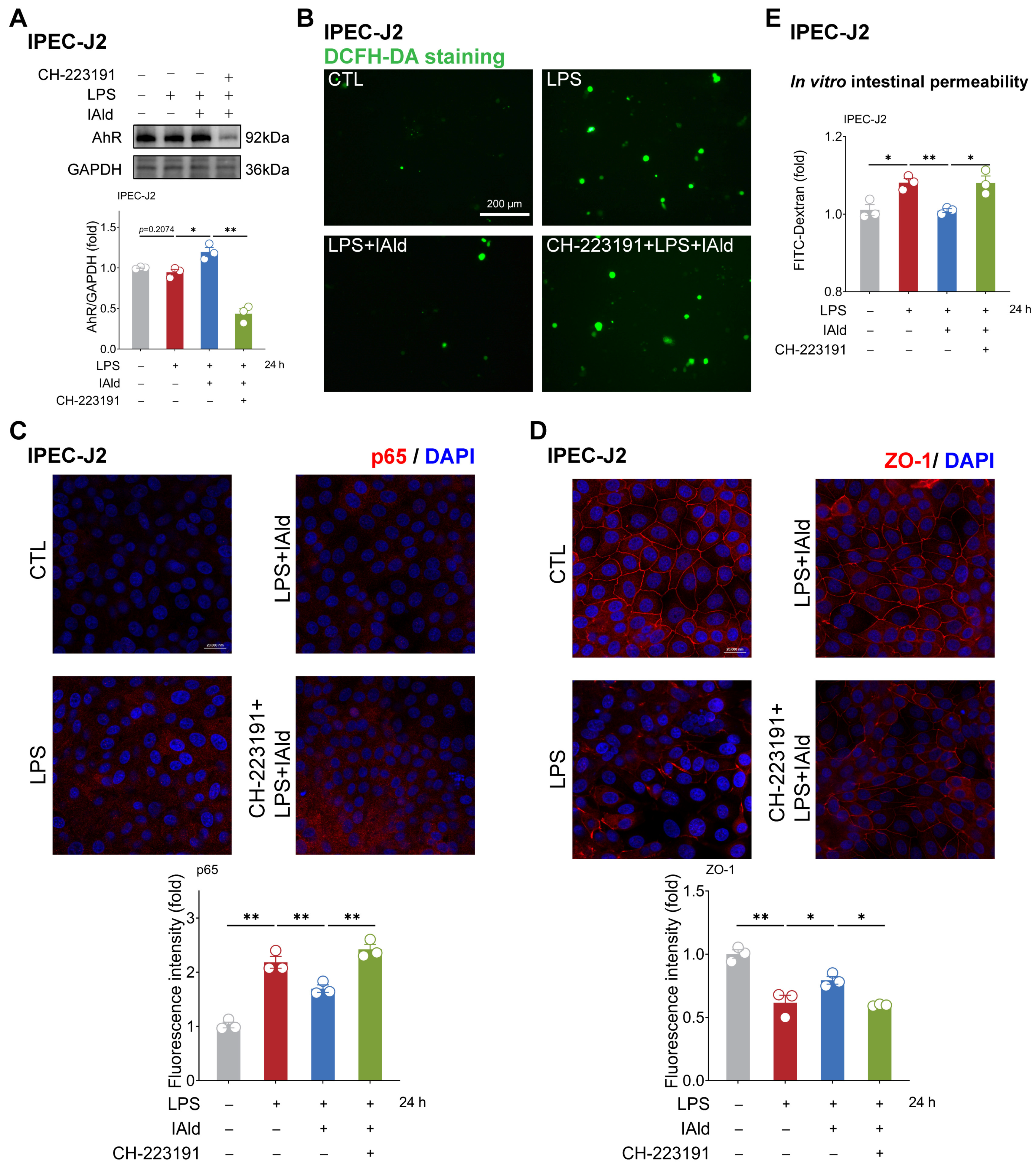
3.1. IAld inhibits LPS-Induced NLRP3 Inflammasome Activation in Intestinal Epithelial Cells
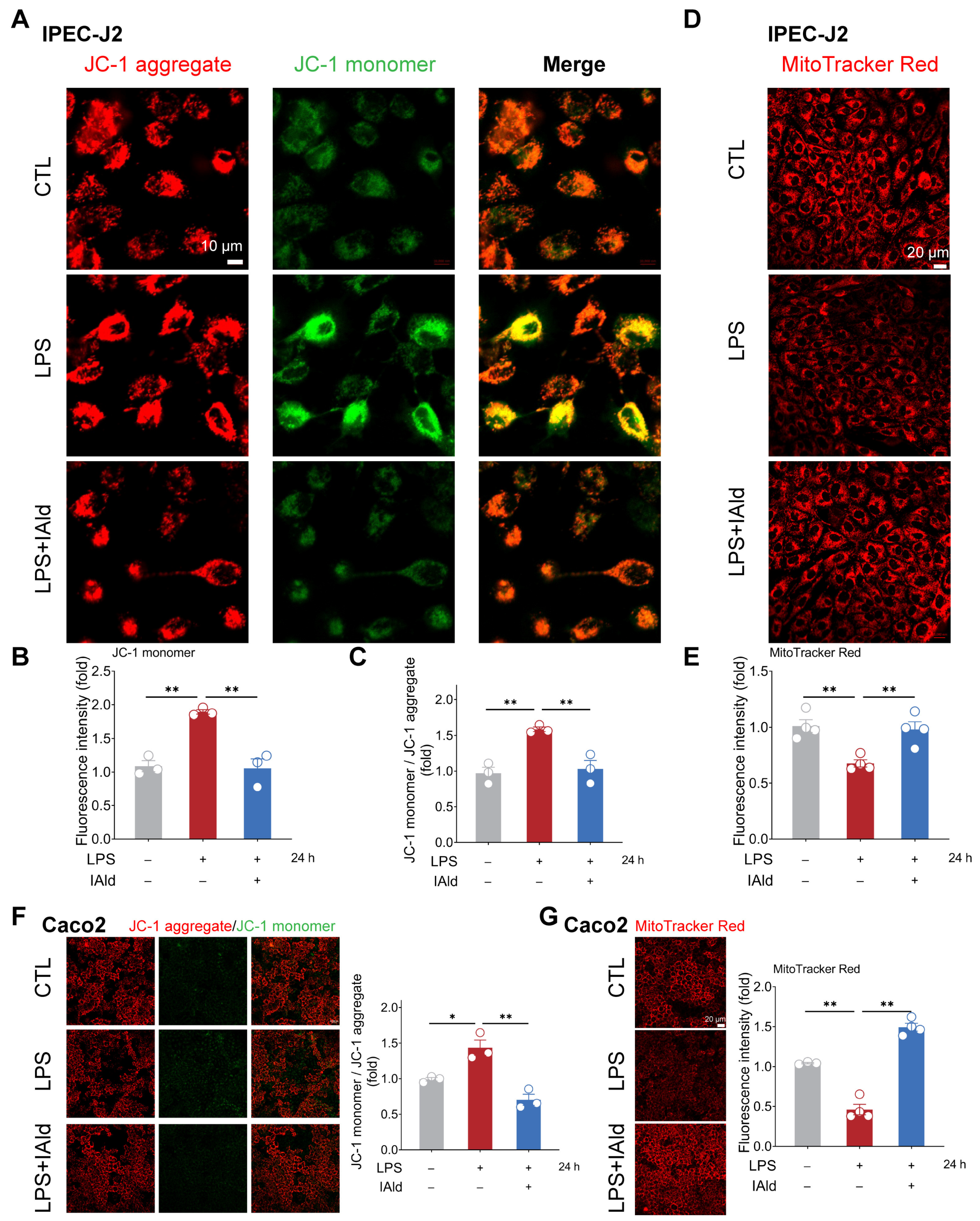
3.2. IAld Prevents LPS-Induced Mitochondrial Dysfunction in Intestinal Epithelial Cells
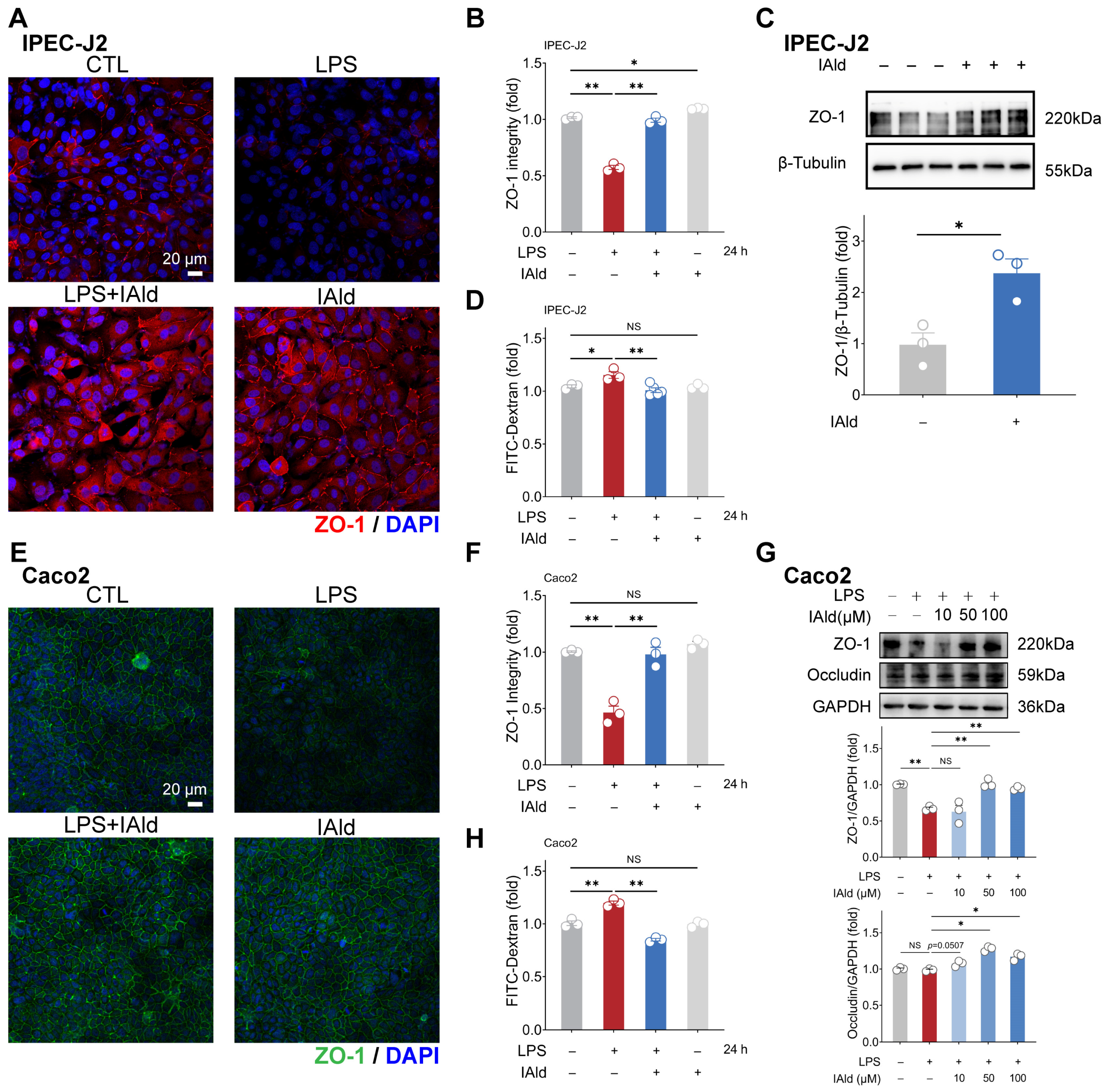
3.3. IAld Alleviates LPS-Induced Barrier Dysfunction in Intestinal Epithelial Cells
3.4. IAld Alleviates Intestinal Inflammatory Injury in an AhR-Dependent Manner
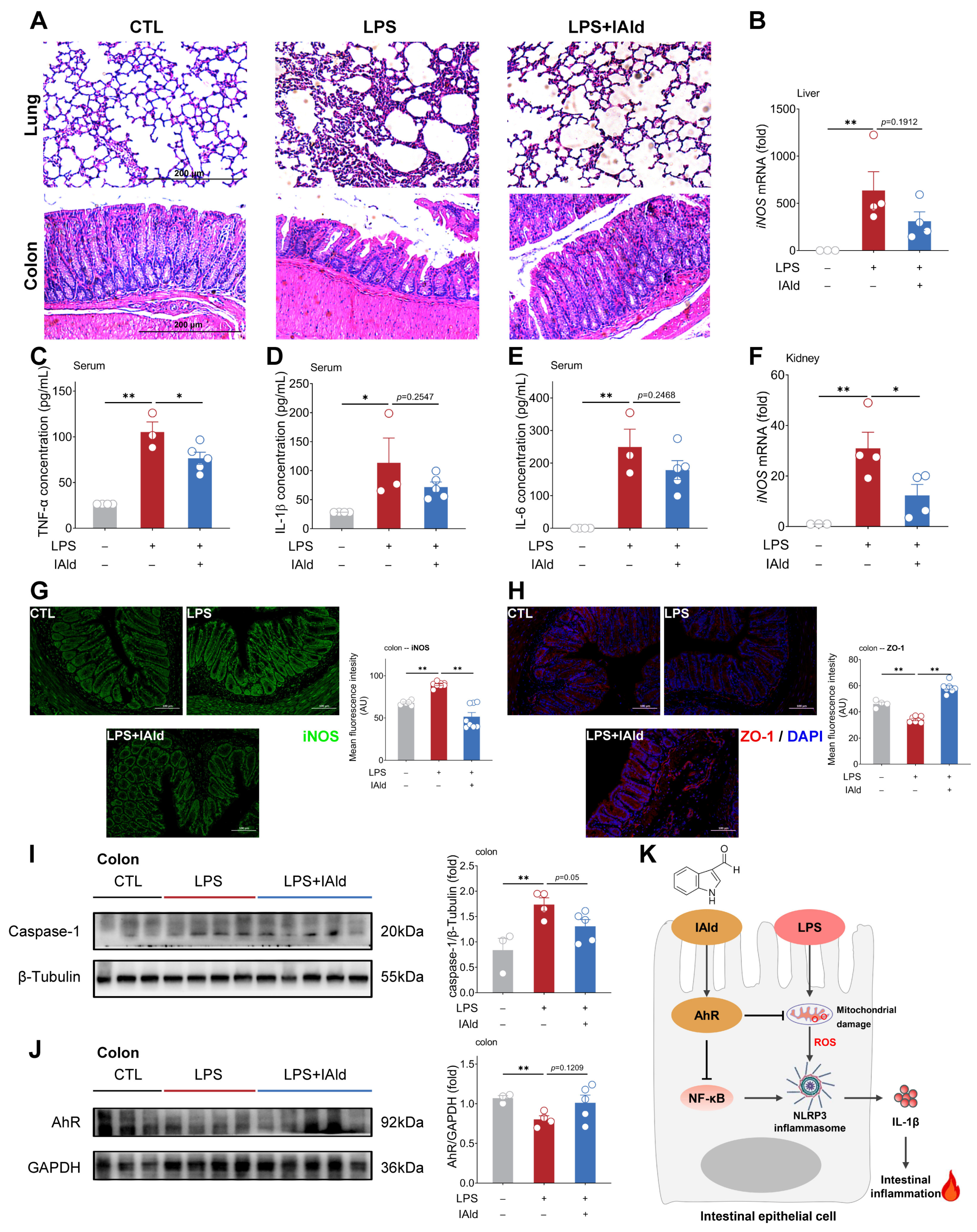
3.5. IAld Alleviates LPS-Induced Intestinal Inflammatory Response in Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhao, L.; Zhang, T.; Zhang, K. Pharmacological effects of ginseng and ginsenosides on intestinal inflammation and the immune system. Front. Immunol. 2024, 15, 1353614. [Google Scholar] [CrossRef]
- Kwon, S.J.; Khan, M.S.; Kim, S.G. Intestinal Inflammation and Regeneration-Interdigitating Processes Controlled by Dietary Lipids in Inflammatory Bowel Disease. Int. J. Mol. Sci. 2024, 25, 1311. [Google Scholar] [CrossRef]
- Zhang, X.; Akhtar, M.; Chen, Y.; Ma, Z.; Liang, Y.; Shi, D.; Cheng, R.; Cui, L.; Hu, Y.; Nafady, A.A.; et al. Chicken jejunal microbiota improves growth performance by mitigating intestinal inflammation. Microbiome 2022, 10, 107. [Google Scholar]
- Pi, Y.; Wu, Y.; Zhang, X.; Lu, D.; Han, D.; Zhao, J.; Zheng, X.; Zhang, S.; Ye, H.; Lian, S.; et al. Gut microbiota-derived ursodeoxycholic acid alleviates low birth weight-induced colonic inflammation by enhancing M2 macrophage polarization. Microbiome 2023, 11, 19. [Google Scholar] [CrossRef]
- Chen, H.; Jia, Z.; He, M.; Chen, A.; Zhang, X.; Xu, J.; Wang, C. Arula-7 powder improves diarrhea and intestinal epithelial tight junction function associated with its regulation of intestinal flora in calves infected with pathogenic Escherichia coli O(1). Microbiome 2023, 11, 172. [Google Scholar] [CrossRef]
- Foppa, C.; Rizkala, T.; Repici, A.; Hassan, C.; Spinelli, A. Microbiota and IBD: Current knowledge and future perspectives. Dig. Liver Dis. Off. J. Ital. Soc. Gastroenterol. Ital. Assoc. Study Liver 2024, 56, 911–922. [Google Scholar] [CrossRef]
- Heller, C.; Moss, A.C.; Rubin, D.T. Overview to Challenges in IBD 2024–2029. Inflamm. Bowel Dis. 2024, 30 (Suppl. S2), S1–S4. [Google Scholar] [CrossRef]
- Zhan, X.; Li, Q.; Xu, G.; Xiao, X.; Bai, Z. The mechanism of NLRP3 inflammasome activation and its pharmacological inhibitors. Front. Immunol. 2022, 13, 1109938. [Google Scholar] [CrossRef]
- Zeng, B.; Huang, Y.; Chen, S.; Xu, R.; Xu, L.; Qiu, J.; Shi, F.; Liu, S.; Zha, Q.; Ouyang, D.; et al. Dextran sodium sulfate potentiates NLRP3 inflammasome activation by modulating the KCa3.1 potassium channel in a mouse model of colitis. Cell. Mol. Immunol. 2022, 19, 925–943. [Google Scholar] [CrossRef]
- Chen, Y.; He, H.; Lin, B.; Chen, Y.; Deng, X.; Jiang, W.; Zhou, R. RRx-001 ameliorates inflammatory diseases by acting as a potent covalent NLRP3 inhibitor. Cell. Mol. Immunol. 2021, 18, 1425–1436. [Google Scholar] [CrossRef]
- Rawat, M.; Nighot, M.; Al-Sadi, R.; Gupta, Y.; Viszwapriya, D.; Yochum, G.; Koltun, W.; Ma, T.Y. IL1B Increases Intestinal Tight Junction Permeability by Up-regulation of MIR200C-3p, Which Degrades Occludin mRNA. Gastroenterology 2020, 159, 1375–1389. [Google Scholar] [CrossRef]
- Nowarski, R.; Jackson, R.; Gagliani, N.; de Zoete, M.R.; Palm, N.W.; Bailis, W.; Low, J.S.; Harman, C.C.; Graham, M.; Elinav, E.; et al. Epithelial IL-18 Equilibrium Controls Barrier Function in Colitis. Cell 2015, 163, 1444–1456. [Google Scholar] [CrossRef]
- Xia, B.; Zhong, R.; Wu, W.; Luo, C.; Meng, Q.; Gao, Q.; Zhao, Y.; Chen, L.; Zhang, S.; Zhao, X.; et al. Mucin O-glycan-microbiota axis orchestrates gut homeostasis in a diarrheal pig model. Microbiome 2022, 10, 139. [Google Scholar] [CrossRef]
- Cao, J.; Lu, M.; Yan, W.; Li, L.; Ma, H. Dehydroepiandrosterone alleviates intestinal inflammatory damage via GPR30-mediated Nrf2 activation and NLRP3 inflammasome inhibition in colitis mice. Free Radic. Biol. Med. 2021, 172, 386–402. [Google Scholar] [CrossRef]
- Miyamoto, K.; Sujino, T.; Kanai, T. The tryptophan metabolic pathway of the microbiome and host cells in health and disease. Int. Immunol. 2024, dxae035. [Google Scholar] [CrossRef]
- Metidji, A.; Omenetti, S.; Crotta, S.; Li, Y.; Nye, E.; Ross, E.; Li, V.; Maradana, M.R.; Schiering, C.; Stockinger, B. The Environmental Sensor AHR Protects from Inflammatory Damage by Maintaining Intestinal Stem Cell Homeostasis and Barrier Integrity. Immunity 2018, 49, 353–362. [Google Scholar] [CrossRef]
- Zhao, X.; Pang, J.; Zhang, W.; Peng, X.; Yang, Z.; Bai, G.; Xia, Y. Tryptophan metabolism and piglet diarrhea: Where we stand and the challenges ahead. Anim. Nutr. 2024, 17, 123–133. [Google Scholar] [CrossRef]
- Monteleone, I.; Rizzo, A.; Sarra, M.; Sica, G.; Sileri, P.; Biancone, L.; MacDonald, T.T.; Pallone, F.; Monteleone, G. Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology 2011, 141, 237–248. [Google Scholar] [CrossRef]
- Furumatsu, K.; Nishiumi, S.; Kawano, Y.; Ooi, M.; Yoshie, T.; Shiomi, Y.; Kutsumi, H.; Ashida, H.; Fujii-Kuriyama, Y.; Azuma, T.; et al. A role of the aryl hydrocarbon receptor in attenuation of colitis. Dig. Dis. Sci. 2011, 56, 2532–2544. [Google Scholar] [CrossRef]
- Scott, S.A.; Fu, J.; Chang, P.V. Microbial tryptophan metabolites regulate gut barrier function via the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA 2020, 117, 19376–19387. [Google Scholar] [CrossRef]
- Ma, M.; Wang, Y.; Fan, S.; Huang, Y.; Su, X.; Lu, C. Urolithin A Alleviates Colitis in Mice by Improving Gut Microbiota Dysbiosis, Modulating Microbial Tryptophan Metabolism, and Triggering AhR Activation. J. Agric. Food Chem. 2023, 71, 7710–7722. [Google Scholar] [CrossRef]
- Wang, M.; Guo, J.; Hart, A.L.; Li, J.V. Indole-3-Aldehyde Reduces Inflammatory Responses and Restores Intestinal Epithelial Barrier Function Partially via Aryl Hydrocarbon Receptor (AhR) in Experimental Colitis Models. J. Inflamm. Res. 2023, 16, 5845–5864. [Google Scholar] [CrossRef]
- Li, Z.; Wang, K.; Ji, X.; Wang, H.; Zhang, Y. ACE2 suppresses the inflammatory response in LPS-induced porcine intestinal epithelial cells via regulating the NF-κB and MAPK pathways. Peptides 2022, 149, 170717. [Google Scholar] [CrossRef]
- Cheng, L.; Wu, H.; Cai, X.; Zhang, Y.; Yu, S.; Hou, Y.; Yin, Z.; Yan, Q.; Wang, Q.; Sun, T.; et al. A Gpr35-tuned gut microbe-brain metabolic axis regulates depressive-like behavior. Cell Host Microbe 2024, 32, 227–243. [Google Scholar] [CrossRef]
- Swimm, A.; Giver, C.R.; DeFilipp, Z.; Rangaraju, S.; Sharma, A.; Ulezko Antonova, A.; Sonowal, R.; Capaldo, C.; Powell, D.; Qayed, M.; et al. Indoles derived from intestinal microbiota act via type I interferon signaling to limit graft-versus-host disease. Blood 2018, 132, 2506–2519. [Google Scholar] [CrossRef]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; De Luca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef]
- Chen, X.; Wu, R.; Li, L.; Zeng, Y.; Chen, J.; Wei, M.; Feng, Y.; Chen, G.; Wang, Y.; Lin, L.; et al. Pregnancy-induced changes to the gut microbiota drive macrophage pyroptosis and exacerbate septic inflammation. Immunity 2023, 56, 336–352. [Google Scholar] [CrossRef]
- Hao, H.; Cao, L.; Jiang, C.; Che, Y.; Zhang, S.; Takahashi, S.; Wang, G.; Gonzalez, F.J. Farnesoid X Receptor Regulation of the NLRP3 Inflammasome Underlies Cholestasis-Associated Sepsis. Cell Metab. 2017, 25, 856–867. [Google Scholar] [CrossRef]
- Cao, J.; Li, L.; Yao, Y.; Xing, Y.; Ma, H. Dehydroepiandrosterone exacerbates nigericin-induced abnormal autophagy and pyroptosis via GPER activation in LPS-primed macrophages. Cell Death Dis. 2022, 13, 372. [Google Scholar] [CrossRef]
- Cao, J.; Li, Q.; Shen, X.; Yao, Y.; Li, L.; Ma, H. Dehydroepiandrosterone attenuates LPS-induced inflammatory responses via activation of Nrf2 in RAW264.7 macrophages. Mol. Immunol. 2021, 131, 97–111. [Google Scholar] [CrossRef]
- Coll, R.C.; Schroder, K.; Pelegrín, P. NLRP3 and pyroptosis blockers for treating inflammatory diseases. Trends Pharmacol. Sci. 2022, 43, 653–668. [Google Scholar] [CrossRef] [PubMed]
- Arrè, V.; Scialpi, R.; Centonze, M.; Giannelli, G.; Scavo, M.P.; Negro, R. The ‘speck’-tacular oversight of the NLRP3-pyroptosis pathway on gastrointestinal inflammatory diseases and tumorigenesis. J. Biomed. Sci. 2023, 30, 90. [Google Scholar] [CrossRef]
- Yang, Y.; Li, S.; Liu, K.; Zhang, Y.; Zhu, F.; Ben, T.; Chen, Z.; Zhi, F. Lipocalin-2-mediated intestinal epithelial cells pyroptosis via NF-κB/NLRP3/GSDMD signaling axis adversely affects inflammation in colitis. Biochim. Biophys. Acta Mol. Basis Dis. 2024, 1870, 167279. [Google Scholar] [CrossRef]
- Du, L.; Chen, C.; Yang, Y.H.; Zheng, Y.; Li, H.; Wu, Z.J.; Wu, H.; Miyashita, K.; Su, G.H. Fucoxanthin alleviates lipopolysaccharide-induced intestinal barrier injury in mice. Food Funct. 2024, 15, 6359–6373. [Google Scholar] [CrossRef]
- Qiu, W.; Zhang, X.; Pang, X.; Huang, J.; Zhou, S.; Wu, R.; Wang, R.; Tang, Z.; Su, R. Tert-butylhydroquinone attenuates LPS-induced pyroptosis of IPEC-J2 cells via downregulating HMGB1/TLR4/NF-κB axis. J. Anim. Physiol. Anim. Nutr. 2024, 108, 194–205. [Google Scholar] [CrossRef]
- Wang, Z.; Guo, L.; Yuan, C.; Zhu, C.; Li, J.; Zhong, H.; Mao, P.; Li, J.; Cui, L.; Dong, J.; et al. Staphylococcus pseudintermedius induces pyroptosis of canine corneal epithelial cells by activating the ROS-NLRP3 signalling pathway. Virulence 2024, 15, 2333271. [Google Scholar] [CrossRef]
- Wang, F.; Liang, Q.; Ma, Y.; Sun, M.; Li, T.; Lin, L.; Sun, Z.; Duan, J. Silica nanoparticles induce pyroptosis and cardiac hypertrophy via ROS/NLRP3/Caspase-1 pathway. Free Radic. Biol. Med. 2022, 182, 171–181. [Google Scholar] [CrossRef]
- Qiao, H.; Yang, B.; Lv, X.; Liu, Y. Exposure to TCBPA stimulates the growth of arterial smooth muscle cells through the activation of the ROS/NF-κB/NLRP3 signaling pathway. Toxicology 2024, 503, 153759. [Google Scholar] [CrossRef]
- Lavelle, A.; Sokol, H. Gut microbiota-derived metabolites as key actors in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 223–237. [Google Scholar] [CrossRef]
- Suez, J.; Elinav, E. The path towards microbiome-based metabolite treatment. Nat. Microbiol. 2017, 2, 17075. [Google Scholar] [CrossRef]
- Liu, X.; Fang, Y.; Lv, X.; Hu, C.; Chen, G.; Zhang, L.; Jin, B.; Huang, L.; Luo, W.; Liang, G.; et al. Deubiquitinase OTUD6A in macrophages promotes intestinal inflammation and colitis via deubiquitination of NLRP3. Cell Death Differ. 2023, 30, 1457–1471. [Google Scholar] [CrossRef] [PubMed]
- Xiao, N.; He, W.; Chen, S.; Yao, Y.; Wu, N.; Xu, M.; Du, H.; Zhao, Y.; Tu, Y. Protective Effect of Egg Yolk Lipids against Dextran Sulfate Sodium-Induced Colitis: The Key Role of Gut Microbiota and Short-Chain Fatty Acids. Mol. Nutr. Food Res. 2024, 68, e2400048. [Google Scholar] [CrossRef] [PubMed]
- Tagé, B.S.S.; Gonzatti, M.B.; Vieira, R.P.; Keller, A.C.; Bortoluci, K.R.; Aimbire, F. Three Main SCFAs Mitigate Lung Inflammation and Tissue Remodeling Nlrp3-Dependent in Murine HDM-Induced Neutrophilic Asthma. Inflammation 2024, 47, 1386–1402. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Wu, K.; Hao, H.; Zhao, Y.; Bao, L.; Qiu, M.; He, Y.; He, Z.; Zhang, N.; Hu, X.; et al. Gut microbiota-mediated secondary bile acid alleviates Staphylococcus aureus-induced mastitis through the TGR5-cAMP-PKA-NF-κB/NLRP3 pathways in mice. NPJ Biofilms Microbiomes 2023, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhang, Y.; Kong, Y.; Ye, T.; Yu, Q.; Kumaran Satyanarayanan, S.; Su, K.P.; Liu, J. Microbiota-derived metabolite Indoles induced aryl hydrocarbon receptor activation and inhibited neuroinflammation in APP/PS1 mice. Brain Behav. Immun. 2022, 106, 76–88. [Google Scholar] [CrossRef]
- Liu, M.; Wang, Y.; Xiang, H.; Guo, M.; Li, S.; Liu, M.; Yao, J. The Tryptophan Metabolite Indole-3-Carboxaldehyde Alleviates Mice with DSS-Induced Ulcerative Colitis by Balancing Amino Acid Metabolism, Inhibiting Intestinal Inflammation, and Improving Intestinal Barrier Function. Molecules 2023, 28, 3704. [Google Scholar] [CrossRef]
- Zhen, Y.; Zhang, H. NLRP3 Inflammasome and Inflammatory Bowel Disease. Front. Immunol. 2019, 10, 276. [Google Scholar] [CrossRef]
- An, Y.; Zhang, H.; Wang, C.; Jiao, F.; Xu, H.; Wang, X.; Luan, W.; Ma, F.; Ni, L.; Tang, X.; et al. Activation of ROS/MAPKs/NF-κB/NLRP3 and inhibition of efferocytosis in osteoclast-mediated diabetic osteoporosis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 12515–12527. [Google Scholar] [CrossRef]
- Wu, J.; Han, Y.; Xu, H.; Sun, H.; Wang, R.; Ren, H.; Wang, G. Deficient chaperone-mediated autophagy facilitates LPS-induced microglial activation via regulation of the p300/NF-κB/NLRP3 pathway. Sci. Adv. 2023, 9, eadi8343. [Google Scholar] [CrossRef]
- Kumar, M.; Murugesan, S.; Ibrahim, N.; Elawad, M.; Al Khodor, S. Predictive biomarkers for anti-TNF alpha therapy in IBD patients. J. Transl. Med. 2024, 22, 284. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Rovira-Llopis, S.; Apostolova, N.; Bañuls, C.; Muntané, J.; Rocha, M.; Victor, V.M. Mitochondria, the NLRP3 inflammasome and sirtuins in type 2 diabetes: New therapeutic targets. Antioxid. Redox Signal. 2018, 29, 749–791. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; He, W.; Tian, H.; Zhan, P.; Liu, J. Thyme (Thymus vulgaris L.) polyphenols ameliorate DSS-induced ulcerative colitis of mice by mitigating intestinal barrier damage, regulating gut microbiota, and suppressing TLR4/NF-κB-NLRP3 inflammasome pathways. Food Funct. 2023, 14, 1113–1132. [Google Scholar] [CrossRef]
- Haque, P.S.; Kapur, N.; Barrett, T.A.; Theiss, A.L. Mitochondrial function and gastrointestinal diseases. Nat. Rev. Gastroenterol. Hepatol. 2024, 21, 537–555. [Google Scholar] [CrossRef]
- Jackson, D.N.; Theiss, A.L. Gut bacteria signaling to mitochondria in intestinal inflammation and cancer. Gut microbes 2020, 11, 285–304. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, J.; Duan, L. The role of microbiota-mitochondria crosstalk in pathogenesis and therapy of intestinal diseases. Pharmacol. Res. 2022, 186, 106530. [Google Scholar] [CrossRef]
- Ge, Y.; Zadeh, M.; Mohamadzadeh, M. Vitamin B12 coordinates ileal epithelial cell and microbiota functions to resist Salmonella infection in mice. J. Exp. Med. 2022, 219, e20220057. [Google Scholar] [CrossRef]
- Wei, W.; Liu, Y.; Hou, Y.; Cao, S.; Chen, Z.; Zhang, Y.; Cai, X.; Yan, Q.; Li, Z.; Yuan, Y.; et al. Psychological stress-induced microbial metabolite indole-3-acetate disrupts intestinal cell lineage commitment. Cell Metab. 2024, 36, 466–483. [Google Scholar] [CrossRef]
- Wiggins, B.G.; Wang, Y.F.; Burke, A.; Grunberg, N.; Vlachaki Walker, J.M.; Dore, M.; Chahrour, C.; Pennycook, B.R.; Sanchez-Garrido, J.; Vernia, S.; et al. Endothelial sensing of AHR ligands regulates intestinal homeostasis. Nature 2023, 621, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Huai, W.; Zhao, R.; Song, H.; Zhao, J.; Zhang, L.; Zhang, L.; Gao, C.; Han, L.; Zhao, W. Aryl hydrocarbon receptor negatively regulates NLRP3 inflammasome activity by inhibiting NLRP3 transcription. Nat. Commun. 2014, 5, 4738. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; Zhang, L.; Wu, D.; Jiang, S.; Wu, Q.; Dai, M. Paeonol attenuates nonalcoholic steatohepatitis by regulating intestinal flora and AhR/NLRP3/Caspase-1 metabolic pathway. J. Ethnopharmacol. 2024, 329, 118147. [Google Scholar] [CrossRef]
- Cui, Q.; Zhang, Z.; Tian, X.; Liang, X.; Lu, Y.; Shi, Y.; Kuerman, M.; Wang, R.; Yu, Z.; Gong, P.; et al. Bifidobacterium bifidum Ameliorates DSS-Induced Colitis in Mice by Regulating AHR/NRF2/NLRP3 Inflammasome Pathways through Indole-3-lactic Acid Production. J. Agric. Food Chem. 2023, 71, 1970–1981. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Lin, T.; Zhu, Q.; Zhang, Y.; Song, Z.; Pan, X. Naringenin protects against acute pancreatitis-associated intestinal injury by inhibiting NLRP3 inflammasome activation via AhR signaling. Front. Pharmacol. 2023, 14, 1090261. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.H.; Tang, L.L.; Sun, X.H.; Zhang, Q.; Liu, C.Y.; Zhang, X.N.; Yu, K.Y.; Yang, Y.; Hu, J.; Shi, X.L.; et al. Qufeng Xuanbi Formula inhibited benzo[a]pyrene-induced aggravated asthma airway mucus secretion by AhR/ROS/ERK pathway. J. Ethnopharmacol. 2024, 319, 117203. [Google Scholar] [CrossRef]
- Renga, G.; Nunzi, E.; Pariano, M.; Puccetti, M.; Bellet, M.M.; Pieraccini, G.; D’Onofrio, F.; Santarelli, I.; Stincardini, C.; Aversa, F.; et al. Optimizing therapeutic outcomes of immune checkpoint blockade by a microbial tryptophan metabolite. J. Immunother. Cancer 2022, 10, e003725. [Google Scholar] [CrossRef]
- Scott, S.A.; Fu, J.; Chang, P.V. Dopamine receptor D2 confers colonization resistance via microbial metabolites. Nature 2024, 628, 180–185. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, J.; Bao, Q.; Hao, H. Indole-3-Carboxaldehyde Alleviates LPS-Induced Intestinal Inflammation by Inhibiting ROS Production and NLRP3 Inflammasome Activation. Antioxidants 2024, 13, 1107. https://doi.org/10.3390/antiox13091107
Cao J, Bao Q, Hao H. Indole-3-Carboxaldehyde Alleviates LPS-Induced Intestinal Inflammation by Inhibiting ROS Production and NLRP3 Inflammasome Activation. Antioxidants. 2024; 13(9):1107. https://doi.org/10.3390/antiox13091107
Chicago/Turabian StyleCao, Ji, Qiuyu Bao, and Haiping Hao. 2024. "Indole-3-Carboxaldehyde Alleviates LPS-Induced Intestinal Inflammation by Inhibiting ROS Production and NLRP3 Inflammasome Activation" Antioxidants 13, no. 9: 1107. https://doi.org/10.3390/antiox13091107
APA StyleCao, J., Bao, Q., & Hao, H. (2024). Indole-3-Carboxaldehyde Alleviates LPS-Induced Intestinal Inflammation by Inhibiting ROS Production and NLRP3 Inflammasome Activation. Antioxidants, 13(9), 1107. https://doi.org/10.3390/antiox13091107