

The Role of Hydrogen Sulfide in the Regulation of the Pulmonary Vasculature in Health and Disease


Abstract
:1. Introduction
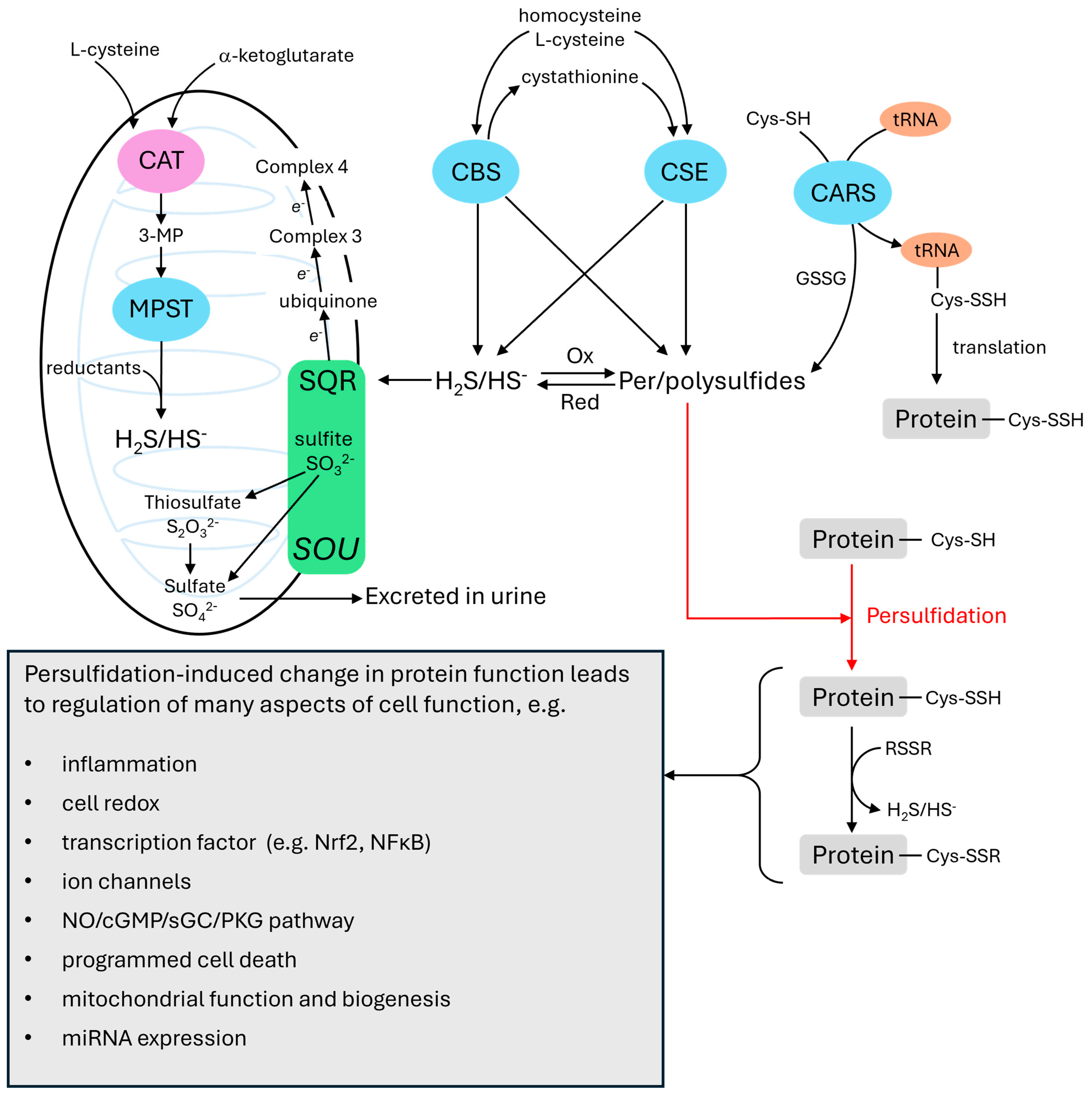
2. Hydrogen Sulfide Metabolism and Signaling
3. Hydrogen Sulfide as an O2 Sensor
3.1. Sulfide and HPV
3.2. Per-/Polysulfides and HPV
3.3. Mechanisms of Sulfide-Induced Contraction
4. Pulmonary Hypertension
4.1. Classification of Pulmonary Hypertension
4.2. Mechanisms of Pulmonary Vascular Remodeling in PH
5. Hydrogen Sulfide and PH
5.1. Evidence That the Plasma Concentration of Hydrogen Sulfide Is Decreased in Human Pulmonary Hypertension
5.2. Sulfide in Animal Models of PH
5.3. CSE Expression, Plasma Sulfide Levels, and Lung Tissue Sulfide Production in PH
6. Mechanisms by Which Sulfide May Inhibit the Development of PH (See Also Table 2, Table 3 and Table 4 for Summaries)
6.1. Involvement of Sulfide in Extracellular Matrix Remodeling of PA
6.2. H2S and NFκB in PH
6.3. H2S, Nrf2 and Hemoxygenase in PH
6.4. Other Potential Beneficial Effects of Sulfide on PH Pathogenesis
7. Summary and Conclusions
Funding
Conflicts of Interest
References
- Wang, R. Physiological implications of hydrogen sulfide: A whiff exploration that blossomed. Physiol. Rev. 2012, 92, 791–896. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Du, J.; Huang, Y.; Tang, C.; Jin, H. Hydrogen Sulfide Regulates Macrophage Function in Cardiovascular Diseases. Antioxid. Redox Signal. 2023, 38, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Yang, G. H2S signaling and extracellular matrix remodeling in cardiovascular diseases: A tale of tense relationship. Nitric Oxide 2021, 116, 14–26. [Google Scholar] [CrossRef]
- Olson, K.R.; Dombkowski, R.A.; Russell, M.J.; Doellman, M.M.; Head, S.K.; Whitfield, N.L.; Madden, J.A. Hydrogen sulfide as an oxygen sensor/transducer in vertebrate hypoxic vasoconstriction and hypoxic vasodilation. J. Exp. Biol. 2006, 209, 4011–4023. [Google Scholar] [CrossRef] [PubMed]
- Chunyu, Z.; Junbao, D.; Dingfang, B.; Hui, Y.; Xiuying, T.; Chaoshu, T. The regulatory effect of hydrogen sulfide on hypoxic pulmonary hypertension in rats. Biochem. Biophys. Res. Commun. 2003, 302, 810–816. [Google Scholar] [CrossRef]
- Roubenne, L.; Marthan, R.; Le Grand, B.; Guibert, C. Hydrogen Sulfide Metabolism and Pulmonary Hypertension. Cells 2021, 10, 1477. [Google Scholar] [CrossRef]
- Ubuka, T. Assay methods and biological roles of labile sulfur in animal tissues. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2002, 781, 227–249. [Google Scholar] [CrossRef]
- Iciek, M.; Bilska-Wilkosz, A.; Gorny, M. Sulfane sulfur—New findings on an old topic. Acta Biochim. Pol. 2019, 66, 533–544. [Google Scholar] [CrossRef]
- Vignane, T.; Filipovic, M.R. Emerging Chemical Biology of Protein Persulfidation. Antioxid. Redox Signal. 2023, 39, 19–39. [Google Scholar] [CrossRef]
- Filipovic, M.R.; Zivanovic, J.; Alvarez, B.; Banerjee, R. Chemical Biology of H2S Signaling through Persulfidation. Chem. Rev. 2018, 118, 1253–1337. [Google Scholar] [CrossRef]
- Fukuto, J.M.; Ignarro, L.J.; Nagy, P.; Wink, D.A.; Kevil, C.G.; Feelisch, M.; Cortese-Krott, M.M.; Bianco, C.L.; Kumagai, Y.; Hobbs, A.J.; et al. Biological hydropersulfides and related polysulfides—A new concept and perspective in redox biology. FEBS. Lett. 2018, 592, 2140–2152. [Google Scholar] [CrossRef] [PubMed]
- Kolluru, G.K.; Shen, X.; Kevil, C.G. Reactive Sulfur Species: A New Redox Player in Cardiovascular Pathophysiology. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 874–884. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.R. Are Reactive Sulfur Species the New Reactive Oxygen Species? Antioxid. Redox Signal. 2020, 33, 1125–1142. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Kong, C.; Ye, D.; Liu, X.; Wang, Y.; Meng, G.; Han, Y.; Xie, L.; Ji, Y. Protein Persulfidation: Recent Progress and Future Directions. Antioxid. Redox Signal. 2023, 39, 829–852. [Google Scholar] [CrossRef]
- Furne, J.; Saeed, A.; Levitt, M.D. Whole tissue hydrogen sulfide concentrations are orders of magnitude lower than presently accepted values. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R1479–R1485. [Google Scholar] [CrossRef]
- Levitt, M.D.; Abdel-Rehim, M.S.; Furne, J. Free and acid-labile hydrogen sulfide concentrations in mouse tissues: Anomalously high free hydrogen sulfide in aortic tissue. Antioxid. Redox Signal. 2011, 15, 373–378. [Google Scholar] [CrossRef]
- Ida, T.; Sawa, T.; Ihara, H.; Tsuchiya, Y.; Watanabe, Y.; Kumagai, Y.; Suematsu, M.; Motohashi, H.; Fujii, S.; Matsunaga, T.; et al. Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 7606–7611. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Zhang, W.; Wu, L.; Yang, G.; Li, H.; Wang, R. Hydrogen sulfide (H2S) metabolism in mitochondria and its regulatory role in energy production. Proc. Natl. Acad. Sci. USA 2012, 109, 2943–2948. [Google Scholar] [CrossRef]
- Teng, H.; Wu, B.; Zhao, K.; Yang, G.; Wu, L.; Wang, R. Oxygen-sensitive mitochondrial accumulation of cystathionine beta-synthase mediated by Lon protease. Proc. Natl. Acad. Sci. USA 2013, 110, 12679–12684. [Google Scholar] [CrossRef]
- Kolluru, G.K.; Shackelford, R.E.; Shen, X.; Dominic, P.; Kevil, C.G. Sulfide regulation of cardiovascular function in health and disease. Nat. Rev. Cardiol. 2023, 20, 109–125. [Google Scholar] [CrossRef]
- Olson, K.R.; Whitfield, N.L.; Bearden, S.E.; St Leger, J.; Nilson, E.; Gao, Y.; Madden, J.A. Hypoxic pulmonary vasodilation: A paradigm shift with a hydrogen sulfide mechanism. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R51–R60. [Google Scholar] [CrossRef] [PubMed]
- Xiaohui, L.; Junbao, D.; Lin, S.; Jian, L.; Xiuying, T.; Jianguang, Q.; Bing, W.; Hongfang, J.; Chaoshu, T. Down-regulation of endogenous hydrogen sulfide pathway in pulmonary hypertension and pulmonary vascular structural remodeling induced by high pulmonary blood flow in rats. Circ. J. 2005, 69, 1418–1424. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Lloret, J.; Snetkov, V.A.; Shaifta, Y.; Docio, I.; Connolly, M.J.; MacKay, C.E.; Knock, G.A.; Ward, J.P.T.; Aaronson, P.I. Role of reactive oxygen species and sulfide-quinone oxoreductase in hydrogen sulfide-induced contraction of rat pulmonary arteries. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L670–L685. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, G.; Wondimu, T.; Ross, B.; Wang, R. Hydrogen sulfide and asthma. Exp. Physiol. 2011, 96, 847–852. [Google Scholar] [CrossRef]
- Sun, Y.; Tang, C.S.; Jin, H.F.; Du, J.B. The vasorelaxing effect of hydrogen sulfide on isolated rat aortic rings versus pulmonary artery rings. Acta Pharmacol. Sin. 2011, 32, 456–464. [Google Scholar] [CrossRef]
- Luo, L.; Liu, D.; Tang, C.; Du, J.; Liu, A.D.; Holmberg, L.; Jin, H. Sulfur dioxide upregulates the inhibited endogenous hydrogen sulfide pathway in rats with pulmonary hypertension induced by high pulmonary blood flow. Biochem. Biophys. Res. Commun. 2013, 433, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Madden, J.A.; Ahlf, S.B.; Dantuma, M.W.; Olson, K.R.; Roerig, D.L. Precursors and inhibitors of hydrogen sulfide synthesis affect acute hypoxic pulmonary vasoconstriction in the intact lung. J. Appl. Physiol. 2012, 112, 411–418. [Google Scholar] [CrossRef]
- Prieto-Lloret, J.; Shaifta, Y.; Ward, J.P.; Aaronson, P.I. Hypoxic pulmonary vasoconstriction in isolated rat pulmonary arteries is not inhibited by antagonists of H2S-synthesizing pathways. J. Physiol. 2015, 593, 385–401. [Google Scholar] [CrossRef]
- Rudyk, O.; Rowan, A.; Prysyazhna, O.; Krasemann, S.; Hartmann, K.; Zhang, M.; Shah, A.M.; Ruppert, C.; Weiss, A.; Schermuly, R.T.; et al. Oxidation of PKGIalpha mediates an endogenous adaptation to pulmonary hypertension. Proc. Natl. Acad. Sci. USA 2019, 116, 13016–13025. [Google Scholar] [CrossRef] [PubMed]
- Akaike, T.; Ida, T.; Wei, F.Y.; Nishida, M.; Kumagai, Y.; Alam, M.M.; Ihara, H.; Sawa, T.; Matsunaga, T.; Kasamatsu, S.; et al. Cysteinyl-tRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics. Nat. Commun. 2017, 8, 1177. [Google Scholar] [CrossRef]
- Matsunaga, T.; Sano, H.; Takita, K.; Morita, M.; Yamanaka, S.; Ichikawa, T.; Numakura, T.; Ida, T.; Jung, M.; Ogata, S.; et al. Supersulphides provide airway protection in viral and chronic lung diseases. Nat. Commun. 2023, 14, 4476. [Google Scholar] [CrossRef] [PubMed]
- Greiner, R.; Palinkas, Z.; Basell, K.; Becher, D.; Antelmann, H.; Nagy, P.; Dick, T.P. Polysulfides link H2S to protein thiol oxidation. Antioxid. Redox Signal. 2013, 19, 1749–1765. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.R.; Gao, Y.; Arif, F.; Arora, K.; Patel, S.; DeLeon, E.R.; Sutton, T.R.; Feelisch, M.; Cortese-Krott, M.M.; Straub, K.D. Metabolism of hydrogen sulfide (H2S) and Production of Reactive Sulfur Species (RSS) by superoxide dismutase. Redox Biol. 2018, 15, 74–85. [Google Scholar] [CrossRef]
- Alvarez, L.; Bianco, C.L.; Toscano, J.P.; Lin, J.; Akaike, T.; Fukuto, J.M. Chemical Biology of Hydropersulfides and Related Species: Possible Roles in Cellular Protection and Redox Signaling. Antioxid. Redox Signal. 2017, 27, 622–633. [Google Scholar] [CrossRef]
- Borisov, V.B.; Forte, E. Impact of Hydrogen Sulfide on Mitochondrial and Bacterial Bioenergetics. Int. J. Mol. Sci. 2021, 22, 12688. [Google Scholar] [CrossRef]
- Olson, K.R.; Deleon, E.R.; Gao, Y.; Hurley, K.; Sadauskas, V.; Batz, C.; Stoy, G.F. Thiosulfate: A readily accessible source of hydrogen sulfide in oxygen sensing. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R592–R603. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Landry, A.P.; Guha, A.; Vitvitsky, V.; Lee, H.J.; Seike, K.; Reddy, P.; Lyssiotis, C.A.; Banerjee, R. A redox cycle with complex II prioritizes sulfide quinone oxidoreductase-dependent H2S oxidation. J. Biol. Chem. 2022, 298, 101435. [Google Scholar] [CrossRef] [PubMed]
- Hanna, D.; Kumar, R.; Banerjee, R. A Metabolic Paradigm for Hydrogen Sulfide Signaling via Electron Transport Chain Plasticity. Antioxid. Redox Signal. 2023, 38, 57–67. [Google Scholar] [CrossRef]
- Dombkowski, R.A.; Doellman, M.M.; Head, S.K.; Olson, K.R. Hydrogen sulfide mediates hypoxia-induced relaxation of trout urinary bladder smooth muscle. J. Exp. Biol. 2006, 209, 3234–3240. [Google Scholar] [CrossRef]
- Olson, K.R.; Healy, M.J.; Qin, Z.; Skovgaard, N.; Vulesevic, B.; Duff, D.W.; Whitfield, N.L.; Yang, G.; Wang, R.; Perry, S.F. Hydrogen sulfide as an oxygen sensor in trout gill chemoreceptors. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R669–R680. [Google Scholar] [CrossRef]
- Olson, K.R.; Whitfield, N.L. Hydrogen sulfide and oxygen sensing in the cardiovascular system. Antioxid. Redox Signal. 2010, 12, 1219–1234. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.R. A Case for Hydrogen Sulfide Metabolism as an Oxygen Sensing Mechanism. Antioxidants 2021, 10, 1650. [Google Scholar] [CrossRef] [PubMed]
- Waypa, G.B.; Marks, J.D.; Guzy, R.; Mungai, P.T.; Schriewer, J.; Dokic, D.; Schumacker, P.T. Hypoxia triggers subcellular compartmental redox signaling in vascular smooth muscle cells. Circ. Res. 2010, 106, 526–535. [Google Scholar] [CrossRef]
- Gallego-Martin, T.; Prieto-Lloret, J.; Aaronson, P.I.; Rocher, A.; Obeso, A. Hydroxycobalamin Reveals the Involvement of Hydrogen Sulfide in the Hypoxic Responses of Rat Carotid Body Chemoreceptor Cells. Antioxidants 2019, 8, 62. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Sun, B.; Wang, X.; Jin, Z.; Zhou, Y.; Dong, L.; Jiang, L.H.; Rong, W. A crucial role for hydrogen sulfide in oxygen sensing via modulating large conductance calcium-activated potassium channels. Antioxid. Redox Signal. 2010, 12, 1179–1189. [Google Scholar] [CrossRef]
- Makarenko, V.V.; Nanduri, J.; Raghuraman, G.; Fox, A.P.; Gadalla, M.M.; Kumar, G.K.; Snyder, S.H.; Prabhakar, N.R. Endogenous H2S is required for hypoxic sensing by carotid body glomus cells. Am. J. Physiol. Cell. Physiol. 2012, 303, C916–C923. [Google Scholar] [CrossRef]
- Peng, Y.J.; Nanduri, J.; Raghuraman, G.; Souvannakitti, D.; Gadalla, M.M.; Kumar, G.K.; Snyder, S.H.; Prabhakar, N.R. H2S mediates O2 sensing in the carotid body. Proc. Natl. Acad. Sci. USA 2010, 107, 10719–10724. [Google Scholar] [CrossRef]
- Prabhakar, N.R. Hydrogen sulfide (H2S): A physiologic mediator of carotid body response to hypoxia. Adv. Exp. Med. Biol. 2012, 758, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Yuan, G.; Vasavda, C.; Peng, Y.J.; Makarenko, V.V.; Raghuraman, G.; Nanduri, J.; Gadalla, M.M.; Semenza, G.L.; Kumar, G.K.; Snyder, S.H.; et al. Protein kinase G-regulated production of H2S governs oxygen sensing. Sci. Signal. 2015, 8, ra37. [Google Scholar] [CrossRef]
- Wang, J.; Hogan, J.O.; Wang, R.; White, C.; Kim, D. Role of cystathionine-gamma-lyase in hypoxia-induced changes in TASK activity, intracellular [Ca(2+)] and ventilation in mice. Respir. Physiol. Neurobiol. 2017, 246, 98–106. [Google Scholar] [CrossRef]
- Kim, D.; Kim, I.; Wang, J.; White, C.; Carroll, J.L. Hydrogen sulfide and hypoxia-induced changes in TASK (K2P3/9) activity and intracellular Ca(2+) concentration in rat carotid body glomus cells. Respir. Physiol. Neurobiol. 2015, 215, 30–38. [Google Scholar] [CrossRef]
- Peng, Y.J.; Nanduri, J.; Wang, N.; Kumar, G.K.; Bindokas, V.; Paul, B.D.; Chen, X.; Fox, A.P.; Vignane, T.; Filipovic, M.R.; et al. Hypoxia sensing requires H2S-dependent persulfidation of olfactory receptor 78. Sci. Adv. 2023, 9, eadf3026. [Google Scholar] [CrossRef] [PubMed]
- Sylvester, J.T.; Shimoda, L.A.; Aaronson, P.I.; Ward, J.P. Hypoxic pulmonary vasoconstriction. Physiol. Rev. 2012, 92, 367–520. [Google Scholar] [CrossRef] [PubMed]
- Dombkowski, R.A.; Russell, M.J.; Schulman, A.A.; Doellman, M.M.; Olson, K.R. Vertebrate phylogeny of hydrogen sulfide vasoactivity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R243–R252. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Frazier, M.; McMahon, T.J.; Eu, J.P. Redox activation of intracellular calcium release channels (ryanodine receptors) in the sustained phase of hypoxia-induced pulmonary vasoconstriction. Chest 2005, 128, 556S–558S. [Google Scholar] [CrossRef]
- Whitfield, N.L.; Kreimier, E.L.; Verdial, F.C.; Skovgaard, N.; Olson, K.R. Reappraisal of H2S/sulfide concentration in vertebrate blood and its potential significance in ischemic preconditioning and vascular signaling. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R1930–R1937. [Google Scholar] [CrossRef]
- Krause, N.C.; Kutsche, H.S.; Santangelo, F.; DeLeon, E.R.; Dittrich, N.P.; Olson, K.R.; Althaus, M. Hydrogen sulfide contributes to hypoxic inhibition of airway transepithelial sodium absorption. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 311, R607–R617. [Google Scholar] [CrossRef]
- Wondimu, E.T.; Zhang, Q.; Jin, Z.; Fu, M.; Torregrossa, R.; Whiteman, M.; Yang, G.; Wu, L.; Wang, R. Effect of hydrogen sulfide on glycolysis-based energy production in mouse erythrocytes. J. Cell. Physiol. 2022, 237, 763–773. [Google Scholar] [CrossRef]
- Stubbert, D.; Prysyazhna, O.; Rudyk, O.; Scotcher, J.; Burgoyne, J.R.; Eaton, P. Protein kinase G Ialpha oxidation paradoxically underlies blood pressure lowering by the reductant hydrogen sulfide. Hypertension 2014, 64, 1344–1351. [Google Scholar] [CrossRef]
- Olson, K.R.; Gao, Y.; DeLeon, E.R.; Markel, T.A.; Drucker, N.; Boone, D.; Whiteman, M.; Steiger, A.K.; Pluth, M.D.; Tessier, C.R.; et al. Extended hypoxia-mediated H2S production provides for long-term oxygen sensing. Acta Physiol. 2020, 228, e13368. [Google Scholar] [CrossRef]
- Alam, S.; Pardue, S.; Shen, X.; Glawe, J.D.; Yagi, T.; Bhuiyan, M.A.N.; Patel, R.P.; Dominic, P.S.; Virk, C.S.; Bhuiyan, M.S.; et al. Hypoxia increases persulfide and polysulfide formation by AMP kinase dependent cystathionine gamma lyase phosphorylation. Redox Biol. 2023, 68, 102949. [Google Scholar] [CrossRef] [PubMed]
- Dongo, E.; Beliczai-Marosi, G.; Dybvig, A.S.; Kiss, L. The mechanism of action and role of hydrogen sulfide in the control of vascular tone. Nitric. Oxide. 2018, 81, 75–87. [Google Scholar] [CrossRef]
- Dongo, E.; Harasztos, L.; Nadasy, G.L.; Kiss, L. The effect of hydrogen sulfide on the contractility of cerebral arterioles. A pilot study. Physiol. Int. 2022. ahead of print. [Google Scholar] [CrossRef]
- Li, S.; Ping, N.N.; Cao, L.; Mi, Y.N.; Cao, Y.X. H2S induces vasoconstriction of rat cerebral arteries via cAMP/adenylyl cyclase pathway. Toxicol. Appl. Pharmacol. 2015, 289, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Mitidieri, E.; Vellecco, V.; Brancaleone, V.; Vanacore, D.; Manzo, O.L.; Martin, E.; Sharina, I.; Krutsenko, Y.; Monti, M.C.; Morretta, E.; et al. Involvement of 3′,5′-cyclic inosine monophosphate in cystathionine gamma-lyase-dependent regulation of the vascular tone. Br. J. Pharmacol. 2021, 178, 3765–3782. [Google Scholar] [CrossRef] [PubMed]
- Ariyaratnam, P.; Loubani, M.; Morice, A.H. Hydrogen sulphide vasodilates human pulmonary arteries: A possible role in pulmonary hypertension? Microvasc. Res. 2013, 90, 135–137. [Google Scholar] [CrossRef]
- Munteanu, C.; Popescu, C.; Vladulescu-Trandafir, A.I.; Onose, G. Signaling Paradigms of H2S-Induced Vasodilation: A Comprehensive Review. Antioxidants 2024, 13, 1158. [Google Scholar] [CrossRef]
- Kang, M.; Hashimoto, A.; Gade, A.; Akbarali, H.I. Interaction between hydrogen sulfide-induced sulfhydration and tyrosine nitration in the KATP channel complex. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G532–G539. [Google Scholar] [CrossRef]
- Cui, Y.; Tran, S.; Tinker, A.; Clapp, L.H. The molecular composition of K(ATP) channels in human pulmonary artery smooth muscle cells and their modulation by growth. Am. J. Respir. Cell. Mol. Biol. 2002, 26, 135–143. [Google Scholar] [CrossRef]
- Nalli, A.D.; Wang, H.; Bhattacharya, S.; Blakeney, B.A.; Murthy, K.S. Inhibition of RhoA/Rho kinase pathway and smooth muscle contraction by hydrogen sulfide. Pharmacol. Res. Perspect. 2017, 5, e00343. [Google Scholar] [CrossRef]
- Fresquez, A.M.; White, C. Extracellular cysteines C226 and C232 mediate hydrogen sulfide-dependent inhibition of Orai3-mediated store-operated calcium entry. Am. J. Physiol. Cell. Physiol. 2022, 322, C38–C48. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Qian, Y.; Zhou, J.; Zhu, C.; Jin, L.; Li, S. Hydrogen sulfide inhibited L-type calcium channels (CaV1.2) via up-regulation of the channel sulfhydration in vascular smooth muscle cells. Eur. J. Pharmacol. 2019, 858, 172455. [Google Scholar] [CrossRef]
- Chai, Q.; Lu, T.; Wang, X.L.; Lee, H.C. Hydrogen sulfide impairs shear stress-induced vasodilation in mouse coronary arteries. Pflugers Arch. 2015, 467, 329–340. [Google Scholar] [CrossRef]
- Martelli, A.; Testai, L.; Breschi, M.C.; Lawson, K.; McKay, N.G.; Miceli, F.; Taglialatela, M.; Calderone, V. Vasorelaxation by hydrogen sulphide involves activation of Kv7 potassium channels. Pharmacol. Res. 2013, 70, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.J.; Liu, Y.H.; Khin, E.S.; Bian, J.S. Vasoconstrictive effect of hydrogen sulfide involves downregulation of cAMP in vascular smooth muscle cells. Am. J. Physiol. Cell. Physiol. 2008, 295, C1261–C1270. [Google Scholar] [CrossRef]
- Kubo, S.; Kajiwara, M.; Kawabata, A. Dual modulation of the tension of isolated gastric artery and gastric mucosal circulation by hydrogen sulfide in rats. Inflammopharmacology 2007, 15, 288–292. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, X.; Ying, L.; Dou, D.; Li, Y.; Bai, Y.; Liu, J.; Liu, L.; Feng, H.; Yu, X.; et al. cIMP synthesized by sGC as a mediator of hypoxic contraction of coronary arteries. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H328–H336. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Chen, Z.; Leung, S.W.; Vanhoutte, P.M. Hypoxic Vasospasm Mediated by cIMP: When Soluble Guanylyl Cyclase Turns Bad. J. Cardiovasc. Pharmacol. 2015, 65, 545–548. [Google Scholar] [CrossRef]
- Detremmerie, C.M.S.; Leung, S.W.S.; Vanhoutte, P.M. Activation of NQO-1 mediates the augmented contractions of isolated arteries due to biased activity of soluble guanylyl cyclase in their smooth muscle. Naunyn. Schmiedebergs. Arch. Pharmacol. 2018, 391, 1221–1235. [Google Scholar] [CrossRef]
- Nan, Y.; Zeng, X.; Jin, Z.; Li, N.; Chen, Z.; Chen, J.; Wang, D.; Wang, Y.; Lin, Z.; Ying, L. PDE1 or PDE5 inhibition augments NO-dependent hypoxic constriction of porcine coronary artery via elevating inosine 3′,5′-cyclic monophosphate level. J. Cell. Mol. Med. 2020, 24, 14514–14524. [Google Scholar] [CrossRef]
- Fagan, K.A.; Tyler, R.C.; Sato, K.; Fouty, B.W.; Morris, K.G., Jr.; Huang, P.L.; McMurtry, I.F.; Rodman, D.M. Relative contributions of endothelial, inducible, and neuronal NOS to tone in the murine pulmonary circulation. Am. J. Physiol. 1999, 277, L472–L478. [Google Scholar] [CrossRef] [PubMed]
- Fouty, B.; Komalavilas, P.; Muramatsu, M.; Cohen, A.; McMurtry, I.F.; Lincoln, T.M.; Rodman, D.M. Protein kinase G is not essential to NO-cGMP modulation of basal tone in rat pulmonary circulation. Am. J. Physiol. 1998, 274, H672–H678. [Google Scholar] [CrossRef]
- Mazmanian, G.M.; Baudet, B.; Brink, C.; Cerrina, J.; Kirkiacharian, S.; Weiss, M. Methylene blue potentiates vascular reactivity in isolated rat lungs. J. Appl. Physiol. 1989, 66, 1040–1045. [Google Scholar] [CrossRef]
- Koenitzer, J.R.; Isbell, T.S.; Patel, H.D.; Benavides, G.A.; Dickinson, D.A.; Patel, R.P.; Darley-Usmar, V.M.; Lancaster, J.R., Jr.; Doeller, J.E.; Kraus, D.W. Hydrogen sulfide mediates vasoactivity in an O2-dependent manner. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1953–H1960. [Google Scholar] [CrossRef]
- Cheng, Y.; Ndisang, J.F.; Tang, G.; Cao, K.; Wang, R. Hydrogen sulfide-induced relaxation of resistance mesenteric artery beds of rats. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2316–H2323. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Yang, G.; Jiang, B.; Ju, Y.; Wu, L.; Wang, R. H2S is an endothelium-derived hyperpolarizing factor. Antioxid. Redox Signal. 2013, 19, 1634–1646. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Lloret, J.; Aaronson, P.I. Hydrogen Sulfide as an O2 Sensor: A Critical Analysis. Adv. Exp. Med. Biol. 2017, 967, 261–276. [Google Scholar] [CrossRef]
- Skovgaard, N.; Olson, K.R. Hydrogen sulfide mediates hypoxic vasoconstriction through a production of mitochondrial ROS in trout gills. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, R487–R494. [Google Scholar] [CrossRef]
- Waypa, G.B.; Chandel, N.S.; Schumacker, P.T. Model for hypoxic pulmonary vasoconstriction involving mitochondrial oxygen sensing. Circ. Res. 2001, 88, 1259–1266. [Google Scholar] [CrossRef]
- Connolly, M.J.; Prieto-Lloret, J.; Becker, S.; Ward, J.P.; Aaronson, P.I. Hypoxic pulmonary vasoconstriction in the absence of pretone: Essential role for intracellular Ca2+ release. J. Physiol. 2013, 591, 4473–4498. [Google Scholar] [CrossRef]
- Leach, R.M.; Hill, H.M.; Snetkov, V.A.; Robertson, T.P.; Ward, J.P. Divergent roles of glycolysis and the mitochondrial electron transport chain in hypoxic pulmonary vasoconstriction of the rat: Identity of the hypoxic sensor. J. Physiol. 2001, 536, 211–224. [Google Scholar] [CrossRef]
- Robertson, T.P.; Hague, D.; Aaronson, P.I.; Ward, J.P. Voltage-independent calcium entry in hypoxic pulmonary vasoconstriction of intrapulmonary arteries of the rat. J. Physiol. 2000, 525 Pt 3, 669–680. [Google Scholar] [CrossRef]
- Jone, P.N.; Ivy, D.D.; Hauck, A.; Karamlou, T.; Truong, U.; Coleman, R.D.; Sandoval, J.P.; Del Cerro Marin, M.J.; Eghtesady, P.; Tillman, K.; et al. Pulmonary Hypertension in Congenital Heart Disease: A Scientific Statement From the American Heart Association. Circ. Heart Fail. 2023, 16, e00080. [Google Scholar] [CrossRef]
- Dickinson, M.G.; Bartelds, B.; Borgdorff, M.A.; Berger, R.M. The role of disturbed blood flow in the development of pulmonary arterial hypertension: Lessons from preclinical animal models. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2013, 305, L1–L14. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, T.; Morio, Y.; Casanova, N.; Bauer, N.; Gebb, S.; McMurtry, I.; Oka, M. Rho/Rho kinase signaling mediates increased basal pulmonary vascular tone in chronically hypoxic rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L665–L672. [Google Scholar] [CrossRef] [PubMed]
- Odagiri, K.; Watanabe, H. Effects of the Rho-kinase inhibitor, fasudil, on pulmonary hypertension. Circ. J. 2015, 79, 1213–1214. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.; Zhang, Y.; Liu, R.; Yang, X. The acute effects of 30 mg vs 60 mg of intravenous Fasudil on patients with congenital heart defects and severe pulmonary arterial hypertension. Congenit. Heart Dis. 2019, 14, 645–650. [Google Scholar] [CrossRef]
- Tajsic, T.; Morrell, N.W. Smooth muscle cell hypertrophy, proliferation, migration and apoptosis in pulmonary hypertension. Compr. Physiol. 2011, 1, 295–317. [Google Scholar] [CrossRef]
- Cober, N.D.; VandenBroek, M.M.; Ormiston, M.L.; Stewart, D.J. Evolving Concepts in Endothelial Pathobiology of Pulmonary Arterial Hypertension. Hypertension 2022, 79, 1580–1590. [Google Scholar] [CrossRef]
- Sakao, S.; Taraseviciene-Stewart, L.; Wood, K.; Cool, C.D.; Voelkel, N.F. Apoptosis of pulmonary microvascular endothelial cells stimulates vascular smooth muscle cell growth. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L362–L368. [Google Scholar] [CrossRef]
- Guignabert, C.; Aman, J.; Bonnet, S.; Dorfmuller, P.; Olschewski, A.J.; Pullamsetti, S.; Rabinovitch, M.; Schermuly, R.T.; Humbert, M.; Stenmark, K.R. Pathology and pathobiology of pulmonary hypertension: Current insights and future directions. Eur. Respir. J. 2024, 64, 2401095. [Google Scholar] [CrossRef]
- Jia, Z.; Wang, S.; Yan, H.; Cao, Y.; Zhang, X.; Wang, L.; Zhang, Z.; Lin, S.; Wang, X.; Mao, J. Pulmonary Vascular Remodeling in Pulmonary Hypertension. J. Pers. Med. 2023, 13, 366. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.J.; Vorla, M.; Kalra, D.K. Molecular Pathways in Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2022, 23, 1. [Google Scholar] [CrossRef]
- Bertero, T.; Oldham, W.M.; Cottrill, K.A.; Pisano, S.; Vanderpool, R.R.; Yu, Q.; Zhao, J.; Tai, Y.; Tang, Y.; Zhang, Y.Y.; et al. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J. Clin. Investig. 2016, 126, 3313–3335. [Google Scholar] [CrossRef]
- Maruyama, K.; Ye, C.L.; Woo, M.; Venkatacharya, H.; Lines, L.D.; Silver, M.M.; Rabinovitch, M. Chronic hypoxic pulmonary hypertension in rats and increased elastolytic activity. Am. J. Physiol. 1991, 261, H1716–H1726. [Google Scholar] [CrossRef]
- Nave, A.H.; Mizikova, I.; Niess, G.; Steenbock, H.; Reichenberger, F.; Talavera, M.L.; Veit, F.; Herold, S.; Mayer, K.; Vadasz, I.; et al. Lysyl oxidases play a causal role in vascular remodeling in clinical and experimental pulmonary arterial hypertension. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1446–1458. [Google Scholar] [CrossRef] [PubMed]
- Thenappan, T.; Chan, S.Y.; Weir, E.K. Role of extracellular matrix in the pathogenesis of pulmonary arterial hypertension. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1322–H1331. [Google Scholar] [CrossRef]
- Hoffmann, J.; Marsh, L.M.; Pieper, M.; Stacher, E.; Ghanim, B.; Kovacs, G.; Konig, P.; Wilkens, H.; Haitchi, H.M.; Hoefler, G.; et al. Compartment-specific expression of collagens and their processing enzymes in intrapulmonary arteries of IPAH patients. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L1002–L1013. [Google Scholar] [CrossRef] [PubMed]
- Bertero, T.; Cottrill, K.A.; Lu, Y.; Haeger, C.M.; Dieffenbach, P.; Annis, S.; Hale, A.; Bhat, B.; Kaimal, V.; Zhang, Y.Y.; et al. Matrix Remodeling Promotes Pulmonary Hypertension through Feedback Mechanoactivation of the YAP/TAZ-miR-130/301 Circuit. Cell. Rep. 2015, 13, 1016–1032. [Google Scholar] [CrossRef]
- Kurakula, K.; Smolders, V.; Tura-Ceide, O.; Jukema, J.W.; Quax, P.H.A.; Goumans, M.J. Endothelial Dysfunction in Pulmonary Hypertension: Cause or Consequence? Biomedicines 2021, 9, 57. [Google Scholar] [CrossRef]
- Durham, G.A.; Palmer, T.M. Is there a role for prostanoid-mediated inhibition of IL-6 trans-signalling in the management of pulmonary arterial hypertension? Biochem. Soc. Trans. 2019, 47, 1143–1156. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Birukov, K. Endothelial Cell Mechano-Metabolomic Coupling to Disease States in the Lung Microvasculature. Front. Bioeng. Biotechnol. 2019, 7, 172. [Google Scholar] [CrossRef]
- Pokharel, M.D.; Marciano, D.P.; Fu, P.; Franco, M.C.; Unwalla, H.; Tieu, K.; Fineman, J.R.; Wang, T.; Black, S.M. Metabolic reprogramming, oxidative stress, and pulmonary hypertension. Redox Biol. 2023, 64, 102797. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Dasgupta, A.; Read, A.D.; Bentley, R.E.T.; Motamed, M.; Chen, K.H.; Al-Qazazi, R.; Mewburn, J.D.; Dunham-Snary, K.J.; Alizadeh, E.; et al. Oxygen sensing, mitochondrial biology and experimental therapeutics for pulmonary hypertension and cancer. Free Radic. Biol. Med. 2021, 170, 150–178. [Google Scholar] [CrossRef] [PubMed]
- Leopold, J.A.; Maron, B.A. Molecular Mechanisms of Pulmonary Vascular Remodeling in Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2016, 17, 761. [Google Scholar] [CrossRef]
- Cao, Y.; Zhang, X.; Wang, L.; Yang, Q.; Ma, Q.; Xu, J.; Wang, J.; Kovacs, L.; Ayon, R.J.; Liu, Z.; et al. PFKFB3-mediated endothelial glycolysis promotes pulmonary hypertension. Proc. Natl. Acad. Sci. USA 2019, 116, 13394–13403. [Google Scholar] [CrossRef]
- Fallon, M.B.; Abrams, G.A.; Abdel-Razek, T.T.; Dai, J.; Chen, S.J.; Chen, Y.F.; Luo, B.; Oparil, S.; Ku, D.D. Garlic prevents hypoxic pulmonary hypertension in rats. Am. J. Physiol. 1998, 275, L283–L287. [Google Scholar] [CrossRef]
- Almatroodi, S.A.; Alsahli, M.A.; Almatroudi, A.; Rahmani, A.H. Garlic and its Active Compounds: A Potential Candidate in The Prevention of Cancer by Modulating Various Cell Signalling Pathways. Anticancer Agents Med. Chem. 2019, 19, 1314–1324. [Google Scholar] [CrossRef]
- Morris, D.M.; Beloni, R.K.; Wheeler, H.E. Effects of garlic consumption on physiological variables and performance during exercise in hypoxia. Appl. Physiol. Nutr. Metab. 2013, 38, 363–367. [Google Scholar] [CrossRef]
- Sun, L.; Sun, S.; Li, Y.; Pan, W.; Xie, Y.; Wang, S.; Zhang, Z. Potential biomarkers predicting risk of pulmonary hypertension in congenital heart disease: The role of homocysteine and hydrogen sulfide. Chin. Med. J. 2014, 127, 893–899. [Google Scholar] [CrossRef]
- Tan, Y.; Wang, S.; Ren, X.; Zhang, C.; Xu, F. The prognostic implications of perioperative endogenous hydrogen sulfide and nitric oxide levels in children with congenital heart disease complicated by pulmonary arterial hypertension. Eur. J. Pediatr. 2021, 180, 1915–1922. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Yao, W.Z.; Geng, B.; Ding, Y.L.; Lu, M.; Zhao, M.W.; Tang, C.S. Endogenous hydrogen sulfide in patients with COPD. Chest 2005, 128, 3205–3211. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wang, K.; Li, M.X.; He, W.; Chang, J.R.; Liao, C.C.; Lin, F.; Qi, Y.F.; Wang, R.; Chen, Y.H. Metabolic changes of H2S in smokers and patients of COPD which might involve in inflammation, oxidative stress and steroid sensitivity. Sci. Rep. 2015, 5, 14971. [Google Scholar] [CrossRef]
- Liao, Y.X.; Wang, X.H.; Bai, Y.; Lin, F.; Li, M.X.; Mi, W.J.; Sun, W.L.; Chen, Y.H. Relationship Between Endogenous Hydrogen Sulfide and Pulmonary Vascular Indexes on High-Resolution Computed Tomography in Patients with Chronic Obstructive Pulmonary Disease. Int. J. Chron Obstruct. Pulmon. Dis. 2021, 16, 2279–2289. [Google Scholar] [CrossRef] [PubMed]
- Raymond, T.E.; Khabbaza, J.E.; Yadav, R.; Tonelli, A.R. Significance of main pulmonary artery dilation on imaging studies. Ann. Am. Thorac. Soc. 2014, 11, 1623–1632. [Google Scholar] [CrossRef]
- Gomez-Arroyo, J.G.; Farkas, L.; Alhussaini, A.A.; Farkas, D.; Kraskauskas, D.; Voelkel, N.F.; Bogaard, H.J. The monocrotaline model of pulmonary hypertension in perspective. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L363–L369. [Google Scholar] [CrossRef]
- Stenmark, K.R.; Meyrick, B.; Galie, N.; Mooi, W.J.; McMurtry, I.F. Animal models of pulmonary arterial hypertension: The hope for etiological discovery and pharmacological cure. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L1013–L1032. [Google Scholar] [CrossRef]
- Ryan, J.J.; Marsboom, G.; Archer, S.L. Rodent models of group 1 pulmonary hypertension. Handb. Exp. Pharmacol. 2013, 218, 105–149. [Google Scholar] [CrossRef]
- Rai, P.R.; Cool, C.D.; King, J.A.; Stevens, T.; Burns, N.; Winn, R.A.; Kasper, M.; Voelkel, N.F. The cancer paradigm of severe pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2008, 178, 558–564. [Google Scholar] [CrossRef]
- Boucherat, O.; Agrawal, V.; Lawrie, A.; Bonnet, S. The Latest in Animal Models of Pulmonary Hypertension and Right Ventricular Failure. Circ. Res. 2022, 130, 1466–1486. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, B.K.; Liu, D.; Nie, W.; Yuan, J.M.; Wang, Z.; Guo, Y.M. Sodium hydrosulfide prevents hypoxia-induced pulmonary arterial hypertension in broilers. Br. Poult. Sci. 2012, 53, 608–615. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lin, Y.; Ma, Y.; Zhang, J.; Wang, C.; Zhang, H. Protective effect of hydrogen sulfide on monocrotaline-induced pulmonary arterial hypertension via inhibition of the endothelial mesenchymal transition. Int. J. Mol. Med. 2019, 44, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Chen, S.; Yu, W.; Zhang, D.; Zhang, C.; Tang, C.; Du, J.; Jin, H. H2S inhibits pulmonary arterial endothelial cell inflammation in rats with monocrotaline-induced pulmonary hypertension. Lab. Investig. 2017, 97, 268–278. [Google Scholar] [CrossRef]
- Li, X.H.; Du, J.B.; Bu, D.F.; Tang, X.Y.; Tang, C.S. Sodium hydrosulfide alleviated pulmonary vascular structural remodeling induced by high pulmonary blood flow in rats. Acta Pharmacol. Sin. 2006, 27, 971–980. [Google Scholar] [CrossRef]
- Qingyou, Z.; Junbao, D.; Weijin, Z.; Hui, Y.; Chaoshu, T.; Chunyu, Z. Impact of hydrogen sulfide on carbon monoxide/heme oxygenase pathway in the pathogenesis of hypoxic pulmonary hypertension. Biochem. Biophys. Res. Commun. 2004, 317, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Hongfang, J.; Cong, B.; Zhao, B.; Zhang, C.; Liu, X.; Zhou, W.; Shi, Y.; Tang, C.; Junbao, D. Effects of hydrogen sulfide on hypoxic pulmonary vascular structural remodeling. Life Sci. 2006, 78, 1299–1309. [Google Scholar] [CrossRef]
- Wei, H.L.; Zhang, C.Y.; Jin, H.F.; Tang, C.S.; Du, J.B. Hydrogen sulfide regulates lung tissue-oxidized glutathione and total antioxidant capacity in hypoxic pulmonary hypertensive rats. Acta Pharmacol. Sin. 2008, 29, 670–679. [Google Scholar] [CrossRef]
- Wu, J.; Pan, W.; Wang, C.; Dong, H.; Xing, L.; Hou, J.; Fang, S.; Li, H.; Yang, F.; Yu, B. H2S attenuates endoplasmic reticulum stress in hypoxia-induced pulmonary artery hypertension. Biosci. Rep. 2019, 39, BSR20190304. [Google Scholar] [CrossRef]
- Yanfei, W.; Lin, S.; Junbao, D.; Chaoshu, T. Impact of L-arginine on hydrogen sulfide/cystathionine-gamma-lyase pathway in rats with high blood flow-induced pulmonary hypertension. Biochem. Biophys. Res. Commun. 2006, 345, 851–857. [Google Scholar] [CrossRef]
- Li, X.; Du, J.; Jin, H.; Geng, B.; Tang, C. Sodium hydrosulfide alleviates pulmonary artery collagen remodeling in rats with high pulmonary blood flow. Heart Vessels 2008, 23, 409–419. [Google Scholar] [CrossRef]
- Li, W.; Jin, H.F.; Liu, D.; Sun, J.H.; Jian, P.J.; Li, X.H.; Tang, C.S.; Du, J.B. Hydrogen sulfide induces apoptosis of pulmonary artery smooth muscle cell in rats with pulmonary hypertension induced by high pulmonary blood flow. Chin. Med. J. 2009, 122, 3032–3038. [Google Scholar] [PubMed]
- Li, X.; Du, J.; Jin, H.; Tang, X.; Bu, D.; Tang, C. The regulatory effect of endogenous hydrogen sulfide on pulmonary vascular structure and gasotransmitters in rats with high pulmonary blood flow. Life Sci. 2007, 81, 841–849. [Google Scholar] [CrossRef]
- Turhan, K.; Alan, E.; Yetik-Anacak, G.; Sevin, G. H2S releasing sodium sulfide protects against pulmonary hypertension by improving vascular responses in monocrotaline-induced pulmonary hypertension. Eur. J. Pharmacol. 2022, 931, 175182. [Google Scholar] [CrossRef]
- Zhang, D.; Wang, X.; Tian, X.; Zhang, L.; Yang, G.; Tao, Y.; Liang, C.; Li, K.; Yu, X.; Tang, X.; et al. The Increased Endogenous Sulfur Dioxide Acts as a Compensatory Mechanism for the Downregulated Endogenous Hydrogen Sulfide Pathway in the Endothelial Cell Inflammation. Front. Immunol. 2018, 9, 882. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, X.; Chen, S.; Chen, S.; Yu, W.; Liu, X.; Yang, G.; Tao, Y.; Tang, X.; Bu, D.; et al. Endogenous hydrogen sulfide sulfhydrates IKKbeta at cysteine 179 to control pulmonary artery endothelial cell inflammation. Clin. Sci. 2019, 133, 2045–2059. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Hao, L.Z.; Pan, J.A.; Gao, Q.; Zhang, J.F.; Kankala, R.K.; Wang, S.B.; Chen, A.Z.; Zhang, H.L. Microfluidic fabrication of inhalable large porous microspheres loaded with H2S-releasing aspirin derivative for pulmonary arterial hypertension therapy. J. Control. Release 2021, 329, 286–298. [Google Scholar] [CrossRef]
- Han, W.; Dong, Z.; Dimitropoulou, C.; Su, Y. Hydrogen sulfide ameliorates tobacco smoke-induced oxidative stress and emphysema in mice. Antioxid. Redox Signal. 2011, 15, 2121–2134. [Google Scholar] [CrossRef]
- Numakura, T.; Sugiura, H.; Akaike, T.; Ida, T.; Fujii, S.; Koarai, A.; Yamada, M.; Onodera, K.; Hashimoto, Y.; Tanaka, R.; et al. Production of reactive persulfide species in chronic obstructive pulmonary disease. Thorax 2017, 72, 1074–1083. [Google Scholar] [CrossRef]
- Zimmer, A.; Teixeira, R.B.; Constantin, R.L.; Campos-Carraro, C.; Aparicio Cordero, E.A.; Ortiz, V.D.; Donatti, L.; Gonzalez, E.; Bahr, A.C.; Visioli, F.; et al. The progression of pulmonary arterial hypertension induced by monocrotaline is characterized by lung nitrosative and oxidative stress, and impaired pulmonary artery reactivity. Eur. J. Pharmacol. 2021, 891, 173699. [Google Scholar] [CrossRef]
- Shen, X.; Kolluru, G.K.; Yuan, S.; Kevil, C.G. Measurement of H2S in vivo and in vitro by the monobromobimane method. Methods Enzymol. 2015, 554, 31–45. [Google Scholar] [CrossRef]
- Van de Louw, A.; Haouzi, P. Oxygen deficit and H2S in hemorrhagic shock in rats. Crit. Care 2012, 16, R178. [Google Scholar] [CrossRef] [PubMed]
- Rajpal, S.; Katikaneni, P.; Deshotels, M.; Pardue, S.; Glawe, J.; Shen, X.; Akkus, N.; Modi, K.; Bhandari, R.; Dominic, P.; et al. Total sulfane sulfur bioavailability reflects ethnic and gender disparities in cardiovascular disease. Redox Biol. 2018, 15, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Peter, E.A.; Bir, S.; Wang, R.; Kevil, C.G. Analytical measurement of discrete hydrogen sulfide pools in biological specimens. Free Radic. Biol. Med. 2012, 52, 2276–2283. [Google Scholar] [CrossRef] [PubMed]
- Wintner, E.A.; Deckwerth, T.L.; Langston, W.; Bengtsson, A.; Leviten, D.; Hill, P.; Insko, M.A.; Dumpit, R.; VandenEkart, E.; Toombs, C.F.; et al. A monobromobimane-based assay to measure the pharmacokinetic profile of reactive sulphide species in blood. Br. J. Pharmacol. 2010, 160, 941–957. [Google Scholar] [CrossRef]
- Polhemus, D.J.; Li, Z.; Pattillo, C.B.; Gojon, G., Sr.; Gojon, G., Jr.; Giordano, T.; Krum, H. A novel hydrogen sulfide prodrug, SG1002, promotes hydrogen sulfide and nitric oxide bioavailability in heart failure patients. Cardiovasc. Ther. 2015, 33, 216–226. [Google Scholar] [CrossRef]
- Kondo, K.; Bhushan, S.; King, A.L.; Prabhu, S.D.; Hamid, T.; Koenig, S.; Murohara, T.; Predmore, B.L.; Gojon, G., Sr.; Gojon, G., Jr.; et al. H2S protects against pressure overload-induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation 2013, 127, 1116–1127. [Google Scholar] [CrossRef]
- Barr, L.A.; Shimizu, Y.; Lambert, J.P.; Nicholson, C.K.; Calvert, J.W. Hydrogen sulfide attenuates high fat diet-induced cardiac dysfunction via the suppression of endoplasmic reticulum stress. Nitric Oxide 2015, 46, 145–156. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, F.; Munske, G.; Zhang, H.; Xian, M. Isotope dilution mass spectrometry for the quantification of sulfane sulfurs. Free Radic. Biol. Med. 2014, 76, 200–207. [Google Scholar] [CrossRef]
- Wallace, J.L.; Nagy, P.; Feener, T.D.; Allain, T.; Ditroi, T.; Vaughan, D.J.; Muscara, M.N.; de Nucci, G.; Buret, A.G. A proof-of-concept, Phase 2 clinical trial of the gastrointestinal safety of a hydrogen sulfide-releasing anti-inflammatory drug. Br. J. Pharmacol. 2020, 177, 769–777. [Google Scholar] [CrossRef]
- Stipanuk, M.H.; Beck, P.W. Characterization of the enzymic capacity for cysteine desulphhydration in liver and kidney of the rat. Biochem. J. 1982, 206, 267–277. [Google Scholar] [CrossRef]
- Zhao, W.; Zhang, J.; Lu, Y.; Wang, R. The vasorelaxant effect of H2S as a novel endogenous gaseous K(ATP) channel opener. EMBO. J. 2001, 20, 6008–6016. [Google Scholar] [CrossRef]
- Olson, K.R.; DeLeon, E.R.; Liu, F. Controversies and conundrums in hydrogen sulfide biology. Nitric Oxide 2014, 41, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Jurkowska, H.; Roman, H.B.; Hirschberger, L.L.; Sasakura, K.; Nagano, T.; Hanaoka, K.; Krijt, J.; Stipanuk, M.H. Primary hepatocytes from mice lacking cysteine dioxygenase show increased cysteine concentrations and higher rates of metabolism of cysteine to hydrogen sulfide and thiosulfate. Amino Acids 2014, 46, 1353–1365. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Chen, L.; Muh, R.W.; Li, P.L.; Li, N. Production and actions of hydrogen sulfide, a novel gaseous bioactive substance, in the kidneys. J. Pharmacol. Exp. Ther. 2009, 329, 1056–1062. [Google Scholar] [CrossRef]
- Doiron, J.E.; Elbatreek, M.H.; Xia, H.; Yu, X.; Wilson Tang, W.H.; LaPenna, K.B.; Sharp, T.E.; Goodchild, T.T.; Xian, M.; Xu, S.; et al. Reduced Hydrogen Sulfide Bioavailability Contributes to Cardiometabolic Heart Failure with Preserved Ejection Fraction. bioRxiv 2024. [Google Scholar] [CrossRef]
- Shen, X.; Pattillo, C.B.; Pardue, S.; Bir, S.C.; Wang, R.; Kevil, C.G. Measurement of plasma hydrogen sulfide in vivo and in vitro. Free Radic. Biol. Med. 2011, 50, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Kovacic, D.; Glavnik, N.; Marinsek, M.; Zagozen, P.; Rovan, K.; Goslar, T.; Mars, T.; Podbregar, M. Total plasma sulfide in congestive heart failure. J. Card. Fail. 2012, 18, 541–548. [Google Scholar] [CrossRef]
- Polhemus, D.J.; Calvert, J.W.; Butler, J.; Lefer, D.J. The cardioprotective actions of hydrogen sulfide in acute myocardial infarction and heart failure. Scientifica 2014, 2014, 768607. [Google Scholar] [CrossRef]
- Xiao, L.; Dong, J.H.; Teng, X.; Jin, S.; Xue, H.M.; Liu, S.Y.; Guo, Q.; Shen, W.; Ni, X.C.; Wu, Y.M. Hydrogen sulfide improves endothelial dysfunction in hypertension by activating peroxisome proliferator-activated receptor delta/endothelial nitric oxide synthase signaling. J. Hypertens. 2018, 36, 651–665. [Google Scholar] [CrossRef]
- Ahmad, F.U.; Sattar, M.A.; Rathore, H.A.; Abdullah, M.H.; Tan, S.; Abdullah, N.A.; Johns, E.J. Exogenous hydrogen sulfide (H2S) reduces blood pressure and prevents the progression of diabetic nephropathy in spontaneously hypertensive rats. Ren. Fail. 2012, 34, 203–210. [Google Scholar] [CrossRef]
- Sen, U.; Vacek, T.P.; Hughes, W.M.; Kumar, M.; Moshal, K.S.; Tyagi, N.; Metreveli, N.; Hayden, M.R.; Tyagi, S.C. Cardioprotective role of sodium thiosulfate on chronic heart failure by modulating endogenous H2S generation. Pharmacology 2008, 82, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Hourihan, J.M.; Kenna, J.G.; Hayes, J.D. The gasotransmitter hydrogen sulfide induces nrf2-target genes by inactivating the keap1 ubiquitin ligase substrate adaptor through formation of a disulfide bond between cys-226 and cys-613. Antioxid. Redox Signal. 2013, 19, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Pushpakumar, S.; Kundu, S.; Weber, G.; Sen, U. Exogenous hydrogen sulfide and miR-21 antagonism attenuates macrophage-mediated inflammation in ischemia reperfusion injury of the aged kidney. Geroscience 2021, 43, 1349–1367. [Google Scholar] [CrossRef] [PubMed]
- Cindrova-Davies, T.; Herrera, E.A.; Niu, Y.; Kingdom, J.; Giussani, D.A.; Burton, G.J. Reduced cystathionine gamma-lyase and increased miR-21 expression are associated with increased vascular resistance in growth-restricted pregnancies: Hydrogen sulfide as a placental vasodilator. Am. J. Pathol. 2013, 182, 1448–1458. [Google Scholar] [CrossRef]
- Yang, G.; Pei, Y.; Cao, Q.; Wang, R. MicroRNA-21 represses human cystathionine gamma-lyase expression by targeting at specificity protein-1 in smooth muscle cells. J. Cell. Physiol. 2012, 227, 3192–3200. [Google Scholar] [CrossRef]
- Nassar, S.Z.; Abdelmonsif, D.A.; Ali, R.G.; Badae, N.M. Sodium hydrosulfide and bone marrow derived mesenchymal stem cells combined therapy for bleomycin induced pulmonary fibrosis in rats: Implication of micro RNA-21 and Lnc GAS5. Life Sci. 2022, 309, 120988. [Google Scholar] [CrossRef]
- Liang, B.; Xiao, T.; Long, J.; Liu, M.; Li, Z.; Liu, S.; Yang, J. Hydrogen sulfide alleviates myocardial fibrosis in mice with alcoholic cardiomyopathy by downregulating autophagy. Int. J. Mol. Med. 2017, 40, 1781–1791. [Google Scholar] [CrossRef]
- Combi, Z.; Potor, L.; Nagy, P.; Sikura, K.E.; Ditroi, T.; Juranyi, E.P.; Galambos, K.; Szerafin, T.; Gergely, P.; Whiteman, M.; et al. Hydrogen sulfide as an anti-calcification stratagem in human aortic valve: Altered biogenesis and mitochondrial metabolism of H2S lead to H2S deficiency in calcific aortic valve disease. Redox Biol. 2023, 60, 102629. [Google Scholar] [CrossRef]
- Karaman, Y.; Kaya-Yasar, Y.; Eylem, C.C.; Onder, S.C.; Nemutlu, E.; Bozkurt, T.E.; Sahin-Erdemli, I. The effect of mitochondria-targeted slow hydrogen sulfide releasing donor AP39-treatment on airway inflammation. Eur. J. Pharmacol. 2023, 946, 175619. [Google Scholar] [CrossRef]
- Magierowska, K.; Korbut, E.; Wojcik-Grzybek, D.; Bakalarz, D.; Sliwowski, Z.; Cieszkowski, J.; Szetela, M.; Torregrossa, R.; Whiteman, M.; Magierowski, M. Mitochondria-targeted hydrogen sulfide donors versus acute oxidative gastric mucosal injury. J. Control. Release 2022, 348, 321–334. [Google Scholar] [CrossRef]
- Stachowicz, A.; Czepiel, K.; Wisniewska, A.; Stachyra, K.; Ulatowska-Bialas, M.; Kusnierz-Cabala, B.; Surmiak, M.; Majka, G.; Kus, K.; Wood, M.E.; et al. Mitochondria-targeted hydrogen sulfide donor reduces fatty liver and obesity in mice fed a high fat diet by inhibiting de novo lipogenesis and inflammation via mTOR/SREBP-1 and NF-kappaB signaling pathways. Pharmacol. Res. 2024, 209, 107428. [Google Scholar] [CrossRef] [PubMed]
- Kieronska-Rudek, A.; Ascencao, K.; Chlopicki, S.; Szabo, C. Increased hydrogen sulfide turnover serves a cytoprotective role during the development of replicative senescence. Biochem. Pharmacol. 2024, 230, 116595. [Google Scholar] [CrossRef] [PubMed]
- Latorre, E.; Torregrossa, R.; Wood, M.E.; Whiteman, M.; Harries, L.W. Mitochondria-targeted hydrogen sulfide attenuates endothelial senescence by selective induction of splicing factors HNRNPD and SRSF2. Aging 2018, 10, 1666–1681. [Google Scholar] [CrossRef] [PubMed]
- Tozzi, C.A.; Poiani, G.J.; McHugh, N.A.; Shakarjian, M.P.; Grove, B.H.; Samuel, C.S.; Unemori, E.N.; Riley, D.J. Recombinant human relaxin reduces hypoxic pulmonary hypertension in the rat. Pulm. Pharmacol. Ther. 2005, 18, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Chi, P.L.; Cheng, C.C.; Hung, C.C.; Wang, M.T.; Liu, H.Y.; Ke, M.W.; Shen, M.C.; Lin, K.C.; Kuo, S.H.; Hsieh, P.P.; et al. MMP-10 from M1 macrophages promotes pulmonary vascular remodeling and pulmonary arterial hypertension. Int. J. Biol. Sci. 2022, 18, 331–348. [Google Scholar] [CrossRef]
- Ding, S.; Cui, J.; Yan, L.; Ru, C.; He, F.; Chen, A. Safflower Alleviates Pulmonary Arterial Hypertension by Inactivating NLRP3: A Combined Approach of Network Pharmacology and Experimental Verification. Clin. Respir. J. 2024, 18, e13826. [Google Scholar] [CrossRef]
- Zhang, E.; Maruyama, J.; Yokochi, A.; Mitani, Y.; Sawada, H.; Nishikawa, M.; Ma, N.; Maruyama, K. Sarpogrelate hydrochloride, a serotonin 5HT2A receptor antagonist, ameliorates the development of chronic hypoxic pulmonary hypertension in rats. J. Anesth. 2015, 29, 715–723. [Google Scholar] [CrossRef]
- Lepetit, H.; Eddahibi, S.; Fadel, E.; Frisdal, E.; Munaut, C.; Noel, A.; Humbert, M.; Adnot, S.; D’Ortho, M.P.; Lafuma, C. Smooth muscle cell matrix metalloproteinases in idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2005, 25, 834–842. [Google Scholar] [CrossRef]
- Li, G.; Tang, L.; Jia, P.; Zhao, J.; Liu, D.; Liu, B. Elevated Plasma Connective Tissue Growth Factor Levels in Children with Pulmonary Arterial Hypertension Associated with Congenital Heart Disease. Pediatr. Cardiol. 2016, 37, 714–721. [Google Scholar] [CrossRef]
- Pi, L.; Fu, C.; Lu, Y.; Zhou, J.; Jorgensen, M.; Shenoy, V.; Lipson, K.E.; Scott, E.W.; Bryant, A.J. Vascular Endothelial Cell-Specific Connective Tissue Growth Factor (CTGF) Is Necessary for Development of Chronic Hypoxia-Induced Pulmonary Hypertension. Front. Physiol. 2018, 9, 138. [Google Scholar] [CrossRef]
- Li, H.; Chen, S.J.; Chen, Y.F.; Meng, Q.C.; Durand, J.; Oparil, S.; Elton, T.S. Enhanced endothelin-1 and endothelin receptor gene expression in chronic hypoxia. J. Appl. Physiol. 1994, 77, 1451–1459. [Google Scholar] [CrossRef]
- Kubben, F.J.; Peeters-Haesevoets, A.; Engels, L.G.; Baeten, C.G.; Schutte, B.; Arends, J.W.; Stockbrugger, R.W.; Blijham, G.H. Proliferating cell nuclear antigen (PCNA): A new marker to study human colonic cell proliferation. Gut 1994, 35, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Biasin, V.; Chwalek, K.; Wilhelm, J.; Best, J.; Marsh, L.M.; Ghanim, B.; Klepetko, W.; Fink, L.; Schermuly, R.T.; Weissmann, N.; et al. Endothelin-1 driven proliferation of pulmonary arterial smooth muscle cells is c-fos dependent. Int. J. Biochem. Cell Biol. 2014, 54, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Zaghloul, N.; Lin, K.; Liu, S.F.; Miller, E.J.; Ahmed, M. Hypoxia-induced activation of specific members of the NF-kB family and its relevance to pulmonary vascular remodeling. Int. J. Biochem. Cell. Biol. 2017, 92, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Farkas, D.; Alhussaini, A.A.; Kraskauskas, D.; Kraskauskiene, V.; Cool, C.D.; Nicolls, M.R.; Natarajan, R.; Farkas, L. Nuclear factor kappaB inhibition reduces lung vascular lumen obliteration in severe pulmonary hypertension in rats. Am. J. Respir. Cell. Mol. Biol. 2014, 51, 413–425. [Google Scholar] [CrossRef]
- Li, L.; Wei, C.; Kim, I.K.; Janssen-Heininger, Y.; Gupta, S. Inhibition of nuclear factor-kappaB in the lungs prevents monocrotaline-induced pulmonary hypertension in mice. Hypertension 2014, 63, 1260–1269. [Google Scholar] [CrossRef]
- Price, L.C.; Caramori, G.; Perros, F.; Meng, C.; Gambaryan, N.; Dorfmuller, P.; Montani, D.; Casolari, P.; Zhu, J.; Dimopoulos, K.; et al. Nuclear factor kappa-B is activated in the pulmonary vessels of patients with end-stage idiopathic pulmonary arterial hypertension. PLoS ONE 2013, 8, e75415. [Google Scholar] [CrossRef]
- Foley, A.; Steinberg, B.E.; Goldenberg, N.M. Inflammasome Activation in Pulmonary Arterial Hypertension. Front. Med. 2021, 8, 826557. [Google Scholar] [CrossRef]
- Sakurai, H.; Chiba, H.; Miyoshi, H.; Sugita, T.; Toriumi, W. IkappaB kinases phosphorylate NF-kappaB p65 subunit on serine 536 in the transactivation domain. J. Biol. Chem. 1999, 274, 30353–30356. [Google Scholar] [CrossRef]
- Du, J.; Huang, Y.; Yan, H.; Zhang, Q.; Zhao, M.; Zhu, M.; Liu, J.; Chen, S.X.; Bu, D.; Tang, C.; et al. Hydrogen sulfide suppresses oxidized low-density lipoprotein (ox-LDL)-stimulated monocyte chemoattractant protein 1 generation from macrophages via the nuclear factor kappaB (NF-kappaB) pathway. J. Biol. Chem. 2014, 289, 9741–9753. [Google Scholar] [CrossRef]
- Luo, K. Signaling Cross Talk between TGF-beta/Smad and Other Signaling Pathways. Cold Spring Harb. Perspect. Biol. 2017, 9, a022137. [Google Scholar] [CrossRef] [PubMed]
- Sawada, H.; Mitani, Y.; Maruyama, J.; Jiang, B.H.; Ikeyama, Y.; Dida, F.A.; Yamamoto, H.; Imanaka-Yoshida, K.; Shimpo, H.; Mizoguchi, A.; et al. A nuclear factor-kappaB inhibitor pyrrolidine dithiocarbamate ameliorates pulmonary hypertension in rats. Chest 2007, 132, 1265–1274. [Google Scholar] [CrossRef] [PubMed]
- Arciniegas, E.; Frid, M.G.; Douglas, I.S.; Stenmark, K.R. Perspectives on endothelial-to-mesenchymal transition: Potential contribution to vascular remodeling in chronic pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L1–L8. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell. Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Nikitopoulou, I.; Orfanos, S.E.; Kotanidou, A.; Maltabe, V.; Manitsopoulos, N.; Karras, P.; Kouklis, P.; Armaganidis, A.; Maniatis, N.A. Vascular endothelial-cadherin downregulation as a feature of endothelial transdifferentiation in monocrotaline-induced pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L352–L363. [Google Scholar] [CrossRef]
- Shi, R.; Zhu, D.; Wei, Z.; Fu, N.; Wang, C.; Liu, L.; Zhang, H.; Liang, Y.; Xing, J.; Wang, X.; et al. Baicalein attenuates monocrotaline-induced pulmonary arterial hypertension by inhibiting endothelial-to-mesenchymal transition. Life Sci. 2018, 207, 442–450. [Google Scholar] [CrossRef]
- Li, L.; Rose, P.; Moore, P.K. Hydrogen sulfide and cell signaling. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 169–187. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.; Paul, B.D.; Gadalla, M.M.; Mustafa, A.K.; Sen, T.; Xu, R.; Kim, S.; Snyder, S.H. Hydrogen sulfide-linked sulfhydration of NF-kappaB mediates its antiapoptotic actions. Mol. Cell. 2012, 45, 13–24. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, C.; Xu, C.; Feng, L.; Li, A.; Jin, X.; Guo, S.; Jiao, X.; Liu, J.; Guo, Y.; et al. H2S mediates apoptosis in response to inflammation through PI3K/Akt/NFkappaB signaling pathway. Biotechnol. Lett. 2020, 42, 375–387. [Google Scholar] [CrossRef]
- Wang, Y.H.; Huang, J.T.; Chen, W.L.; Wang, R.H.; Kao, M.C.; Pan, Y.R.; Chan, S.H.; Tsai, K.W.; Kung, H.J.; Lin, K.T.; et al. Dysregulation of cystathionine gamma-lyase promotes prostate cancer progression and metastasis. EMBO. Rep. 2019, 20, e45986. [Google Scholar] [CrossRef]
- Zhen, Y.; Pan, W.; Hu, F.; Wu, H.; Feng, J.; Zhang, Y.; Chen, J. Exogenous hydrogen sulfide exerts proliferation/anti-apoptosis/angiogenesis/migration effects via amplifying the activation of NF-kappaB pathway in PLC/PRF/5 hepatoma cells. Int. J. Oncol. 2015, 46, 2194–2204. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, M.; Gaddam, R.R. Hydrogen Sulfide in Inflammation: A Novel Mediator and Therapeutic Target. Antioxid. Redox Signal. 2021, 34, 1368–1377. [Google Scholar] [CrossRef]
- Wang, H.; Shi, X.; Qiu, M.; Lv, S.; Zheng, H.; Niu, B.; Liu, H. Hydrogen Sulfide Plays an Important Role by Influencing NLRP3 inflammasome. Int. J. Biol. Sci. 2020, 16, 2752–2760. [Google Scholar] [CrossRef] [PubMed]
- Dilek, N.; Papapetropoulos, A.; Toliver-Kinsky, T.; Szabo, C. Hydrogen sulfide: An endogenous regulator of the immune system. Pharmacol. Res. 2020, 161, 105119. [Google Scholar] [CrossRef]
- Cornwell, A.; Fedotova, S.; Cowan, S.; Badiei, A. Cystathionine gamma-lyase and hydrogen sulfide modulates glucose transporter Glut1 expression via NF-kappaB and PI3k/Akt in macrophages during inflammation. PLoS ONE 2022, 17, e0278910. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Ma, S.; Yang, J.; Luo, K.; Qian, Q.; Pan, J.; Liang, K.; Wang, Y.; Gao, Y.; Li, M. Sodium Hydrosulfide Protects Rats from Hypobaric-Hypoxia-Induced Acute Lung Injury. Int. J. Mol. Sci. 2024, 25, 10734. [Google Scholar] [CrossRef]
- Xie, L.; Gu, Y.; Wen, M.; Zhao, S.; Wang, W.; Ma, Y.; Meng, G.; Han, Y.; Wang, Y.; Liu, G.; et al. Hydrogen Sulfide Induces Keap1 S-sulfhydration and Suppresses Diabetes-Accelerated Atherosclerosis via Nrf2 Activation. Diabetes 2016, 65, 3171–3184. [Google Scholar] [CrossRef]
- Yang, G.; Zhao, K.; Ju, Y.; Mani, S.; Cao, Q.; Puukila, S.; Khaper, N.; Wu, L.; Wang, R. Hydrogen sulfide protects against cellular senescence via S-sulfhydration of Keap1 and activation of Nrf2. Antioxid. Redox Signal. 2013, 18, 1906–1919. [Google Scholar] [CrossRef]
- Qin, Y.; Qiao, Y.; Wang, D.; Li, L.; Li, M.; Yan, G.; Tang, C. Target Nuclear Factor Erythroid 2-Related Factor 2 in Pulmonary Hypertension: Molecular Insight into Application. Oxid. Med. Cell. Longev. 2022, 2022, 7845503. [Google Scholar] [CrossRef]
- Vomund, S.; Schafer, A.; Parnham, M.J.; Brune, B.; von Knethen, A. Nrf2, the Master Regulator of Anti-Oxidative Responses. Int. J. Mol. Sci. 2017, 18, 2772. [Google Scholar] [CrossRef]
- Roberts, J.A.; Rainbow, R.D.; Sharma, P. Mitigation of Cardiovascular Disease and Toxicity through NRF2 Signalling. Int. J. Mol. Sci. 2023, 24, 6723. [Google Scholar] [CrossRef] [PubMed]
- Jamaluddin, M.; Haas de Mello, A.; Tapryal, N.; Hazra, T.K.; Garofalo, R.P.; Casola, A. NRF2 Regulates Cystathionine Gamma-Lyase Expression and Activity in Primary Airway Epithelial Cells Infected with Respiratory Syncytial Virus. Antioxidants 2022, 11, 1582. [Google Scholar] [CrossRef]
- Ramos, K.S.; Lin, H.; McGrath, J.J. Modulation of cyclic guanosine monophosphate levels in cultured aortic smooth muscle cells by carbon monoxide. Biochem. Pharmacol. 1989, 38, 1368–1370. [Google Scholar] [CrossRef]
- Morita, T.; Kourembanas, S. Endothelial cell expression of vasoconstrictors and growth factors is regulated by smooth muscle cell-derived carbon monoxide. J. Clin. Investig. 1995, 96, 2676–2682. [Google Scholar] [CrossRef]
- Morita, T.; Mitsialis, S.A.; Koike, H.; Liu, Y.; Kourembanas, S. Carbon monoxide controls the proliferation of hypoxic vascular smooth muscle cells. J. Biol. Chem. 1997, 272, 32804–32809. [Google Scholar] [CrossRef] [PubMed]
- Stanford, S.J.; Walters, M.J.; Hislop, A.A.; Haworth, S.G.; Evans, T.W.; Mann, B.E.; Motterlini, R.; Mitchell, J.A. Heme oxygenase is expressed in human pulmonary artery smooth muscle where carbon monoxide has an anti-proliferative role. Eur. J. Pharmacol. 2003, 473, 135–141. [Google Scholar] [CrossRef]
- Stone, J.R.; Marletta, M.A. Soluble guanylate cyclase from bovine lung: Activation with nitric oxide and carbon monoxide and spectral characterization of the ferrous and ferric states. Biochemistry 1994, 33, 5636–5640. [Google Scholar] [CrossRef] [PubMed]
- Tao, B.B.; Zhu, Q.; Zhu, Y.C. Mechanisms Underlying the Hydrogen Sulfide Actions: Target Molecules and Downstream Signaling Pathways. Antioxid. Redox Signal. 2024, 40, 86–109. [Google Scholar] [CrossRef]
- Salazar, M.; Rojo, A.I.; Velasco, D.; de Sagarra, R.M.; Cuadrado, A. Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J. Biol. Chem. 2006, 281, 14841–14851. [Google Scholar] [CrossRef]
- Baird, L.; Lleres, D.; Swift, S.; Dinkova-Kostova, A.T. Regulatory flexibility in the Nrf2-mediated stress response is conferred by conformational cycling of the Keap1-Nrf2 protein complex. Proc. Natl. Acad. Sci. USA 2013, 110, 15259–15264. [Google Scholar] [CrossRef]
- Parfenova, H.; Liu, J.; Hoover, D.T.; Fedinec, A.L. Vasodilator effects of sulforaphane in cerebral circulation: A critical role of endogenously produced hydrogen sulfide and arteriolar smooth muscle K(ATP) and BK channels in the brain. J. Cereb. Blood Flow Metab. 2020, 40, 1987–1996. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D.; Hannink, M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef] [PubMed]
- Brampton, J.; Aaronson, P.I. Role of Hydrogen Sulfide in Systemic and Pulmonary Hypertension: Cellular Mechanisms and Therapeutic Implications. Cardiovasc. Hematol. Agents Med. Chem. 2016, 14, 4–22. [Google Scholar] [CrossRef]
- Giuffre, A.; Vicente, J.B. Hydrogen Sulfide Biochemistry and Interplay with Other Gaseous Mediators in Mammalian Physiology. Oxidative Med. Cell. Longev. 2018, 2018, 6290931. [Google Scholar] [CrossRef]
- Chiche, J.D.; Schlutsmeyer, S.M.; Bloch, D.B.; de la Monte, S.M.; Roberts, J.D., Jr.; Filippov, G.; Janssens, S.P.; Rosenzweig, A.; Bloch, K.D. Adenovirus-mediated gene transfer of cGMP-dependent protein kinase increases the sensitivity of cultured vascular smooth muscle cells to the antiproliferative and pro-apoptotic effects of nitric oxide/cGMP. J. Biol. Chem. 1998, 273, 34263–34271. [Google Scholar] [CrossRef]
- Wang, Y.F.; Mainali, P.; Tang, C.S.; Shi, L.; Zhang, C.Y.; Yan, H.; Liu, X.Q.; Du, J.B. Effects of nitric oxide and hydrogen sulfide on the relaxation of pulmonary arteries in rats. Chin. Med. J. 2008, 121, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Knock, G.A. NADPH oxidase in the vasculature: Expression, regulation and signalling pathways; role in normal cardiovascular physiology and its dysregulation in hypertension. Free Radic. Biol. Med. 2019, 145, 385–427. [Google Scholar] [CrossRef]
- Fessel, J.P.; West, J.D. Redox biology in pulmonary arterial hypertension (2013 Grover Conference Series). Pulm. Circ. 2015, 5, 599–609. [Google Scholar] [CrossRef]
- Mittal, M.; Roth, M.; Konig, P.; Hofmann, S.; Dony, E.; Goyal, P.; Selbitz, A.C.; Schermuly, R.T.; Ghofrani, H.A.; Kwapiszewska, G.; et al. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ. Res. 2007, 101, 258–267. [Google Scholar] [CrossRef]
- Veit, F.; Pak, O.; Egemnazarov, B.; Roth, M.; Kosanovic, D.; Seimetz, M.; Sommer, N.; Ghofrani, H.A.; Seeger, W.; Grimminger, F.; et al. Function of NADPH oxidase 1 in pulmonary arterial smooth muscle cells after monocrotaline-induced pulmonary vascular remodeling. Antioxid. Redox Signal. 2013, 19, 2213–2231. [Google Scholar] [CrossRef]
- Ge, X.; Sun, J.; Fei, A.; Gao, C.; Pan, S.; Wu, Z. Hydrogen sulfide treatment alleviated ventilator-induced lung injury through regulation of autophagy and endoplasmic reticulum stress. Int. J. Biol. Sci. 2019, 15, 2872–2884. [Google Scholar] [CrossRef]
- Hu, H.J.; Wang, X.H.; Liu, Y.; Zhang, T.Q.; Chen, Z.R.; Zhang, C.; Tang, Z.H.; Qu, S.L.; Tang, H.F.; Jiang, Z.S. Hydrogen Sulfide Ameliorates Angiotensin II-Induced Atrial Fibrosis Progression to Atrial Fibrillation Through Inhibition of the Warburg Effect and Endoplasmic Reticulum Stress. Front. Pharmacol. 2021, 12, 690371. [Google Scholar] [CrossRef]
- Li, M.H.; Tang, J.P.; Zhang, P.; Li, X.; Wang, C.Y.; Wei, H.J.; Yang, X.F.; Zou, W.; Tang, X.Q. Disturbance of endogenous hydrogen sulfide generation and endoplasmic reticulum stress in hippocampus are involved in homocysteine-induced defect in learning and memory of rats. Behav. Brain Res. 2014, 262, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Liao, C.; Zhang, J.; Sun, Y.; Lu, W.; Bai, Y.; Liao, Y.; Li, M.; Qi, Y.; Chen, Y. Hydrogen Sulfide Inhibits Bronchial Epithelial Cell Epithelial Mesenchymal Transition Through Regulating Endoplasm Reticulum Stress. Front. Mol. Biosci. 2022, 9, 828766. [Google Scholar] [CrossRef]
- Ying, R.; Wang, X.Q.; Yang, Y.; Gu, Z.J.; Mai, J.T.; Qiu, Q.; Chen, Y.X.; Wang, J.F. Hydrogen sulfide suppresses endoplasmic reticulum stress-induced endothelial-to-mesenchymal transition through Src pathway. Life Sci. 2016, 144, 208–217. [Google Scholar] [CrossRef]
- Lu, G.; Xu, C.; Tang, K.; Zhang, J.; Li, Q.; Peng, L.; Wang, Y.; Huang, Z.; Gao, X. H2S inhibits angiotensin II-induced atrial Kv1.5 upregulation by attenuating Nox4-mediated ROS generation during atrial fibrillation. Biochem. Biophys. Res. Commun. 2017, 483, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Pan, L.L.; Long, F.; Wu, W.J.; Yan, D.; Xu, P.; Liu, S.Y.; Qin, M.; Jia, W.W.; Liu, X.H.; et al. Endogenous Hydrogen Sulfide Ameliorates NOX4 Induced Oxidative Stress in LPS-Stimulated Macrophages and Mice. Cell. Physiol. Biochem. 2018, 47, 458–474. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.L.; Liu, X.H.; Shen, Y.Q.; Wang, N.Z.; Xu, J.; Wu, D.; Xiong, Q.H.; Deng, H.Y.; Huang, G.Y.; Zhu, Y.Z. Inhibition of NADPH oxidase 4-related signaling by sodium hydrosulfide attenuates myocardial fibrotic response. Int. J. Cardiol. 2013, 168, 3770–3778. [Google Scholar] [CrossRef]
- Zheng, D.; Dong, S.; Li, T.; Yang, F.; Yu, X.; Wu, J.; Zhong, X.; Zhao, Y.; Wang, L.; Xu, C.; et al. Exogenous Hydrogen Sulfide Attenuates Cardiac Fibrosis Through Reactive Oxygen Species Signal Pathways in Experimental Diabetes Mellitus Models. Cell. Physiol. Biochem. 2015, 36, 917–929. [Google Scholar] [CrossRef]
- Huang, Q.; Sparatore, A.; Del Soldato, P.; Wu, L.; Desai, K. Hydrogen sulfide releasing aspirin, ACS14, attenuates high glucose-induced increased methylglyoxal and oxidative stress in cultured vascular smooth muscle cells. PLoS ONE 2014, 9, e97315. [Google Scholar] [CrossRef]
- Ge, Q.; Zhang, T.; Yu, J.; Lu, X.; Xiao, S.; Zhang, T.; Qing, T.; Xiao, Z.; Zeng, L.; Luo, L. A new perspective on targeting pulmonary arterial hypertension: Programmed cell death pathways (Autophagy, Pyroptosis, Ferroptosis). Biomed. Pharmacother. 2024, 181, 117706. [Google Scholar] [CrossRef]
- Wang, E.; Zhou, S.; Zeng, D.; Wang, R. Molecular regulation and therapeutic implications of cell death in pulmonary hypertension. Cell. Death. Discov. 2023, 9, 239. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, H.; Fu, X.; Wang, K.; Yang, J.; Zhang, X.; Wang, H. The role of hydrogen sulfide regulation of pyroptosis in different pathological processes. Eur. J. Med. Chem. 2024, 268, 116254. [Google Scholar] [CrossRef]
- Ma, J.; Yang, P.; Zhou, Z.; Song, T.; Jia, L.; Ye, X.; Yan, W.; Sun, J.; Ye, T.; Zhu, L. GYY4137-induced p65 sulfhydration protects synovial macrophages against pyroptosis by improving mitochondrial function in osteoarthritis development. J. Adv. Res. 2024, (in press). [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Zhang, X.; Li, Z.; Yan, H.; Tian, X.; Luo, C.; Ma, K.; Li, L.; Zhang, L. Hydrogen sulfide mitigates ox-LDL-induced NLRP3/caspase-1/GSDMD dependent macrophage pyroptosis by S-sulfhydrating caspase-1. Mol. Med. Rep. 2024, 30, 13259. [Google Scholar] [CrossRef]
- Ejikeme, C.; Safdar, Z. Exploring the pathogenesis of pulmonary vascular disease. Front. Med. 2024, 11, 1402639. [Google Scholar] [CrossRef]
- Santos-Ferreira, C.A.; Abreu, M.T.; Marques, C.I.; Goncalves, L.M.; Baptista, R.; Girao, H.M. Micro-RNA Analysis in Pulmonary Arterial Hypertension: Current Knowledge and Challenges. JACC. Basic. Transl. Sci. 2020, 5, 1149–1162. [Google Scholar] [CrossRef]
- Zang, H.; Zhang, Q.; Li, X. Non-Coding RNA Networks in Pulmonary Hypertension. Front. Genet. 2021, 12, 703860. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Cao, S.; Sun, X.; Chen, G. The interplay of hydrogen sulfide and microRNAs in cardiovascular diseases: Insights and future perspectives. Mamm. Genome 2024, 35, 309–323. [Google Scholar] [CrossRef]
- Green, D.E.; Murphy, T.C.; Kang, B.Y.; Bedi, B.; Yuan, Z.; Sadikot, R.T.; Hart, C.M. Peroxisome proliferator-activated receptor-gamma enhances human pulmonary artery smooth muscle cell apoptosis through microRNA-21 and programmed cell death 4. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L371–L383. [Google Scholar] [CrossRef]
- Parikh, V.N.; Jin, R.C.; Rabello, S.; Gulbahce, N.; White, K.; Hale, A.; Cottrill, K.A.; Shaik, R.S.; Waxman, A.B.; Zhang, Y.Y.; et al. MicroRNA-21 integrates pathogenic signaling to control pulmonary hypertension: Results of a network bioinformatics approach. Circulation 2012, 125, 1520–1532. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, Z.; Liang, B.; Li, L.; Liu, S.; Tan, W.; Long, J.; Tang, F.; Chu, C.; Yang, J. Hydrogen sulfide ameliorates rat myocardial fibrosis induced by thyroxine through PI3K/AKT signaling pathway. Endocr. J. 2018, 65, 769–781. [Google Scholar] [CrossRef]
- Zhang, W.; Tao, Z.; Xu, F.; Diao, Q.; Li, J.; Zhou, L.; Miao, Y.; Xie, S.; Wan, J.; Xu, R. An Overview of miRNAs Involved in PASMC Phenotypic Switching in Pulmonary Hypertension. Biomed Res. Int. 2021, 2021, 5765029. [Google Scholar] [CrossRef]
- Guignabert, C.; Tu, L.; Izikki, M.; Dewachter, L.; Zadigue, P.; Humbert, M.; Adnot, S.; Fadel, E.; Eddahibi, S. Dichloroacetate treatment partially regresses established pulmonary hypertension in mice with SM22alpha-targeted overexpression of the serotonin transporter. FASEB J. 2009, 23, 4135–4147. [Google Scholar] [CrossRef] [PubMed]
- Michelakis, E.D.; Gurtu, V.; Webster, L.; Barnes, G.; Watson, G.; Howard, L.; Cupitt, J.; Paterson, I.; Thompson, R.B.; Chow, K.; et al. Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients. Sci. Transl. Med. 2017, 9, aao4583. [Google Scholar] [CrossRef]
- Michelakis, E.D.; McMurtry, M.S.; Wu, X.C.; Dyck, J.R.; Moudgil, R.; Hopkins, T.A.; Lopaschuk, G.D.; Puttagunta, L.; Waite, R.; Archer, S.L. Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: Role of increased expression and activity of voltage-gated potassium channels. Circulation 2002, 105, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Gojon, G.; Morales, G.A. SG1002 and Catenated Divalent Organic Sulfur Compounds as Promising Hydrogen Sulfide Prodrugs. Antioxid. Redox Signal. 2020, 33, 1010–1045. [Google Scholar] [CrossRef]
- Zhang, H.; Dhalla, N.S. The Role of Pro-Inflammatory Cytokines in the Pathogenesis of Cardiovascular Disease. Int. J. Mol. Sci. 2024, 25, 1082. [Google Scholar] [CrossRef]
- Zhang, Y.; Murugesan, P.; Huang, K.; Cai, H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: Novel therapeutic targets. Nat. Rev. Cardiol. 2020, 17, 170–194. [Google Scholar] [CrossRef]
- Chen, R.; Zhang, H.; Tang, B.; Luo, Y.; Yang, Y.; Zhong, X.; Chen, S.; Xu, X.; Huang, S.; Liu, C. Macrophages in cardiovascular diseases: Molecular mechanisms and therapeutic targets. Signal. Transduct. Target. Ther. 2024, 9, 130. [Google Scholar] [CrossRef]
- Inferrera, F.; Marino, Y.; Genovese, T.; Cuzzocrea, S.; Fusco, R.; Di Paola, R. Mitochondrial quality control: Biochemical mechanism of cardiovascular disease. Biochim. Biophys. Acta Mol. Cell. Res. 2025, 1872, 119906. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Polavarapu, R.; Eskla, K.L.; Nicholson, C.K.; Koczor, C.A.; Wang, R.; Lewis, W.; Shiva, S.; Lefer, D.J.; Calvert, J.W. Hydrogen sulfide regulates cardiac mitochondrial biogenesis via the activation of AMPK. J. Mol. Cell. Cardiol. 2018, 116, 29–40. [Google Scholar] [CrossRef]
- Kar, S.; Shahshahan, H.R.; Kambis, T.N.; Yadav, S.K.; Li, Z.; Lefer, D.J.; Mishra, P.K. Hydrogen Sulfide Ameliorates Homocysteine-Induced Cardiac Remodeling and Dysfunction. Front. Physiol. 2019, 10, 598. [Google Scholar] [CrossRef]
- Islam, R.K.; Donnelly, E.; Donnarumma, E.; Hossain, F.; Gardner, J.D.; Islam, K.N. H2S Prodrug, SG-1002, Protects against Myocardial Oxidative Damage and Hypertrophy In Vitro via Induction of Cystathionine beta-Synthase and Antioxidant Proteins. Biomedicines 2023, 11, 612. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.L.; Vaughan, D.; Dicay, M.; MacNaughton, W.K.; de Nucci, G. Hydrogen Sulfide-Releasing Therapeutics: Translation to the Clinic. Antioxid. Redox Signal. 2018, 28, 1533–1540. [Google Scholar] [CrossRef]
- Zhu, Y.; Romero, E.L.; Ren, X.; Sanca, A.J.; Du, C.; Liu, C.; Karim, Z.A.; Alshbool, F.Z.; Khasawneh, F.T.; Zhou, J.; et al. Clopidogrel as a donor probe and thioenol derivatives as flexible promoieties for enabling H2S biomedicine. Nat. Commun. 2018, 9, 3952. [Google Scholar] [CrossRef] [PubMed]
- Bucci, M.; Vellecco, V.; Cantalupo, A.; Brancaleone, V.; Zhou, Z.; Evangelista, S.; Calderone, V.; Papapetropoulos, A.; Cirino, G. Hydrogen sulfide accounts for the peripheral vascular effects of zofenopril independently of ACE inhibition. Cardiovasc. Res. 2014, 102, 138–147. [Google Scholar] [CrossRef]
- Otoo, R.A.; Allen, A.R. Sulforaphane’s Multifaceted Potential: From Neuroprotection to Anticancer Action. Molecules 2023, 28, 6902. [Google Scholar] [CrossRef]
- Pan, Y.; Ye, S.; Yuan, D.; Zhang, J.; Bai, Y.; Shao, C. Hydrogen sulfide (H2S)/cystathionine gamma-lyase (CSE) pathway contributes to the proliferation of hepatoma cells. Mutat. Res. 2014, 763–764, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Conzatti, A.; Colombo, R.; Siqueira, R.; Campos-Carraro, C.; Turck, P.; Luz de Castro, A.; Bello-Klein, A.; Sander da Rosa Araujo, A. Sulforaphane Improves Redox Homeostasis and Right Ventricular Contractility in a Model of Pulmonary Hypertension. J. Cardiovasc. Pharmacol. 2024, 83, 612–620. [Google Scholar] [CrossRef]
- Zhang, G.; Kang, Y.; Cathey, D.; LeBlanc, A.J.; Cai, J.; Cai, L.; Wang, S.; Huang, J.; Keller, B.B. Sulforaphane Does Not Protect Right Ventricular Systolic and Diastolic Functions in Nrf2 Knockout Pulmonary Artery Hypertension Mice. Cardiovasc. Drugs Ther. 2022, 36, 425–436. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| PH Model | mPAP Control mmHg | mPAP 1 PH mmHg | mPAP PH + H2S mmHg | mPAP PH + PPG mmHg | Plasma H2S Control μmol/L | Plasma H2S PH μmol/L | Plasma 2 H2S + PH + NaHS | Plasma 3 H2S + PH + PPG | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| CH 3 weeks Wistar | 16.3 | 23.7 | 16.3 | 301 | 192 | 317 | [5] | ||
| 14.8 | 20.5 | 14.4 | 25.8 | 294 | 196 | 324 | 142 | [135] | |
| 15.7 | 23.7 | 16.3 | 299 | 195 | 271 | [136] | |||
| 16.3 | 23.7 | 16.3 | 300 | 187 | 309 | [137] | |||
| CH 4 weeks Sprague- Dawley | ≈14 4 | ≈34 | ≈26 | ≈152 | ≈96 | ≈130 | [138] | ||
| CH 3 weeks broilers | ≈23 | ≈37 | ≈25 | [131] | |||||
| AC shunt 11 weeks Sprague-Dawley (SD) | 15.9 | 23.6 | 51 | 36 | [22] | ||||
| 15.8 | 23.7 | 51 | 36 | [139] | |||||
| 26.3 | 39.1 | 31.3 | [134] | ||||||
| 17.3 | 27.6 | 24.2 | [140] | ||||||
| 17.1 | 27.5 | 23.2 | [141] | ||||||
| AC shunt 4 weeks SD | 15.5 | 16.4 | 19.5 | [142] | |||||
| 15.5 | 16.2 | 20.3 | [141] | ||||||
| MC 3 weeks Wistar | ≈22 | ≈34 | ≈22 | [143] | |||||
| MC 3 weeks Wistar | ≈15 | ≈51 | ≈37 | ≈14 | ≈9 | ≈12 | [133] | ||
| MC 3 weeks Wistar | ≈18 | ≈38 | ≈25 | [144] | |||||
| MC 3 weeks Wistar | ≈17 | ≈34 | ≈25 | ≈12 | ≈4 | ≈14 | [145] | ||
| MC 3 weeks SD | ≈15 | ≈36 | ≈26 | ≈43 | 30 | 16 | [132] | ||
| MC 3 weeks SD | ≈13 | ≈38 | ≈22 | [146] |
| PH Model | Effect of PH and Sulfide Treatment on CSE mRNA and/or Protein Expression in Lung Tissue. | Lung Sulfide Production nmole/mg Wet wt /min | Effects of Treatment with Sulfide, and Other Agents on Changes in PA Phenotype Induced by CH or Tobacco Smoke | Ref. | ||
|---|---|---|---|---|---|---|
| PH vs. Control | PH + H2S Vs. PH | Control | PH | |||
| CH 3 weeks Wistar rats | ↓ mRNA, protein | ↑ mRNA, protein | 0.278 | 0.127 | [5] | |
| CH ↑ plasma [CO] and PA expression of HO-1. Treatment with 14 mg/kg/day NaHS further ↑ [CO] and HO-1 expression. | [135] | |||||
| CH ↑ PA expression of urotensin 2, collagens 1 & 3, elastin, TGFβ3, PCNA, procollagens 1 & 3. Treating with 14 mg/kg/day NaHS ↓all of these effects. | [136] | |||||
| 0.289 | 0.187 | CH ↑ oxidized glutathione and malonaldehyde and ↓ total antioxidant capacity in lung tissue. 14 mg/kg/day NaHS treatment ↓ the effect of PH on total antioxidant capacity & oxidized glutathione | [137] | |||
| CH 4 weeks SD | CH ↑ expression of endoplasmic reticulum stress-related proteins ATF6 & GRP78. These effects were absent in rats treated with GYY4137 (dose not stated). | [138] | ||||
| CH 3 weeks broilers | ↓ mRNA, protein | ≈0.28 | ≈0.12 | [131] | ||
| PH Model | Effect of PH on CSE mRNA and/or Protein Expression in Lung Tissue. | Lung Sulfide Production in nmol/(g wet wt/min) or Content in μmol/mg 1 | Effects of Treatment with Sulfide, and Other Agents on Changes in PA Phenotype Induced by Aortocaval Shunting | Ref. | |
|---|---|---|---|---|---|
| PH Vs. Control | Control | PH | |||
| AC shunt 11 weeks SD rats | ↓ mRNA in PA, ↓ protein in lung tissue | 0.26 | 0.13 | [22] | |
| ↓ mRNA in lung tissue | 0.26 | 0.13 | L-arginine (1 g/kg body weight) given daily following shunting prevented the ↑ in PAP, PA remodeling and ↓ in lung CSE expression and plasma [sulfide] induced by shunting. | [139] | |
| 30.2 | 20.2 | Shunting ↑ PCNA, phosphorylation of extracellular signal-related kinase (ERK), endothelial nitric oxide synthase (eNOS), and nitric oxide (NO) synthesis by lung tissue. NaHS 56 mg/kg/day ↓ these effects, also ↑ HO-1 expression and CO production in lung tissue. | [134] | ||
| 30.2 | 20.2 | Shunting ↑ expression of collagens 1 and 3, matrix metalloproteinase-13, tissue inhibitor of metalloproteinase 1, connective tissue growth factor in intra-acinar PA. ↑ hydroxyproline content in lung tissue. ↑ plasma [endothelin-1] and endothelin-1 mRNA in lung tissue. NaHS 56 mg/kg/day ↓ all of these effects. | [140] | ||
| 32.8 | 24.2 | Shunting ↓ proportion of apoptotic pulmonary artery smooth muscle cells in lung sections and ↓expression of Fas and caspase-3 and ↑ expression of bcl2. NaHS 56 mg/kg/day following shunting ↑ proportion of apoptotic cells, Fas and caspase 3 expression and ↓ bcl2 expression. | [141] | ||
| AC shunt 4 weeks SD rats | 14.4 | 37.6 | Shunting ↑ NO production and eNOS expression, and ↓ CO production and HO-1 expression in lung tissue. PPG mg/kg/day I.P.) following shunting exaggerated these effects and also ↑ PCNA expression & ERK phosphorylation. | [142] | |
| 23.6 | 32.7 | Shunting ↓ proportion of apoptotic PASMCs in lung sections and ↓ expression of Fas and caspase-3 and ↑ expression of bcl2. PPG (37.5 mg/kg/day injected I.P.) ↓ proportion of apoptotic PASMCs, Fas and caspase 3 expression, ↑ bcl2 expression. | [141] | ||
| PH Model | Effect of PH and Sulfide Treatment on CSE mRNA and/or Protein Expression in Lung Tissue. | Lung Sulfide Production or Lung Sulfide Content | Effects of Treatment with Sulfide and Other Agents on Changes in PA Phenotype Induced by MC | Ref. | ||
|---|---|---|---|---|---|---|
| Effect of PH vs. Control | H2S Effect vs. PH | Control | PH | |||
| MC 3 weeks Wistar rats | 2.28 nmol/mg/ protein/min | 1.55 nmol/mg/ protein/min | MC ↓ vasoconstrictions to high K+ PSS and phenylephrine and also ↓ vasorelaxations to acetylcholine and cysteine. These effects were all greatly ↓ in rats treated with Na2S (2.5 mg/kg/day). | [143] | ||
| MC 3 weeks Wistar rats | ↓ CSE protein in lung tissue | ↑ CSE protein in lung tissue | ≈1.0 μmol/g protein | ≈0.45 μmol/g protein | MC ↑ expression of inflammation markers intercellular adhesion molecule-1 (ICAM-1), tumor necrosis factor α (TNFα), interleukins 6 and 8 (IL-6, IL-8), monocyte chemoattractant protein-1 (MCP-1) in plasma & lung tissue & activated activity of nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) in lung tissue. These effects were all greatly ↓in rats treated with NaHS (56 μmol/kg/day). | [133] |
| MC 3 weeks Wistar rats | ↓ CSE protein and activity in lung tissue | MC↑ expression of ICAM-1, TNFα, IL-6 in lung tissue and plasma; also ↑ NFκB activity in lung tissue. These effects were ↓in rats treated with NaHS (56 μmol/kg/day) and in rats in which a persufidation-resistant variant of NFκB was expressed in PA. | [145] | |||
| MC 3 weeks SD rats | ↓ CSE protein and activity in lung tissue | MC↓ expression of VE-cadherin and ↑ expression of a-smooth muscle actin, TGFβ1 and Snail-1 in PA; also ↑ activity of NFκB. These effects were ↓ in rats treated with NaHS (56 μmol/kg/day) and were ↑ in rats treated with PPG (10 mg/kg/day). | [132] | |||
| MC 3 weeks SD rats | MC↓ expression of VE-cadherin and ↑ expression of a-smooth muscle actin and Snail-1 in PA; also ↑ activity of NFκB. These effects were ↓ in rats treated with porous microspheres containing H2S-releasing drug ACS14. | [146] | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aaronson, P.I. The Role of Hydrogen Sulfide in the Regulation of the Pulmonary Vasculature in Health and Disease. Antioxidants 2025, 14, 341. https://doi.org/10.3390/antiox14030341
Aaronson PI. The Role of Hydrogen Sulfide in the Regulation of the Pulmonary Vasculature in Health and Disease. Antioxidants. 2025; 14(3):341. https://doi.org/10.3390/antiox14030341
Chicago/Turabian StyleAaronson, Philip I. 2025. "The Role of Hydrogen Sulfide in the Regulation of the Pulmonary Vasculature in Health and Disease" Antioxidants 14, no. 3: 341. https://doi.org/10.3390/antiox14030341
APA StyleAaronson, P. I. (2025). The Role of Hydrogen Sulfide in the Regulation of the Pulmonary Vasculature in Health and Disease. Antioxidants, 14(3), 341. https://doi.org/10.3390/antiox14030341