
Copper-Induced Enhancement of Glioblastoma Tumorigenicity via Cytochrome C Oxidase


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Enzymatic Activities
2.3. Measurement of Radioactive Copper Uptake in Cells
2.4. High-Energy Redox Metabolomic Analysis
2.5. ROS Levels
2.6. Xenograft Mouse Model with Intracranial Tumors
2.7. Clonogenic Assays
2.8. Crystal Violet Proliferation Assay
2.9. Determination of Apoptosis
2.10. Statistics
3. Results
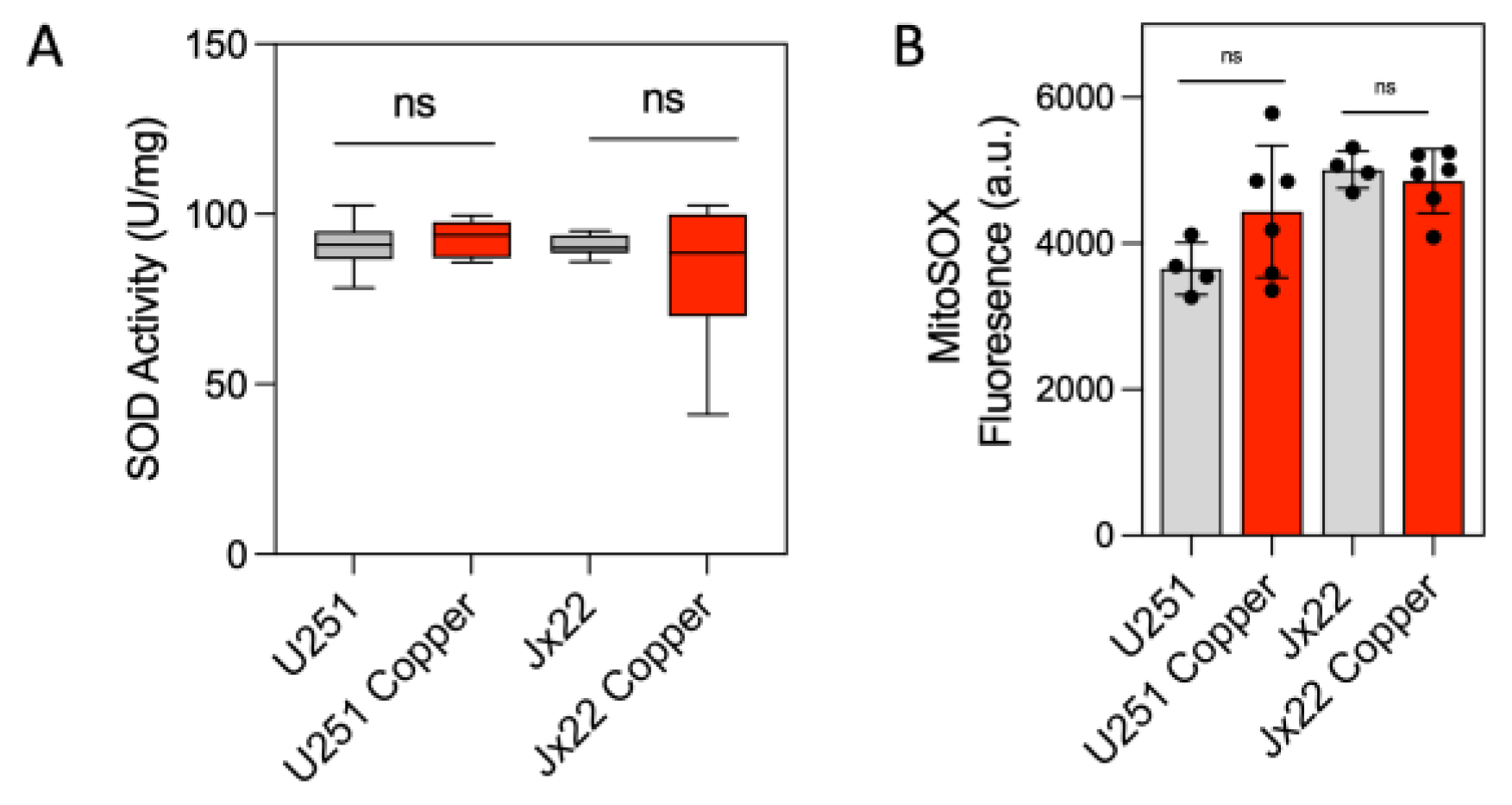
3.1. Bioavailable Copper Increases CcO Activity in GBM Cells
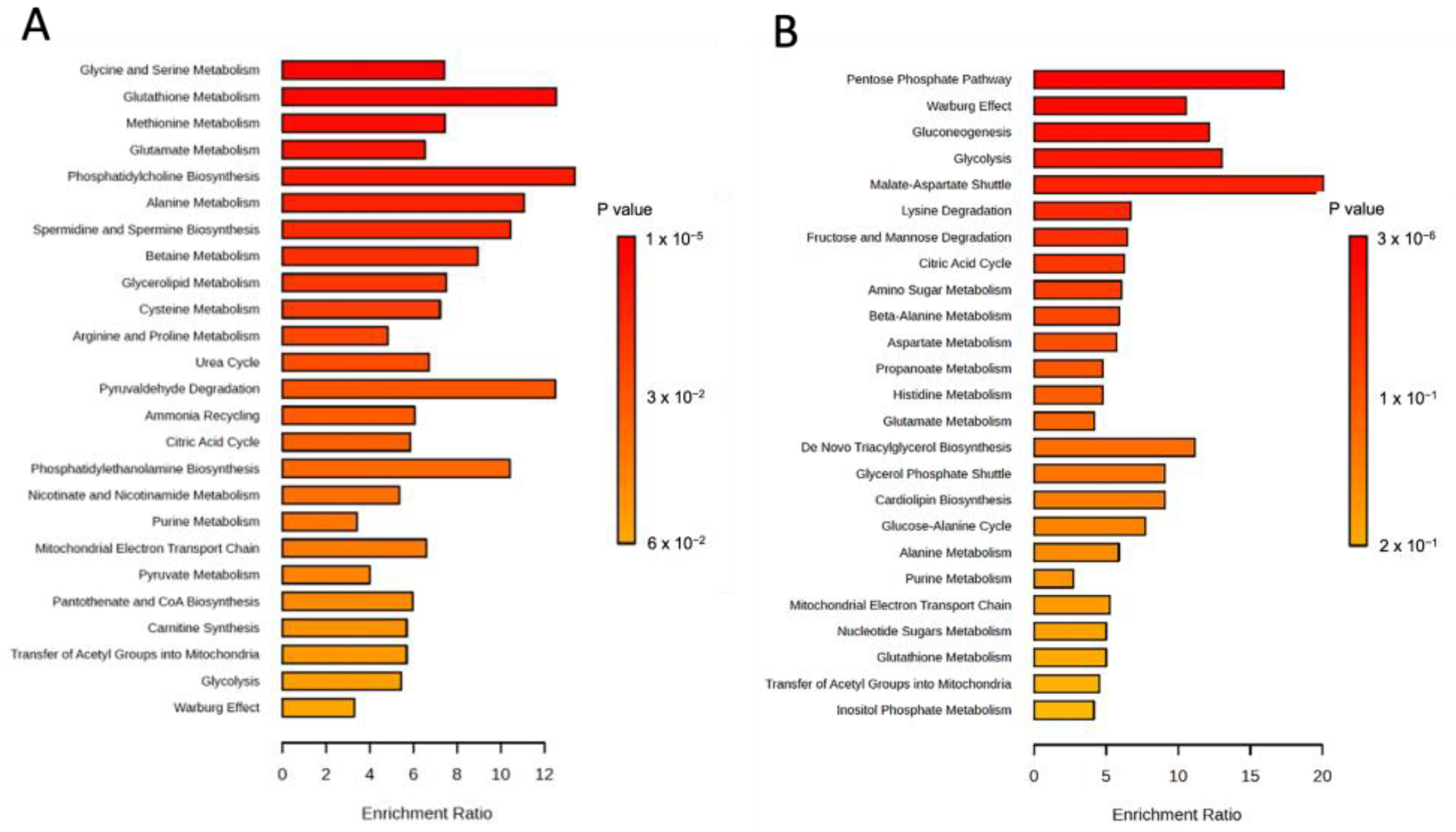
3.2. Bioavailable Copper Rewires GBM Metabolism
3.3. Bioavailable Copper Accelerates GBM Tumor Growth and Reduces Treatment-Induced GBM Cell Death
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Charkiewicz, A.E. Is Copper Still Safe for Us? What Do We Know and What Are the Latest Literature Statements? Curr. Issues Mol. Biol. 2024, 46, 8441–8463. [Google Scholar] [CrossRef] [PubMed]
- Linder, M.C.; Moor, J.R.; Wright, K. Ceruloplasmin assays in diagnosis and treatment of human lung, breast, and gastrointestinal cancers. J. Natl. Cancer Inst. 1981, 67, 263–275. [Google Scholar] [PubMed]
- Pagliardi, E.; Giangrandi, E. Clinical significance of the blood copper in Hodgkin’s disease. Acta Haematol. 1960, 24, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Wang, B.; Yin, W.; Ding, X.; Yu, J.; Kang, Y.J. Disturbance of copper homeostasis is a mechanism for homocysteine-induced vascular endothelial cell injury. PLoS ONE 2013, 8, e76209. [Google Scholar] [CrossRef] [PubMed]
- Gaetke, L.M.; Chow-Johnson, H.S.; Chow, C.K. Copper: Toxicological relevance and mechanisms. Arch. Toxicol. 2014, 88, 1929–1938. [Google Scholar] [CrossRef]
- Gembillo, G.; Labbozzetta, V.; Giuffrida, A.E.; Peritore, L.; Calabrese, V.; Spinella, C.; Stancanelli, M.R.; Spallino, E.; Visconti, L.; Santoro, D. Potential Role of Copper in Diabetes and Diabetic Kidney Disease. Metabolites 2022, 13, 17. [Google Scholar] [CrossRef]
- Guo, F.; Lin, Y.; Meng, L.; Peng, L.; Zhang, H.; Zhang, X.; Jin, M.; Wang, J.; Zhang, Y.; Tang, M.; et al. Association of copper exposure with prevalence of chronic kidney disease in older adults. Clin. Nutr. 2022, 41, 2720–2728. [Google Scholar] [CrossRef]
- Adeoti, M.L.; Oguntola, A.S.; Akanni, E.O.; Agodirin, O.S.; Oyeyemi, G.M. Trace elements; copper, zinc and selenium, in breast cancer afflicted female patients in LAUTECH Osogbo, Nigeria. Indian J. Cancer 2015, 52, 106–109. [Google Scholar] [CrossRef]
- Ding, X.; Jiang, M.; Jing, H.; Sheng, W.; Wang, X.; Han, J.; Wang, L. Analysis of serum levels of 15 trace elements in breast cancer patients in Shandong, China. Environ. Sci. Pollut. Res. Int. 2015, 22, 7930–7935. [Google Scholar] [CrossRef]
- Feng, J.F.; Lu, L.; Zeng, P.; Yang, Y.H.; Luo, J.; Yang, Y.W.; Wang, D. Serum total oxidant/antioxidant status and trace element levels in breast cancer patients. Int. J. Clin. Oncol. 2012, 17, 575–583. [Google Scholar] [CrossRef]
- Kuo, H.W.; Chen, S.F.; Wu, C.C.; Chen, D.R.; Lee, J.H. Serum and tissue trace elements in patients with breast cancer in Taiwan. Biol. Trace Elem. Res. 2002, 89, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Pavithra, V.; Sathisha, T.G.; Kasturi, K.; Mallika, D.S.; Amos, S.J.; Ragunatha, S. Serum levels of metal ions in female patients with breast cancer. J. Clin. Diagn. Res. 2015, 9, BC25-c27. [Google Scholar] [CrossRef]
- Diez, M.; Cerdan, F.J.; Arroyo, M.; Balibrea, J.L. Use of the copper/zinc ratio in the diagnosis of lung cancer. Cancer 1989, 63, 726–730. [Google Scholar] [CrossRef]
- Jin, Y.; Zhang, C.; Xu, H.; Xue, S.; Wang, Y.; Hou, Y.; Kong, Y.; Xu, Y. Combined effects of serum trace metals and polymorphisms of CYP1A1 or GSTM1 on non-small cell lung cancer: A hospital based case-control study in China. Cancer Epidemiol. 2011, 35, 182–187. [Google Scholar] [CrossRef]
- Oyama, T.; Matsuno, K.; Kawamoto, T.; Mitsudomi, T.; Shirakusa, T.; Kodama, Y. Efficiency of serum copper/zinc ratio for differential diagnosis of patients with and without lung cancer. Biol. Trace Elem. Res. 1994, 42, 115–127. [Google Scholar] [CrossRef]
- Margalioth, E.J.; Schenker, J.G.; Chevion, M. Copper and zinc levels in normal and malignant tissues. Cancer 1983, 52, 868–872. [Google Scholar] [CrossRef]
- Nayak, S.B.; Bhat, V.R.; Upadhyay, D.; Udupa, S.L. Copper and ceruloplasmin status in serum of prostate and colon cancer patients. Indian. J. Physiol. Pharmacol. 2003, 47, 108–110. [Google Scholar]
- Ribeiro, S.M.; Moya, A.M.; Braga, C.B.; Domenici, F.A.; Feitosa, M.R.; Feres, O.; Rocha, J.J.; Cunha, S.F. Copper-Zinc ratio and nutritional status in colorectal cancer patients during the perioperative period. Acta Cir. Bras. 2016, 31 (Suppl. S1), 24–28. [Google Scholar] [CrossRef]
- Sohrabi, M.; Gholami, A.; Azar, M.H.; Yaghoobi, M.; Shahi, M.M.; Shirmardi, S.; Nikkhah, M.; Kohi, Z.; Salehpour, D.; Khoonsari, M.R.; et al. Trace Element and Heavy Metal Levels in Colorectal Cancer: Comparison Between Cancerous and Non-cancerous Tissues. Biol. Trace Elem. Res. 2018, 183, 1–8. [Google Scholar] [CrossRef]
- Stepien, M.; Jenab, M.; Freisling, H.; Becker, N.P.; Czuban, M.; Tjonneland, A.; Olsen, A.; Overvad, K.; Boutron-Ruault, M.C.; Mancini, F.R.; et al. Pre-diagnostic copper and zinc biomarkers and colorectal cancer risk in the European Prospective Investigation into Cancer and Nutrition cohort. Carcinogenesis 2017, 38, 699–707. [Google Scholar] [CrossRef]
- Khanna, S.S.; Karjodkar, F.R. Circulating immune complexes and trace elements (Copper, Iron and Selenium) as markers in oral precancer and cancer: A randomised, controlled clinical trial. Head Face Med. 2006, 2, 33. [Google Scholar] [CrossRef] [PubMed]
- Baltaci, A.K.; Dundar, T.K.; Aksoy, F.; Mogulkoc, R. Changes in the Serum Levels of Trace Elements Before and After the Operation in Thyroid Cancer Patients. Biol. Trace Elem. Res. 2017, 175, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Singh, M.K.; Singh, T.B.; Bhartiya, S.K.; Singh, S.P.; Shukla, V.K. Heavy and trace metals in carcinoma of the gallbladder. World J. Surg. 2013, 37, 2641–2646. [Google Scholar] [CrossRef]
- Yaman, M.; Kaya, G.; Yekeler, H. Distribution of trace metal concentrations in paired cancerous and non-cancerous human stomach tissues. World J. Gastroenterol. 2007, 13, 612–618. [Google Scholar] [CrossRef]
- Saleh, S.A.K.; Adly, H.M.; Abdelkhaliq, A.A.; Nassir, A.M. Serum Levels of Selenium, Zinc, Copper, Manganese, and Iron in Prostate Cancer Patients. Curr. Urol. 2020, 14, 44–49. [Google Scholar] [CrossRef]
- Apelgot, S.; Coppey, J.; Fromentin, A.; Guille, E.; Poupon, M.F.; Roussel, A. Altered distribution of copper (64Cu) in tumor-bearing mice and rats. Anticancer Res. 1986, 6, 159–164. [Google Scholar]
- Arslan, M.; Demir, H.; Arslan, H.; Gokalp, A.S.; Demir, C. Trace elements, heavy metals and other biochemical parameters in malignant glioma patients. Asian Pac. J. Cancer Prev. 2011, 12, 447–451. [Google Scholar]
- Bagnall, H.J.; Benda, P.; Brownell, G.L.; Sweet, W.H. Positron-scanning with copper-64 in the diagnosis of intracranial lesions: Partition of copper-64 versenate in, and excretion from, the body. J. Neurosurg. 1958, 15, 411–426. [Google Scholar] [CrossRef]
- Brem, S.; Cotran, R.; Folkman, J. Tumor angiogenesis: A quantitative method for histologic grading. J. Natl. Cancer Inst. 1972, 48, 347–356. [Google Scholar]
- Gaman, L.; Radoi, M.P.; Delia, C.E.; Luzardo, O.P.; Zumbado, M.; Rodriguez-Hernandez, A.; Stoian, I.; Gilca, M.; Boada, L.D.; Henriquez-Hernandez, L.A. Concentration of heavy metals and rare earth elements in patients with brain tumours: Analysis in tumour tissue, non-tumour tissue, and blood. Int. J. Environ. Health Res. 2021, 31, 741–754. [Google Scholar] [CrossRef]
- Kaiser, J.; Gullotta, F. Estimation of the copper content of astrocytomas and glioblastomas by the cuproin method (author’s transl). Neurochirurgia 1980, 23, 20–23. [Google Scholar] [CrossRef] [PubMed]
- Lelievre, P.; Sancey, L.; Coll, J.L.; Deniaud, A.; Busser, B. The Multifaceted Roles of Copper in Cancer: A Trace Metal Element with Dysregulated Metabolism, but Also a Target or a Bullet for Therapy. Cancers 2020, 12, 3594. [Google Scholar] [CrossRef] [PubMed]
- Gupte, A.; Mumper, R.J. Elevated copper and oxidative stress in cancer cells as a target for cancer treatment. Cancer Treat. Rev. 2009, 35, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Ishida, S.; Andreux, P.; Poitry-Yamate, C.; Auwerx, J.; Hanahan, D. Bioavailable copper modulates oxidative phosphorylation and growth of tumors. Proc. Natl. Acad. Sci. USA 2013, 110, 19507–19512. [Google Scholar] [CrossRef]
- Pan, Q.; Kleer, C.G.; van Golen, K.L.; Irani, J.; Bottema, K.M.; Bias, C.; De Carvalho, M.; Mesri, E.A.; Robins, D.M.; Dick, R.D.; et al. Copper deficiency induced by tetrathiomolybdate suppresses tumor growth and angiogenesis. Cancer Res. 2002, 62, 4854–4859. [Google Scholar]
- Ramchandani, D.; Berisa, M.; Tavarez, D.A.; Li, Z.; Miele, M.; Bai, Y.; Lee, S.B.; Ban, Y.; Dephoure, N.; Hendrickson, R.C.; et al. Copper depletion modulates mitochondrial oxidative phosphorylation to impair triple negative breast cancer metastasis. Nat. Commun. 2021, 12, 7311. [Google Scholar] [CrossRef]
- Huttemann, M.; Kadenbach, B.; Grossman, L.I. Mammalian subunit IV isoforms of cytochrome c oxidase. Gene 2001, 267, 111–123. [Google Scholar] [CrossRef]
- Kadenbach, B.; Huttemann, M.; Arnold, S.; Lee, I.; Bender, E. Mitochondrial energy metabolism is regulated via nuclear-coded subunits of cytochrome c oxidase. Free Radic. Biol. Med. 2000, 29, 211–221. [Google Scholar] [CrossRef]
- Horn, D.; Barrientos, A. Mitochondrial copper metabolism and delivery to cytochrome c oxidase. IUBMB Life 2008, 60, 421–429. [Google Scholar] [CrossRef]
- Leary, S.C.; Winge, D.R.; Cobine, P.A. “Pulling the plug” on cellular copper: The role of mitochondria in copper export. Biochim. Biophys. Acta 2009, 1793, 146–153. [Google Scholar] [CrossRef]
- Oliva, C.R.; Ali, M.Y.; Flor, S.; Griguer, C.E. COX4-1 promotes mitochondrial supercomplex assembly and limits reactive oxide species production in radioresistant GBM. Cell Stress 2022, 6, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Oliva, C.R.; Markert, T.; Gillespie, G.Y.; Griguer, C.E. Nuclear-encoded cytochrome c oxidase subunit 4 regulates BMI1 expression and determines proliferative capacity of high-grade gliomas. Oncotarget 2015, 6, 4330–4344. [Google Scholar] [CrossRef] [PubMed]
- Oliva, C.R.; Moellering, D.R.; Gillespie, G.Y.; Griguer, C.E. Acquisition of chemoresistance in gliomas is associated with increased mitochondrial coupling and decreased ROS production. PLoS ONE 2011, 6, e24665. [Google Scholar] [CrossRef]
- Oliva, C.R.; Nozell, S.E.; Diers, A.; McClugage, S.G., 3rd; Sarkaria, J.N.; Markert, J.M.; Darley-Usmar, V.M.; Bailey, S.M.; Gillespie, G.Y.; Landar, A.; et al. Acquisition of temozolomide chemoresistance in gliomas leads to remodeling of mitochondrial electron transport chain. J. Biol. Chem. 2010, 285, 39759–39767. [Google Scholar] [CrossRef]
- Brem, S.S.; Zagzag, D.; Tsanaclis, A.M.; Gately, S.; Elkouby, M.P.; Brien, S.E. Inhibition of angiogenesis and tumor growth in the brain. Suppression of endothelial cell turnover by penicillamine and the depletion of copper, an angiogenic cofactor. Am. J. Pathol. 1990, 137, 1121–1142. [Google Scholar]
- Yoshida, D.; Ikeda, Y.; Nakazawa, S. Suppression of tumor growth in experimental 9L gliosarcoma model by copper depletion. Neurol. Med. Chir. 1995, 35, 133–135. [Google Scholar] [CrossRef]
- Oliva, C.R.; Markert, T.; Ross, L.J.; White, E.L.; Rasmussen, L.; Zhang, W.; Everts, M.; Moellering, D.R.; Bailey, S.M.; Suto, M.J.; et al. Identification of Small Molecule Inhibitors of Human Cytochrome c Oxidase That Target Chemoresistant Glioma Cells. J. Biol. Chem. 2016, 291, 24188–24199. [Google Scholar] [CrossRef]
- Flor, S.; Oliva, C.R.; Ali, M.Y.; Coleman, K.L.; Greenlee, J.D.; Jones, K.A.; Monga, V.; Griguer, C.E. Catalase Overexpression Drives an Aggressive Phenotype in Glioblastoma. Antioxidants 2021, 10, 1988. [Google Scholar] [CrossRef]
- Ali, M.Y.; Oliva, C.R.; Flor, S.; Goswami, P.C.; Griguer, C.E. Cytochrome c oxidase mediates labile iron level and radioresistance in glioblastoma. Free Radic. Biol. Med. 2022, 185, 25–35. [Google Scholar] [CrossRef]
- Cheng, F.; Peng, G.; Lu, Y.; Wang, K.; Ju, Q.; Ju, Y.; Ouyang, M. Relationship between copper and immunity: The potential role of copper in tumor immunity. Front. Oncol. 2022, 12, 1019153. [Google Scholar] [CrossRef]
- De Jorge FB, C.C.; Delascio, D. Biochemical studies on fibroleiomyoma. Matern. E Infanc. 1965, 24, 649–654. [Google Scholar]
- Turecky, L.; Kalina, P.; Uhlikova, E.; Namerova, S.; Krizko, J. Serum ceruloplasmin and copper levels in patients with primary brain tumors. Klin. Wochenschr. 1984, 62, 187–189. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, D.; Ikeda, Y.; Nakazawa, S. Quantitative analysis of copper, zinc and copper/zinc ratio in selected human brain tumors. J. Neurooncol. 1993, 16, 109–115. [Google Scholar] [CrossRef]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef]
- El-Botty, R.; Morriset, L.; Montaudon, E.; Tariq, Z.; Schnitzler, A.; Bacci, M.; Lorito, N.; Sourd, L.; Huguet, L.; Dahmani, A.; et al. Oxidative phosphorylation is a metabolic vulnerability of endocrine therapy and palbociclib resistant metastatic breast cancers. Nat. Commun. 2023, 14, 4221. [Google Scholar] [CrossRef]
- Zhao, Z.; Mei, Y.; Wang, Z.; He, W. The Effect of Oxidative Phosphorylation on Cancer Drug Resistance. Cancers 2022, 15, 62. [Google Scholar] [CrossRef]
- Bansal, A.; Simon, M.C. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef]
- Kennedy, L.; Sandhu, J.K.; Harper, M.E.; Cuperlovic-Culf, M. Role of Glutathione in Cancer: From Mechanisms to Therapies. Biomolecules 2020, 10, 1429. [Google Scholar] [CrossRef]
- Ali-Osman, F.; Caughlan, J.; Gray, G.S. Decreased DNA interstrand cross-linking and cytotoxicity induced in human brain tumor cells by 1,3-bis(2-chloroethyl)-1-nitrosourea after in vitro reaction with glutathione. Cancer Res. 1989, 49, 5954–5958. [Google Scholar]
- Evans, C.G.; Bodell, W.J.; Tokuda, K.; Doane-Setzer, P.; Smith, M.T. Glutathione and related enzymes in rat brain tumor cell resistance to 1,3-bis(2-chloroethyl)-1-nitrosourea and nitrogen mustard. Cancer Res. 1987, 47, 2525–2530. [Google Scholar]
- Friedman, H.S.; Colvin, O.M.; Kaufmann, S.H.; Ludeman, S.M.; Bullock, N.; Bigner, D.D.; Griffith, O.W. Cyclophosphamide resistance in medulloblastoma. Cancer Res. 1992, 52, 5373–5378. [Google Scholar] [PubMed]
- Rocha, C.R.; Garcia, C.C.; Vieira, D.B.; Quinet, A.; de Andrade-Lima, L.C.; Munford, V.; Belizario, J.E.; Menck, C.F. Glutathione depletion sensitizes cisplatin- and temozolomide-resistant glioma cells in vitro and in vivo. Cell Death Dis. 2014, 5, e1505. [Google Scholar] [CrossRef] [PubMed]
- Scheiber, I.F.; Dringen, R. Copper-treatment increases the cellular GSH content and accelerates GSH export from cultured rat astrocytes. Neurosci. Lett. 2011, 498, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Freedman, J.H.; Ciriolo, M.R.; Peisach, J. The role of glutathione in copper metabolism and toxicity. J. Biol. Chem. 1989, 264, 5598–5605. [Google Scholar] [CrossRef]
- Bonet-Aleta, J.P.; Pezacki, A.; Oi, M.; Huesi, J.L.; Santamaria, J.; Chang, C.J. Therapeutic Copper-based Nanoparticles Release Labile Copper(II) and Trigger Cellular Responses in Glutathione and NRF2 Redox Pathways and Metal Homeostasis. Version 1. chemrXiv 2023. [Google Scholar] [CrossRef]
- Cox, C.; Teknos, T.N.; Barrios, M.; Brewer, G.J.; Dick, R.D.; Merajver, S.D. The role of copper suppression as an antiangiogenic strategy in head and neck squamous cell carcinoma. Laryngoscope 2001, 111, 696–701. [Google Scholar] [CrossRef]
- Hassouneh, B.; Islam, M.; Nagel, T.; Pan, Q.; Merajver, S.D.; Teknos, T.N. Tetrathiomolybdate promotes tumor necrosis and prevents distant metastases by suppressing angiogenesis in head and neck cancer. Mol. Cancer Ther. 2007, 6, 1039–1045. [Google Scholar] [CrossRef]
- Ishida, S.; McCormick, F.; Smith-McCune, K.; Hanahan, D. Enhancing tumor-specific uptake of the anticancer drug cisplatin with a copper chelator. Cancer Cell 2010, 17, 574–583. [Google Scholar] [CrossRef]
- Jain, S.; Cohen, J.; Ward, M.M.; Kornhauser, N.; Chuang, E.; Cigler, T.; Moore, A.; Donovan, D.; Lam, C.; Cobham, M.V.; et al. Tetrathiomolybdate-associated copper depletion decreases circulating endothelial progenitor cells in women with breast cancer at high risk of relapse. Ann. Oncol. 2013, 24, 1491–1498. [Google Scholar] [CrossRef]
- Pan, Q.; Bao, L.W.; Kleer, C.G.; Brewer, G.J.; Merajver, S.D. Antiangiogenic tetrathiomolybdate enhances the efficacy of doxorubicin against breast carcinoma. Mol. Cancer Ther. 2003, 2, 617–622. [Google Scholar]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliva, C.R.; Ali, M.Y.; Flor, S.; Griguer, C.E. Copper-Induced Enhancement of Glioblastoma Tumorigenicity via Cytochrome C Oxidase. Antioxidants 2025, 14, 142. https://doi.org/10.3390/antiox14020142
Oliva CR, Ali MY, Flor S, Griguer CE. Copper-Induced Enhancement of Glioblastoma Tumorigenicity via Cytochrome C Oxidase. Antioxidants. 2025; 14(2):142. https://doi.org/10.3390/antiox14020142
Chicago/Turabian StyleOliva, Claudia R., Md Yousuf Ali, Susanne Flor, and Corinne E. Griguer. 2025. "Copper-Induced Enhancement of Glioblastoma Tumorigenicity via Cytochrome C Oxidase" Antioxidants 14, no. 2: 142. https://doi.org/10.3390/antiox14020142
APA StyleOliva, C. R., Ali, M. Y., Flor, S., & Griguer, C. E. (2025). Copper-Induced Enhancement of Glioblastoma Tumorigenicity via Cytochrome C Oxidase. Antioxidants, 14(2), 142. https://doi.org/10.3390/antiox14020142