

Lung EC-SOD Overexpression Prevents Hypoxia-Induced Platelet Activation and Lung Platelet Accumulation

 , , , , ,
, , , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
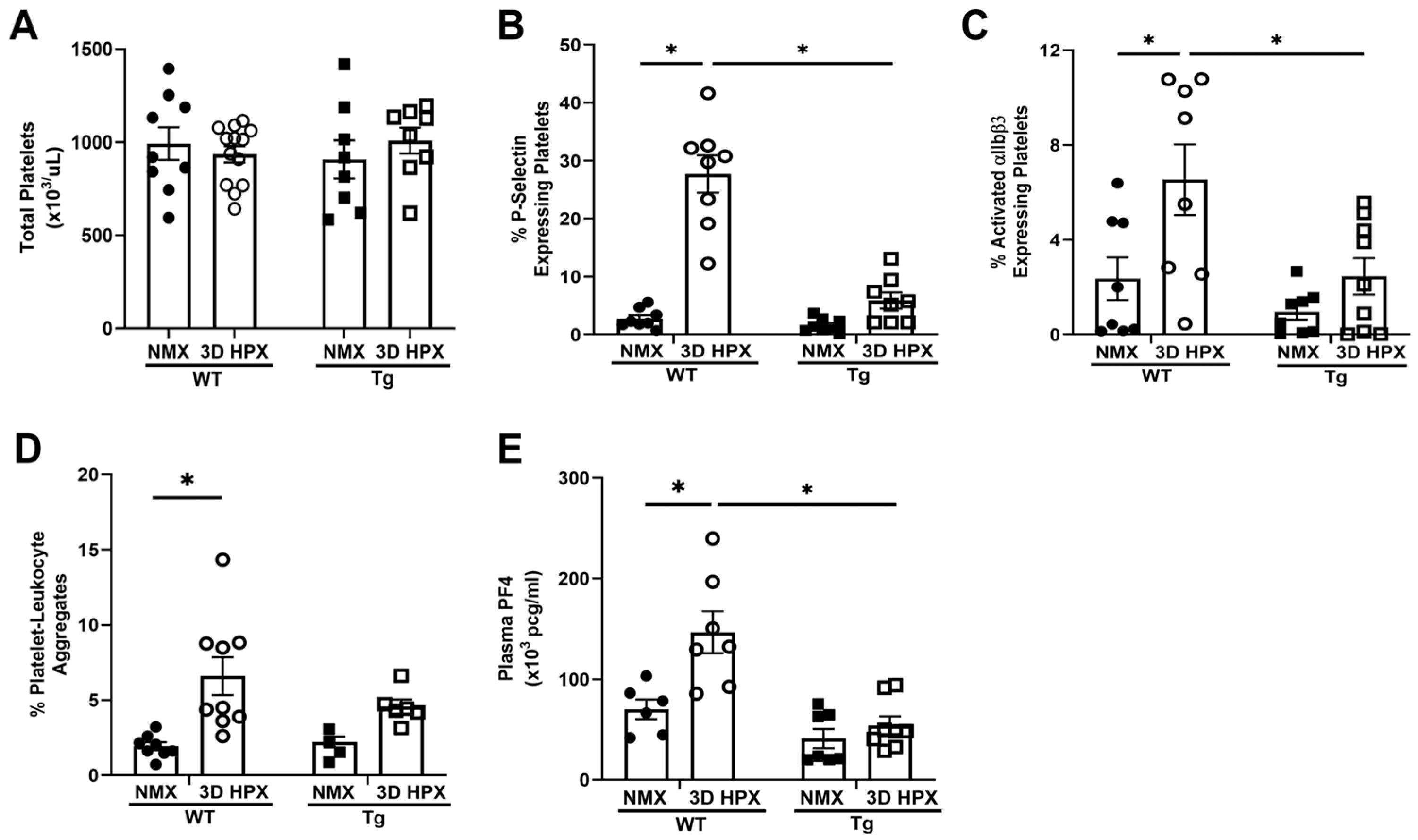
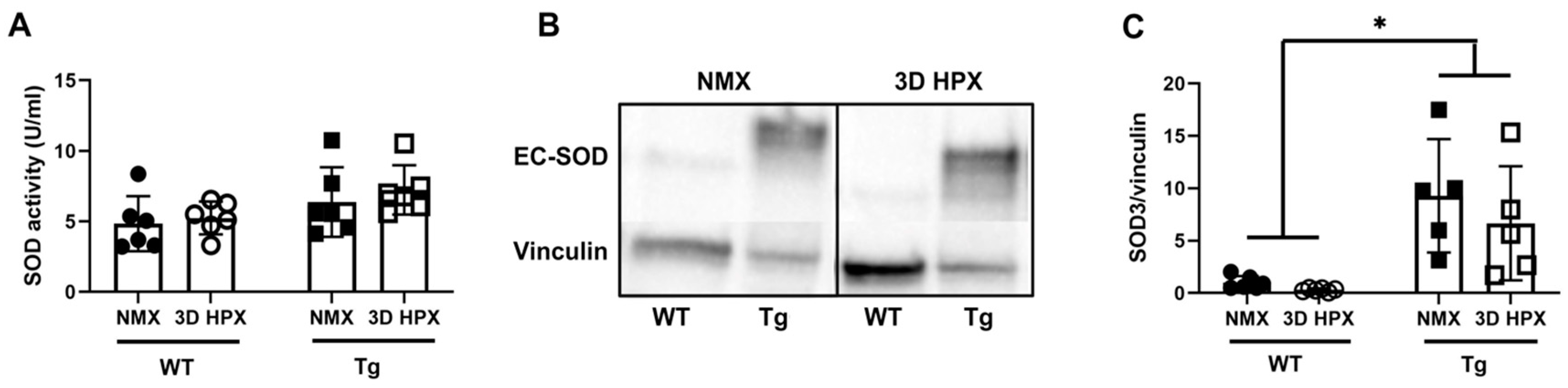
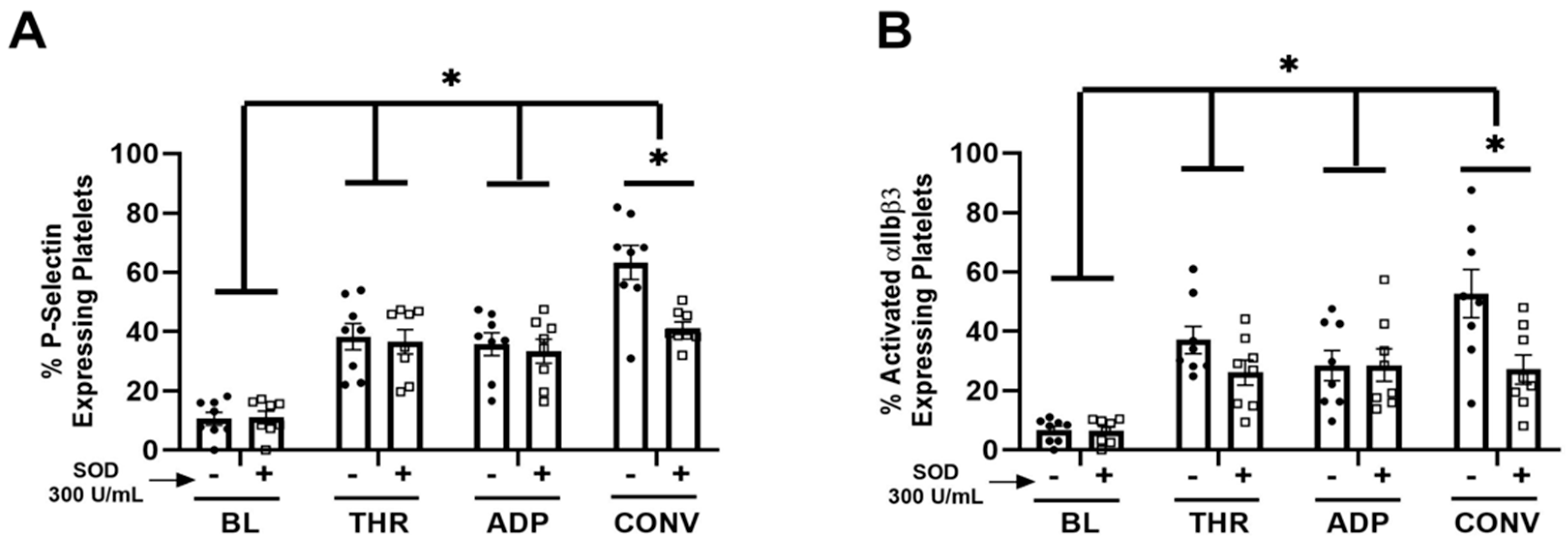
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Taichman, D.B.; Mandel, J. Epidemiology of pulmonary arterial hypertension. Clin. Chest Med. 2013, 34, 619–637. [Google Scholar] [CrossRef]
- Thenappan, T.; Ormiston, M.L.; Ryan, J.J.; Archer, S.L. Pulmonary arterial hypertension: Pathogenesis and clinical management. BMJ 2018, 360, j5492. [Google Scholar] [CrossRef]
- Hassoun, P.M. Pulmonary Arterial Hypertension. N. Engl. J. Med. 2021, 385, 2361–2376. [Google Scholar] [CrossRef] [PubMed]
- Frost, A.; Badesch, D.; Gibbs, J.S.R.; Gopalan, D.; Khanna, D.; Manes, A.; Oudiz, R.; Satoh, T.; Torres, F.; Torbicki, A. Diagnosis of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801904. [Google Scholar] [CrossRef] [PubMed]
- Firth, A.L.; Mandel, J.; Yuan, J.X.-J. Idiopathic pulmonary arterial hypertension. Dis. Model. Mech. 2010, 3, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-F.; Veetil, N.N.; Li, Q.; Kucherenko, M.M.; Knosalla, C.; Kuebler, W.M. Pulmonary hypertension: Linking inflammation and pulmonary arterial stiffening. Front. Immunol. 2022, 13, 959209. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, S.C.; Poth, J.M.; Fini, M.A.; Olschewski, A.; El Kasmi, K.C.; Stenmark, K.R. The role of inflammation in hypoxic pulmonary hypertension: From cellular mechanisms to clinical phenotypes. Am. J. Physiol. Cell. Mol. Physiol. 2015, 308, L229–L252. [Google Scholar] [CrossRef] [PubMed]
- El Kasmi, K.C.; Pugliese, S.C.; Riddle, S.R.; Poth, J.M.; Anderson, A.L.; Frid, M.G.; Li, M.; Pullamsetti, S.S.; Savai, R.; Nagel, M.A.; et al. Adventitial Fibroblasts Induce a Distinct Proinflammatory/Profibrotic Macrophage Phenotype in Pulmonary Hypertension. J. Immunol. 2014, 193, 597–609. [Google Scholar] [CrossRef]
- Liang, S.; Desai, A.A.; Black, S.M.; Tang, H. Cytokines, Chemokines, and Inflammation in Pulmonary Arterial Hypertension. In Lung Inflammation in Health and Disease, Volume I. Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2021; Volume 1303. [Google Scholar] [CrossRef]
- Zhao, H.; Song, J.; Li, X.; Xia, Z.; Wang, Q.; Fu, J.; Miao, Y.; Wang, D.; Wang, X. The role of immune cells and inflammation in pulmonary hypertension: Mechanisms and implications. Front. Immunol. 2024, 15, 1374506. [Google Scholar] [CrossRef] [PubMed]
- Tamosiuniene, R.; Tian, W.; Dhillon, G.; Wang, L.; Sung, Y.K.; Gera, L.; Patterson, A.J.; Agrawal, R.; Rabinovitch, M.; Ambler, K.; et al. Regulatory T cells limit vascular endothelial injury and prevent pulmonary hypertension. Circ. Res. 2011, 109, 867–879. [Google Scholar] [CrossRef]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive oxygen species in metabolic and inflammatory signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Reszka, K.J.; Fukai, T.; Weintraub, N.L. Extracellular superoxide dismutase (ecSOD) in vascular biology: An update on exogenous gene transfer and endogenous regulators of ecSOD. Transl. Res. 2007, 151, 68–78. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [PubMed]
- Sandström, J.; Nilsson, P.; Karlsson, K.; Marklund, S. 10-Fold increase in human plasma extracellular superoxide dismutase content caused by a mutation in heparin-binding domain. J. Biol. Chem. 1994, 269, 19163–19166. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Spaulding, H.R. Extracellular superoxide dismutase, a molecular transducer of health benefits of exercise. Redox Biol. 2020, 32, 101508. [Google Scholar] [CrossRef] [PubMed]
- Mouradian, G.C.; Gaurav, R.; Pugliese, S.; El Kasmi, K.; Hartman, B.; Hernandez-Lagunas, L.; Stenmark, K.R.; Bowler, R.P.; Nozik-Grayck, E. Superoxide dismutase 3 R213G single-nucleotide polymorphism blocks murine bleomycin-induced fibrosis and promotes resolution of inflammation. Am. J. Respir. Cell Mol. Biol. 2017, 56, 362–371. [Google Scholar] [CrossRef]
- Elajaili, H.; Hernandez-Lagunas, L.; Harris, P.; Sparagna, G.C.; Jonscher, R.; Ohlstrom, D.; Sucharov, C.C.; Bowler, R.P.; Suliman, H.; Fritz, K.S.; et al. Extracellular superoxide dismutase (EC-SOD) R213G variant reduces mitochondrial ROS and preserves mitochondrial function in bleomycin-induced lung injury: EC-SOD R213G variant and intracellular redox regulation. Adv. Redox Res. 2022, 5, 100035. [Google Scholar] [CrossRef] [PubMed]
- Sul, C.; Lewis, C.; Dee, N.; Burns, N.; Oshima, K.; Schmidt, E.; Vohwinkel, C.; Nozik, E. Release of extracellular superoxide dismutase into alveolar fluid protects against acute lung injury and inflammation in Staphylococcus aureus pneumonia. Am. J. Physiol. Cell. Mol. Physiol. 2023, 324, L445–L455. [Google Scholar] [CrossRef]
- Nozik-Grayck, E.; Suliman, H.B.; Majka, S.; Albietz, J.; Van Rheen, Z.; Roush, K.; Stenmark, K.R. Lung EC-SOD overexpression attenuates hypoxic induction of Egr-1 and chronic hypoxic pulmonary vascular remodeling. Am. J. Physiol. Cell. Mol. Physiol. 2008, 295, L422–L430. [Google Scholar] [CrossRef]
- Deppermann, C.; Kubes, P. Start a fire, kill the bug: The role of platelets in inflammation and infection. J. Endotoxin Res. 2018, 24, 335–348. [Google Scholar] [CrossRef]
- Rayes, J.; Bourne, J.H.; Brill, A.; Watson, S.P. The dual role of platelet-innate immune cell interactions in thrombo-inflammation. Res. Pract. Thromb. Haemost. 2020, 4, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Scherlinger, M.; Richez, C.; Tsokos, G.C.; Boilard, E.; Blanco, P. The role of platelets in immune-mediated inflammatory diseases. Nat. Rev. Immunol. 2023, 23, 495–510. [Google Scholar] [CrossRef]
- Sonmez, O.; Sonmez, M. Role of platelets in immune system and inflammation. Porto Biomed. J. 2017, 2, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Canobbio, I.; Balduini, C.; Torti, M. Signalling through the platelet glycoprotein Ib-V–IX complex. Cell. Signal. 2004, 16, 1329–1344. [Google Scholar] [CrossRef] [PubMed]
- Estevez, B.; Du, X. New concepts and mechanisms of platelet activation signaling. Physiology 2017, 32, 162–177. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.-H.; Sim, E.-H.; Goh, R.-Y.; Park, J.-I.; Han, J.-Y. Platelet activation: The mechanisms and potential biomarkers. BioMed Res. Int. 2016, 2016, 9060143. [Google Scholar] [CrossRef] [PubMed]
- Lannan, K.L.; Phipps, R.P.; White, R.J. Thrombosis, platelets, microparticles and PAH: More than a clot. Drug Discov. Today 2014, 19, 1230–1235. [Google Scholar] [CrossRef] [PubMed]
- Åberg, M.; Björklund, E.; Wikström, G.; Christersson, C. Platelet-leukocyte aggregate formation and inflammation in patients with pulmonary arterial hypertension and CTEPH. Platelets 2022, 33, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Delaney, C.; Davizon-Castillo, P.; Allawzi, A.; Posey, J.; Gandjeva, A.; Neeves, K.; Tuder, R.M.; Di Paola, J.; Stenmark, K.R.; Nozik, E.S. Platelet activation contributes to hypoxia-induced inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 320, L413–L421. [Google Scholar] [CrossRef]
- Gu, S.X.; Dayal, S. Redox Mechanisms of Platelet Activation in Aging. Antioxidants 2022, 11, 995. [Google Scholar] [CrossRef] [PubMed]
- Pietraforte, D.; Vona, R.; Marchesi, A.; de Jacobis, I.T.; Villani, A.; Del Principe, D.; Straface, E. Redox control of platelet functions in physiology and pathophysiology. Antioxid. Redox Signal. 2014, 21, 177–193. [Google Scholar] [CrossRef]
- Essex, D.W. Redox control of platelet function. Antioxid. Redox Signal. 2009, 11, 1191–1225. [Google Scholar] [CrossRef] [PubMed]
- Nozik-Grayck, E.; Woods, C.; Taylor, J.M.; Benninger, R.K.P.; Johnson, R.D.; Villegas, L.R.; Stenmark, K.R.; Harrison, D.G.; Majka, S.M.; Irwin, D.; et al. Selective depletion of vascular EC-SOD augments chronic hypoxic pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L868–L876. [Google Scholar] [CrossRef]
- Tseng, V.; Ni, K.; Allawzi, A.; Prohaska, C.; Hernandez-Lagunas, L.; Elajaili, H.; Cali, V.; Midura, R.; Hascall, V.; Triggs-Raine, B.; et al. Extracellular Superoxide Dismutase Regulates Early Vascular Hyaluronan Remodeling in Hypoxic Pulmonary Hypertension. Sci. Rep. 2020, 10, 280. [Google Scholar] [CrossRef] [PubMed]
- Hartney, J.M.; Stidham, T.; Goldstrohm, D.A.; Oberley-Deegan, R.E.; Weaver, M.R.; Valnickova-Hansen, Z.; Scavenius, C.; Benninger, R.K.; Leahy, K.F.; Johnson, R.; et al. A common polymorphism in extracellular superoxide dismutase affects cardiopulmonary disease risk by altering protein distribution. Circ. Cardiovasc. Genet. 2014, 7, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Delaney, C.; Wright, R.H.; Tang, J.-R.; Woods, C.; Villegas, L.; Sherlock, L.; Savani, R.C.; Abman, S.H.; Nozik-Grayck, E. Lack of EC-SOD worsens alveolar and vascular development in a neonatal mouse model of bleomycin-induced bronchopulmonary dysplasia and pulmonary hypertension. Pediatr. Res. 2015, 78, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Davizon-Castillo, P.; Allawzi, A.; Sorrells, M.; Fisher, S.; Baltrunaite, K.; Neeves, K.; Nozik-Grayck, E.; DiPaola, J.; Delaney, C. Platelet activation in experimental murine neonatal pulmonary hypertension. Physiol. Rep. 2020, 8, e14386. [Google Scholar] [CrossRef]
- Stacher, E.; Graham, B.B.; Hunt, J.M.; Gandjeva, A.; Groshong, S.D.; McLaughlin, V.V.; Jessup, M.; Grizzle, W.E.; Aldred, M.A.; Cool, C.D.; et al. Modern Age Pathology of Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 261–272. [Google Scholar] [CrossRef]
- Oliveira, T.; Calderaro, D.; Piloto, B.; Castro, M.; Hoette, S.; Jardim, C.; Souza, R.; Fernandes, C.J.C. Platelets and pulmonary arterial hypertension (PAH). Eur. Respir. J. 2019, 54 (Suppl. S63), PA4756. [Google Scholar] [CrossRef]
- Zanjani, K.S. Platelets in pulmonary hypertension: A causative role or a simple association? Iran. J. Pediatr. 2012, 22, 145–157. [Google Scholar] [PubMed]
- Lopes, A.A.B.; Maeda, N.Y.; Ebaid, M.; Chamone, D.A. Aggregation of platelets in whole blood from children with pulmonary hypertension. Int. J. Cardiol. 1990, 28, 173–178. [Google Scholar] [CrossRef]
- Nicoleau, S.; Wojciak-Stothard, B. Beyond Thrombosis: The Role of Platelets in Pulmonary Hypertension. SciMedicine J. 2020, 2, 243–271. [Google Scholar] [CrossRef]
- Ferguson, S.K.; Redinius, K.; Yalamanoglu, A.; Harral, J.W.; Baek, J.H.; Pak, D.; Loomis, Z.; Hassell, D.; Eigenberger, P.; Nozik-Grayck, E.; et al. Effects of living at moderate altitude on pulmonary vascular function and exercise capacity in mice with sickle cell anaemia. J. Physiol. 2019, 597, 1073–1085. [Google Scholar] [CrossRef] [PubMed]
- Cullivan, S.; Murphy, C.A.; Weiss, L.; Comer, S.P.; Kevane, B.; McCullagh, B.; Maguire, P.B.; Ainle, F.N.; Gaine, S.P. Platelets, extracellular vesicles and coagulation in pulmonary arterial hypertension. Pulm. Circ. 2021, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Herve, P.; Humbert, M.; Sitbon, O.; Parent, F.; Nunes, H.; Legal, C.; Garcia, G.; Simonneau, G. Pathobiology of pulmonary hypertension: The role of platelets and thrombosis. Clin. Chest Med. 2001, 22, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Blaine, K.P.; Cullinane, A.; Hall, C.; Dulau-Florea, A.; Sun, J.; Chenwi, H.F.; Graninger, G.M.; Harper, B.; Thompson, K.; et al. Pulmonary arterial hypertension patients display normal kinetics of clot formation using thrombelastography. Pulm. Circ. 2021, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Awad, A.; Elnemr, S.; Hodeib, H.; El Amrousy, D. Platelet Activation Markers in Children with Pulmonary Arterial Hypertension Associated with Congenital Heart Disease. Pediatr. Cardiol. 2022, 43, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Q.L.; Corey, C.; White, P.; Watson, A.; Gladwin, M.T.; Simon, M.A.; Shiva, S. Platelets from pulmonary hypertension patients show increased mitochondrial reserve capacity. J. Clin. Insight 2017, 2, e91415. [Google Scholar] [CrossRef]
- Krötz, F.; Sohn, H.Y.; Gloe, T.; Zahler, S.; Riexinger, T.; Schiele, T.M.; Becker, B.F.; Theisen, K.; Klauss, V.; Pohl, U. NAD(P)H oxidase–dependent platelet superoxide anion release increases platelet recruitment. Blood 2002, 100, 917–924. [Google Scholar] [CrossRef]
- Sonkar, V.K.; Eustes, A.S.; Ahmed, A.; Jensen, M.; Solanki, M.V.; Swamy, J.; Kumar, R.; Fidler, T.P.; Houtman, J.C.; Allen, B.G.; et al. Endogenous SOD2 (Superoxide Dismutase) Regulates Platelet-Dependent Thrombin Generation and Thrombosis During Aging. Arter. Thromb. Vasc. Biol. 2023, 43, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Polgár, J.; Clemetson, J.M.; Kehrel, B.E.; Wiedemann, M.; Magnenat, E.M.; Wells, T.N.C.; Clemetson, K.J. Platelet activation and signal transduction by convulxin, a C-type lectin from Crotalus durissus terrificus (Tropical rattlesnake) venom via the p62/GPVI collagen receptor. J. Biol. Chem. 1997, 272, 13576–13583. [Google Scholar] [CrossRef] [PubMed]
- Rayes, J.; Watson, S.P.; Nieswandt, B. Functional significance of the platelet immune receptors GPVI and CLEC-2. J. Clin. Investig. 2019, 129, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Stegner, D.; Nieswandt, B. Platelet receptor signaling in thrombus formation. J. Mol. Med. 2011, 89, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, K.; Bhattacharyya, M. Overview of platelet physiology: Its hemostatic and nonhemostatic role in disease pathogenesis. Sci. World J. 2014, 2014, 781857. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Heemskerk, J.W.M.; Baaten, C.C.F.M.J. Platelet Membrane Receptor Proteolysis: Implications for Platelet Function. Front. Cardiovasc. Med. 2021, 7, 608391. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.K.; Karunakaran, D.; Gardiner, E.E.; Berndt, M.C. Platelet Receptor Proteolysis. Arter. Thromb. Vasc. Biol. 2007, 27, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- Bender, M.; Hofmann, S.; Stegner, D.; Chalaris, A.; Bösl, M.; Braun, A.; Scheller, J.; Rose-John, S.; Nieswandt, B. Differentially regulated GPVI ectodomain shedding by multiple platelet–expressed proteinases. Blood 2010, 116, 3347–3355. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, E.; Solouki, A.; Roudsari, Z.O.; Kargar, F.; Ghasemzadeh, M. Reducing state attenuates ectodomain shedding of GPVI while restoring adhesion capacities of stored platelets: Evidence addressing the controversy around the effects of redox condition on thrombosis. J. Thromb. Thrombolysis 2020, 50, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Montague, S.J.; Andrews, R.K.; Gardiner, E.E. Mechanisms of receptor shedding in platelets. Blood 2018, 132, 2535–2545. [Google Scholar] [CrossRef]
- Jiang, H.; Nechipurenko, D.Y.; Panteleev, M.A.; Xu, K.; Qiao, J. Redox regulation of platelet function and thrombosis. J. Thromb. Haemost. 2024, 22, 1550–1557. [Google Scholar] [CrossRef] [PubMed]
- Russo, I.; Barale, C.; Melchionda, E.; Penna, C.; Pagliaro, P. Platelets and Cardioprotection: The Role of Nitric Oxide and Carbon Oxide. Int. J. Mol. Sci. 2023, 24, 6107. [Google Scholar] [CrossRef] [PubMed]
- Cheung, P.-Y.; Salas, E.; Schulz, R.; Radomski, M.W. Nitric oxide and platelet function: Implications for neonatology. Semin. Perinatol. 1997, 21, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Gkaliagkousi, E.; Ritter, J.; Ferro, A. Platelet-derived nitric oxide signaling and regulation. Circ. Res. 2007, 101, 654–662. [Google Scholar] [CrossRef]
- Chavez, M.D.; Lakshmanan, N.; Kavdia, M. Impact of superoxide dismutase on nitric oxide and peroxynitrite levels in the microcirculation—A computational model. In Proceedings of the 2007 29th Annual International Conference of the IEEE Engineering in Medicine and Biology Society, Lyon, France, 22–26 August 2007; pp. 1022–1026. [Google Scholar]
- Beckman, J.S.; Koppenol, W.H. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and the ugly. Am. J. Physiol. Cell Physiol. 1996, 271, C1424–C1437. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colon Hidalgo, D.; Jordan, M.; Posey, J.N.; Burciaga, S.D.; Nguyen, T.-T.N.; Sul, C.; Lewis, C.V.; Delaney, C.; Nozik, E.S. Lung EC-SOD Overexpression Prevents Hypoxia-Induced Platelet Activation and Lung Platelet Accumulation. Antioxidants 2024, 13, 975. https://doi.org/10.3390/antiox13080975
Colon Hidalgo D, Jordan M, Posey JN, Burciaga SD, Nguyen T-TN, Sul C, Lewis CV, Delaney C, Nozik ES. Lung EC-SOD Overexpression Prevents Hypoxia-Induced Platelet Activation and Lung Platelet Accumulation. Antioxidants. 2024; 13(8):975. https://doi.org/10.3390/antiox13080975
Chicago/Turabian StyleColon Hidalgo, Daniel, Mariah Jordan, Janelle N. Posey, Samuel D. Burciaga, Thi-Tina N. Nguyen, Christina Sul, Caitlin V. Lewis, Cassidy Delaney, and Eva S. Nozik. 2024. "Lung EC-SOD Overexpression Prevents Hypoxia-Induced Platelet Activation and Lung Platelet Accumulation" Antioxidants 13, no. 8: 975. https://doi.org/10.3390/antiox13080975
APA StyleColon Hidalgo, D., Jordan, M., Posey, J. N., Burciaga, S. D., Nguyen, T.-T. N., Sul, C., Lewis, C. V., Delaney, C., & Nozik, E. S. (2024). Lung EC-SOD Overexpression Prevents Hypoxia-Induced Platelet Activation and Lung Platelet Accumulation. Antioxidants, 13(8), 975. https://doi.org/10.3390/antiox13080975